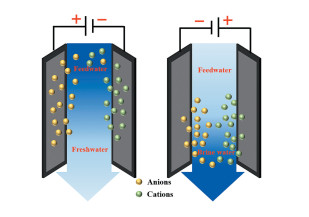
图 1.
电容去离子原理图
Figure 1.
Schematic diagram of capacitive deionization

近几十年中,水资源短缺逐渐成为威胁人类生存的严峻挑战。据统计,目前全球有超过三分之一的人口生活在缺水的地区,到2025年,这一数字预计将增长到三分之二。人口增长、工业化、气候变化,使得获取充足和安全的淡水资源变得愈加困难[1]。由于资源丰富,对海水或微咸水进行脱盐有助于增加淡水供应[2, 3]。
电容去离子(Capacitive deionization,CDI)是一种基于超级电容器原理发展起来的绿色脱盐技术。其技术原理如图 1:当盐离子通过一对或多对平行的涂覆有活性材料的电极时,施加在电极上的偏压电场会使得溶液中的正、负离子被相应地吸附到负、正电极表面,从而实现对溶液的脱盐。离子吸附达到饱和后,对电极施加反向电压或短路,吸附的离子可以解脱下来以实现电极的再生[4]。相比于传统脱盐技术,如多级闪蒸和反渗透等,CDI具有操作简单、无二次污染和能耗低(见表 1)等优势[5~9]。
 下载:
导出CSV
下载:
导出CSV
| 技术种类 | 热能消耗/(MJ/m3) | 盐水温度/℃ | 电能消耗/(kW·h)/m3 | 总体能耗/(kW·h)/m3 |
| 反渗透(RO) | 无 | 与进水温度一致 | 1.5~6 | 1.5~6 |
| 多级闪蒸(MSF) | 190~282 | 90~120 | 2.5~5 | 18.3~28.5 |
| 多效蒸馏(MED) | 145~230 | 50~90 | 2~2.5 | 14.2~21.6 |
| 电渗析(ED) | 无 | < 45 | 0.7~5.5 | 0.7~5.5 |
| 电容去离子(CDI) | 无 | 与进水温度一致 | 0.1~2.03 | 0.1~2.03 |
CDI的除盐性能强烈依赖于电极材料,具有高比电容、高电吸附能力和高脱盐速率的电极材料是提升CDI脱盐性能的核心。有效的CDI电极材料应具有高电导率、较大的比电容、合适的孔径分布、高比表面积以及优异的电化学稳定性等特点[10]。
电极的比电容是开发出具有高除盐性能的电极材料的基础。可在三电极体系中进行循环伏安分析初步得出电极的电容特性。比电容计算公式如下[11]:
|
|
式中,ΔV为电势窗口(V);I为响应电流(A);m为电极材料的质量(g);v为扫描速率(mV/s)。
按照机理可将材料的电容分为双电层电容与赝电容。在进行循环伏安分析或除盐测试时,材料表面可形成双电层,由于外加电压可使离子在电极表面发生氧化还原反应或进入电极材料内部发生嵌入/脱嵌的过程,因而使其具有赝电容特性。可初步根据循环伏安曲线的氧化还原峰来判断电极材料是否发生了氧化还原反应[12]。
在CDI领域,进行电化学测试时通常会选取不同浓度的NaCl作为电解质溶液,即在中性盐溶液中进行电化学表征,以更好地反应电极材料对于Na+的电化学响应特性。比电容计算结果会和超级电容器领域有所不同,这是因为在超级电容器领域,可以选取碱性(例如KOH)、酸性(例如H2SO4)、中性电解质(例如Na2SO4)溶液来进行电化学表征;而Na+的水合半径、扩散速率与其他离子有所不同[13],Na+水合半径较大、极限扩散系数较低致使其反应动力学缓慢,因此用NaCl作为电解质溶液计算出的比电容可能会低于其他体系中计算出的比电容值。
盐吸附容量(salt adsorption capacity,SAC)作为常规的电极材料性能评价指标,常以mg/g为单位,其计算公式为[14]:
|
|
式中,Mw是NaCl的分子量(58.44g/mol),Me是电极质量(g),ci是出水NaCl浓度(mmol/L),cd是进水NaCl浓度(mmol/L),Q为流速(mL/min)。
SAC通常取决于电压、进水浓度、停留时间等。一般来说,电压越大,静电吸引力越强,吸附容量越高。理论上,应该将电压控制在1.23V以下,即不超过水的理论分解电压。但在实际操作中由于电路间存在电阻,施加的电压也会超过1.23V,根据电极材料的不同,可将电压控制在2V以内波动。适当提高进水浓度会提高电吸附容量,这是因为离子间阻力变小,使其到达电极表面或内部的速度变快。充分的停留时间有利于离子和电极材料的充分接触,有利于吸附效果的提升。
除以上所述,SAC也会受到电极孔径分布的影响,一般认为,介孔(2~50 nm)有利于离子的吸附。但也有不同观点,Porada等[15]通过实验分析表明,SAC与多孔碳的孔径分布有很强的线性关系,小于1.1nm的孔隙比较大的孔隙对淡化过程的贡献更大。一般认为,比表面积大有利于提高离子和溶液的接触面积。但在控制平均孔径相似的前提下,对平均孔径相近但比表面积(712、1062和1347 m2/g)不同的碳材料的电吸附性能进行比较发现,电吸附容量随比表面积的增加而缓慢增加,三种碳材料的电吸附容量只有很细微的差异[16]。
在实际测量中,SAC主要根据电导率的变化来用于评价NaCl的去除效果,但考虑到实际水体的复杂性,则无法使用电导率来反映SAC。例如,当给水中同时存在K+、Ca2+等阳离子时,就需要借助离子色谱来分别对不同离子的吸附容量进行评价。同时,对于材料的孔径结构、表面积对SAC的贡献,学者之间有不同的见解,没有统一的标准。因此笔者认为需要将理论分析与大量的实验验证相结合,设计出合理孔隙结构的电极材料以达到优先去除某种离子或达到选择性去除的目的。
SAC只能分析出CDI体系中有多少盐离子被吸附,而盐离子去除速率(salt adsorption rate,SAR,通常以mg·g-1·min-1或mg·g-1·s-1为单位,其中“mg”表示去除的盐的质量,“g”表示两块电极的质量,“min”或“s”指充电时间[17])能反映出盐离子吸附的快慢。SAR受多种因素影响。充电方式会对离子吸附容量或速率产生直接影响。Kang等[18]研究了恒电压模式(CV)和恒电流操作模式(CC)下离子去除容量的区别,结果表明,在CV模式下达到最小电导率水平的时间比CC模式下短。这是因为在CV模式下,在充电步骤开始时便施加了很强的初始电压(1.2V),具有很强的静电吸引力,大量离子被迅速吸附,因此在给定的充电时间下盐吸附速度更快。电极厚度对盐的电吸附和电荷转移速率都有很大的影响。通过增加电极的厚度,CDI脱盐速率减慢,但对SAC几乎没影响[15]。
已有大量文献针对离子去除速率进行了系统研究,这些结果对CDI的实际应用具有重要意义。由于操作模式和电极材料的多样性,因此有必要将不同的操作模式进行耦合,例如CV和CC模式的复合可能会充分发挥CV模式下去除速率高以及CC模式下能耗低的优点。此外,应针对不同特性的电极材料进行大量重复实验,有必要将理论与实际紧密结合,通过建模来更详细地了解电吸附过程,最终达到优化吸附动力学过程的目的。
在传统的CDI中,电极材料一般都以碳基材料为主,例如活性炭(AC)[19]、石墨烯[20]和碳纳米管(CNT)[21]等。然而,由于离子存储容量有限,碳材料不适合于高浓度盐水除盐。此外,由于碳基材料阴极上会生成H2O2腐蚀电极以及阳极上的碳会发生氧化(见式(1)和式(2)[22]),导致碳材料的循环稳定性有限,这阻碍了它们在CDI领域的实际应用。
阴极(充电过程中带负电):
|
|
(1) |
阳极(在充电过程中带正电):
|
|
(2) |
为了解决上述问题,在“脱盐电池”[23]概念的基础上,具有离子嵌入/脱嵌能力的电极材料已成为具有很高比容量的新型CDI电极材料[24, 25]。与基于双电层的吸附机制相比,将离子存储在材料的晶体学位点或原子平面之间可以实现更高的盐存储容量、更高的能量密度,即使在盐浓度较高的情况下也可以实现有效脱盐[26]。该类电极材料在CDI领域的开发缓解了传统碳基电极吸附容量有限的问题。
本文将以过渡金属氧化物、MXenes、NASICON型磷酸盐材料等为分类,介绍近几年具有代表性的基于离子嵌入/脱嵌的电极材料的设计及在CDI领域的应用,以期深入理解构效关系,开发出性能更优的电极材料。
迄今为止,过渡金属氧化物是具有离子嵌入/脱嵌赝电容特性的被研究最广泛的材料[27]。它们具有出色的化学稳定性、良好的润湿性、高比电容以及高活性表面[28]。MnO2由于其低成本、来源丰富、环境友好和理论电容值(1370F/g)高等优势,成为被广泛研究的过渡金属氧化物电极材料之一[29]。
MnO2是一种多晶形化合物,晶体结构的基本骨架单元是锰氧八面体[MnO6]。由于MnO6八面体的互连方式不同,MnO2能以几种晶体形式存在,分别为α、β、γ、δ和λ形式。α、β和γ的形式均具有一维(1D)隧道结构,δ是二维(2D)层状结构,λ是3D尖晶石结构。根据不同的反应条件,MnO2可形成线状、片状、棒状、球状等不同的形态,这些不同的形态使它们在储能领域具有广泛的应用[30~32]。
Lee等[33]首次研究了KCl水性电解质中无定形MnO2·nH2O的电化学性质,从此诱发了MnO2在电化学领域的研究。到目前为止,提出了两种机制来解释MnO2的电荷存储行为。(1)质子(H+)或碱金属阳离子(C+)(例如Li+)嵌入材料的主体中,然后氧化脱嵌;(2)基于电解质阳离子(C+)在MnO2上的表面吸附(其中C+=Na+,K+,Li+等)[34]。
|
|
(1) |
|
|
(2) |
Tan等[35]将MnO2与通过原位化学氧化聚合方法制备的聚吡咯接枝活性炭(PPy/AC)复合材料耦合制备了无膜CDI电极。研究发现,MnO2电极具有较低的电荷转移和离子扩散阻力,此外,由于用PPy/AC代替了离子交换膜,使得两个电极之间的距离缩小,有利于离子的快速传输。10% PPy /AC-MnO2电池的最大除盐能力为52.93mg/g,在50个循环内除盐能力可维持稳定。Li等[14]将α-MnO2原位生长在活性炭布(ACC)基质上,优化后的ACC/α-MnO2比电容高达311F/g,由ACC/α-MnO2和ACC/Ag组成的CDI体系具有83%的充电效率,在1.2V电压下,盐吸附能力为17.8mg/g,500次循环后可维持初始除盐容量的70%。
虽然MnO2理论比电容优越,但实际应用中其比电容与理论值相差甚远,主要是因为MnO2导电性差。而MnO2作为多晶型化合物,其合成方法与形态具有多样性,因此在后续的研究工作中如何通过材料复合的方式将导电基体与MnO2复合以弥补其导电性差的缺陷,对于充分发挥MnO2的赝电容特性具有重要意义。
MAX相是一种三元陶瓷材料,其中M代表过渡金属元素(如Ti、Ta),A代表第三主族或第四主族元素(如Al、Ga、Si或Ge),X代表C或N元素[36]。通常,通过从Mn+1AXn相的层状三元材料中选择性去除A元素可得到比表面积较大的呈2D纳米薄片状的MXenes,其通式为Mn+1Xn或Mn+1XnTx(n=1~3),其中M是过渡金属(如Sc,Ti,Zr,V,Nb,Mo或Cr),X为C或N,Tx代表表面基团-F、-OH或-O等。据报道,MXenes在硫酸锂或硫酸钠水介质中显示出较高的赝电容特性(>300F/cm3),同时,MXenes具有形成无粘结剂电极的独特能力,可作为快速可逆地插入离子的理想的插层材料。自2011年首次由Gogotsi等成功制备以来,MXenes由于其独特的电子、机械、光学等特性而在储能、催化、传感器和其他领域中显示出广阔的应用前景[37~39]。
Chen等[40]用HF蚀刻Ti3AlC2,并将产物用NaOH处理,得到Na+插层的Ti3C2Tx(NaOH-Ti3C2Tx),将其用作阴极,与活性炭(AC)耦合组装成不对称CDI体系。NaOH-Ti3C2Tx上带负电荷的表面基团(-OH、-O和-F)会实现阳离子选择性并会弱化共离子效应;NaOH处理扩大了Ti3C2Tx的层间空间,减小了钠的扩散势垒。NaOH-Ti3C2Tx电极于50mV/s扫速下经过3000次循环,仅有6.46%的电容损失,具有良好的电化学稳定性。不对称CDI系统在1.2V电压下对100mg/L的NaCl溶液具有12.19mg/g的吸附容量,在20个吸附解吸循环过程后,电吸附容量仍可保持为11.54mg/g。
不同的蚀刻方法对MXene的性能具有重要影响。相较于HF蚀刻,用LiF/HCl蚀刻Ti3C2Tx后其层间间距更大,并有利于减少材料中-F的含量和提高亲水性。当进料浓度为10mmol,该材料在20mA/g恒定电流密度和1.2V电压窗口下具有68mg/g的脱盐能力,并具有相对较低的能耗(0.24kWh/kg-NaCl),能量回收率可达5.44%[36]。
操作条件可能会影响MXene的除盐能力。Agartan等[41]系统研究了放电电位、半周期长度(HCL)和流速对SAC和SAR的影响。结果表明,MXene的除盐性能取决于运行条件。较低的放电电位使SAC和SAR增加152%。HCL不会显著影响脱盐性能,当HCL从10min增加到30min时,SAC最多可提高35%,但SAR会减少54%。更快的流速使SAC和SAR降低20%,但是流速减慢会使寄生损耗加剧导致充电效率较低。
MXene这类二维材料的开发为各种电化学工作界面的搭建提供了良好的平台。但是由于层间范德华力的相互作用,导致MXene的片层结构发生堆叠,这是制约着其在CDI领域发展的不利因素。因此,如何巧妙设计MXene防止片层堆积以保证足够的活性表面应该是CDI领域研究的重要主题。
分层的二维材料基于离子插层来存储电荷[42]。二硫化钼(MoS2)是过渡金属二硫族化物(TMDs)中最具有代表性的化合物之一,具有可同时嵌入阳离子和阴离子的能力[43]。其具有类似于石墨烯的独特分层结构,理论容量为670mAh/g,层间间距(0.62nm)约为石墨烯(0.335nm)的两倍,可以用作钠离子的插入基质材料[44]。
Han等[45]利用水热法将MoS2原位生长在石墨烯表面,开发出了具有高体积吸附容量(VAC)的MoS2-G杂化电极。在1.2V直流电压下,MoS2-G电极的VAC高达14.3mg/cm3,重量吸附容量为19.4mg/g。并且,MoS2-G在不同流速下均显示出良好的稳定性。原位拉曼光谱显示Na+可以插入到MoS2层间,利用HAADF-STEM分析可证明随着Na+的嵌入,MoS2可实现从2H相到1T相的转变。这项工作为未来CDI器件的小型化开辟了一条新的途径。
溶剂可能会对微观结构和电化学性能有影响。由水/乙醇(体积比为2∶1)为溶剂制备的MoS2/rGO(还原氧化石墨烯)复合材料用作负极、AC用作正极组装成不对称电极。3D花状MoS2无序缠结到rGO片的结构有助于提供更多的Na+吸附位点。施加1.0V电压,在200mg/L NaCl溶液中的最大脱盐能力为16.82mg/g,NaCl在MoS2/rGO上的电吸附遵循准一级动力学模型,表明电吸附过程中电极和溶液中离子之间的静电作用占主导[46]。
缺陷可能会影响MoS2的电化学性能。Jia等[47]通过热处理制备了富含缺陷的MoS2(T-MoS2)。原子力显微镜结果显示,热处理后MoS2的表面上有许多边缘缺陷,Na+会被优先吸附在MoS2的边缘上。研究表明,T-MoS2的表面带有大量的负电荷,增强了Na+到电极表面的静电吸引力,同时有利于形成更厚的双电层以增强电吸附能力。在100mg/L NaCl溶液中,施加0.8V电压时的脱盐能力为35mg/g,约为普通MoS2(12.8mg/g)的3倍。
MoS2有三种不同的晶体结构,即1T、2H、3R。目前CDI中应用到的几乎都是2H相,2H相具有导电性差、层间堆叠以及循环稳定性差的缺陷;而1T金属相的MoS2电导率比2H相高约105倍,同时其具有高亲水性有利于离子扩散。因此,在后续研究工作中可以考虑将1T相与2H相结合用于CDI体系来弥补2H相在应用中的缺陷。
普鲁士蓝(PB)是最古老的合成配位化合物之一,具有面心立方结构。开放框架结构由位于角共享八面体上的Fe(II)和Fe(III)离子组成,并由小的共轭氰化物阴离子(C≡N—)桥接。在这种结构中,低自旋的亚铁离子与碳原子键合,高自旋的三价铁离子与氮原子键合,键长分别为1.92和2.05 Å[48]。
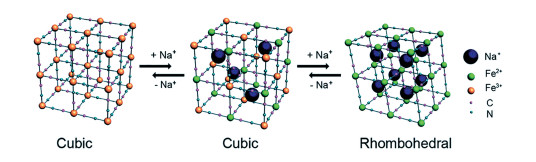
普鲁士蓝类似物(PBAs,Na2M[Fe(CN)6],M=Fe,Co,Mn,Ni,Cu等)理论容量高达170mAh/g。由于其三维框架结构中具有大的离子通道和晶格空隙,易于进行可逆的嵌入反应,PBAs是为数不多的能够容纳较大的碱金属阳离子(如Na+和K+)的主体材料之一。图 2为Na+嵌入/脱嵌过程中PB相变示意图。由于PBAs中包含M2+/3+和Fe2+/3+两个氧化还原中心,使其具有双电子氧化还原容量,相当于每个分子单元可进行2个Na+的可逆储存。其反应机理如下[50, 51]:
|
|
(3) |
|
|
(4) |
Lee等[52]设计了一个基于NaNiHCF和NaFeHCF材料的“摇椅电池”,该系统电极之间由阴离子交换膜隔开,充电时Na+脱嵌,同时Cl-穿过离子交换膜与Na+一同形成浓缩液被去除,放电时,Cl-移动方向相反。该系统的特点是在充放电过程中都能进行除盐,具有59.9mg/g的海水淡化能力,而且能耗较低,Na+去除率为40%时的能耗为0.34Wh/L。
Guo等[53]将FeFe(CN)6(PB)纳米粒子用于电化学去离子(Electrochemical deionization,EDI)系统,在恒流模式下,通过合理设计PB/rGO复合材料,EDI实现了120.0mg/g的去除容量和0.5430mg·g-1·s-1的超高速离子去除率。设置吸附解吸循环为5.5min,经过600次循环后去除容量为43.4mg/g,循环后的溶液中铁离子浓度较低,说明PB纳米立方体可以牢固地附着在多孔石墨烯的表面,表明复合材料具有出色的循环稳定性。该体系除盐机理见式(5)。
|
|
(5) |
Vafakhah等[54]设计了嵌入高导电性还原氧化石墨烯气凝胶中的PB纳米立方体复合材料(PB/rGA),将其用作无粘合剂的阳极,rGA用作阴极。PB和rGA的结合使复合材料在100mA/g的电流密度下产生了130mg/g的高除盐能力。原位XRD测试表明Na+可以插入到PB中;此外,该系统最低能耗仅为0.23Wh/g,在-0.2~1.4V的电压范围内实现了39%的最高能量回收率。
PBAs为新型CDI电极材料的发展开辟了一条全新路径,但在CDI领域还处于起步阶段。PBAs中过渡金属离子种类多样,不同离子的电化学行为非常不同,通过调整金属离子的种类可能是改善PBAs材料电化学性能的有效手段。因此有必要加强这一方面的研究。
NASICON(钠超离子导体)型磷酸盐材料,例如NaTi2(PO4)3(NTP)和Na3V2(PO4)3(NVP)都具有稳定的晶格结构和用于Na+迁移的大型3D隧道,使Na+在循环和快速迁移过程中可实现零应变插层。具体来说,钛离子或钒离子的存在使NTP和NVP具有相当大的Na+扩散系数,从而在钠离子电池中具有高的速率能力和较长的使用寿命[55]。
NASICON型磷酸盐材料简写为AxMy(PO4)3,其中A和M分别代表碱金属和过渡金属原子。MO6八面体和PO4四面体通过角共享构成了一个“灯笼单元”,具有三维框架结构,以促进Na+快速扩散[56]。
近几年,受钠离子电池材料开发的启发,NASICON型磷酸盐在CDI中也有许多应用。
NTP具有两个Na+嵌入/脱嵌的氧化还原平台,理论容量为133mAh/g。即使在高电荷状态下,强共价(PO4)3单元也能维持结构的稳定性。然而,NTP具有较弱的导电性[57]。常见的改善其导电性的方法是与碳基材料复合。
Wang等[60]制作了由金属有机骨架MIL-125前体衍生出的NTP/C材料,并将其用作混合电容去离子(Hybrid capacitive deionization,HCDI)系统的阴极,AC作为阳极。结果表明,相较于NTP,NTP/C复合材料具有较窄的氧化/还原峰和较小的峰电势差,说明碳改善了NTP的导电性。复合材料在1000mg/L溶液中具有167.4mg/g的脱盐能力。在1.8V电压下,HCDI系统在10个循环中的pH变化不明显,表明脱盐过程中水不会发生分解。该系统除盐机理见式(6)和式(7)。
|
|
(6) |
|
|
(7) |
Huang等[61]报道了基于NTP/rGO和AC复合材料的新型EDI系统,该系统在CC模式下运行,考察了电流密度、流速对除盐能力的影响,进料浓度为1000mg/L、电流密度为100mA/g时除盐能力可达到140mg/g。2019年,他们[62]又开发了一种新型双离子电化学去离子(Dual-ion electrochemical deionization,DEDI)系统。其中NTP/rGO用作阴极,Ag/rGO用作阳极。通过EDS能谱测试对Na和P的比例的分析结果表明钠离子可插入到NTP电极中。设置初始盐浓度为0.25%,DEDI系统在-1.2~1.4 V电压范围内,在50个循环中具有105mg/g的稳定且可逆的除盐能力。当施加0~1.4 V电压时,尽管去除能力相对较低(35.8mg/g),但该系统的能量回收率高于30%,能耗低至0.127Wh/g。
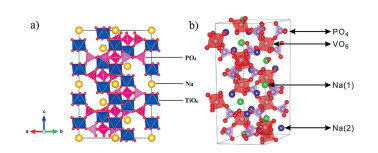
在所有NASICON化合物中,NVP具有较高的理论可逆容量(117.6mAh/g)和良好的热稳定性(450℃)[63]。较大的开放隧道结构有利于Na+扩散,其具有3.4V和1.6 V(vs.Na+/Na)两个电压平台,是一种有前途的钠离子电池阴极材料[64]。NVP结构如图 3(b)所示,其中VO6八面体和PO4四面体通过共用顶点的方式相互连接形成三维框架结构;Na+分布在两种不同的位点上,分别属于六配位(6b)和八配位(18e)的位置,定义为Na(1)位和Na(2)位[59, 65]。
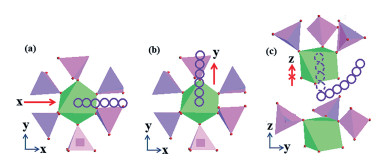
Song等[66]利用第一性原理计算了Na+在NVP晶体中不同扩散路径的活化能,同时得出了三种可能的Na+扩散路径(图 4)。机理A:Na离子将沿x方向通过两个PO4四面体之间的通道迁移;机理B:Na+可通过PO4四面体和VO6八面体之间的空隙沿y方向扩散(两种机制的活化能分别为0.0904和0.11774 eV);机理C:Na+以弯曲的路径传输以绕过八面体并进入相邻的PO4四面体和VO6八面体之间的空隙/通道,弯向z扩散,对应活化能为2.438eV。
Cao等[63]使用溶胶凝胶法制备了NVP@C,将NVP@C/AC作为HCDI系统的电极。当给水浓度为100mmol/L时,该体系具有137.30mg/g的超高盐去除能力和2.16kg(NaCl)/kWh较低的能耗。Zhao等[67]设计了一种新型DEDI系统,该系统由NVP@C纳米线和AgCl组成。以聚多巴胺为涂层碳源的NVP@C复合材料呈纳米线形态,经高温退火处理后,聚多巴胺碳化形成的碳涂层有助于提升复合材料的导电性。在100mA/g的电流密度下,经过50个以上的循环,NVP@C/AgCl电极具有98.0mg/g的超高脱盐能力。
同PBAs材料一样,NASICON型磷酸盐材料在CDI领域还处于起步阶段,在CDI应用中,NASICON型磷酸盐材料存在导电性差的缺陷。有以下两种方式来弥补此缺陷:首先,可减小材料尺寸以缩短Na+迁移距离,例如制备纳米颗粒、纳米纤维,然而几乎所有这些纳米材料都是通过水热法、电纺丝法或其他特定方法合成的,成本高、操作复杂,难以将NASICON型磷酸盐材料用作大型装置的电极材料;其次,导电碳层涂覆被认为是提高电极材料电导率的简单有效的方法,但是大多数材料的煅烧温度通常在1100℃以下,这不足以使碳层完全石墨化,使得碳的导电性不够强。因此,如何设计开发出一种新的合成NASICON型磷酸盐复合材料的方法非常重要[59]。同时,考虑到NASICON型磷酸盐材料与PBAs材料一样具有形成“摇椅电池”的能力,但对于此方面的研究还未曾有报道,因此笔者认为,可以考虑将其应用在“摇椅电池”系统中,并与PBAs材料做比较。
作为基于CDI的技术的一大优势,具有阴阳离子插入行为的电极材料的应用为解决传统的碳基电极电吸附容量低和阳极上的氧化问题提供了可能性。尽管基于阴阳离子插入行为的电极材料在CDI体系中已经取得了重要进展,但仍需不断深化CDI应用研究。虽然离子插层材料具有比容量高的特点,但其导电性差和多次充放电过程中会引起体积膨胀等缺陷限制了其应用,因此如何通过材料复合的方法提高材料的导电性与稳定性至关重要。基于CDI的技术的更多未来研究应转向真实的水条件,例如地表水、地下水和废水。细菌和有机化合物以及某些离子(例如碳酸氢根离子)共存于实际水中,可能会污染CDI电极或使电极结垢,从而对CDI系统产生负面影响,因此有必要研究不同污染物的污染特征以期开发出抗污电极,同时可以考虑探索CDI与其他预处理工艺或其他技术的耦合工艺,以最大程度地减少此类影响。
Elimelech M, Phillip W A. Science, 2011, 333(6043): 712~717. doi: 10.1126/science.1200488
Ying P J, Li M, Yu F L, et al. ACS Appl. Mater. Interf., 2020, 12(29): 32880~32887. doi: 10.1021/acsami.0c09965
Geng Y, Sun W, Ying P J, et al. Adv. Funct. Mater., 2020, 2007648.
Liu Y, Gao X, Wang Z P, et al. Chem. Eng. J., 2021, 403: 126326. doi: 10.1016/j.cej.2020.126326
Nassrullah H, AnisS F, Hashaikeh R, et al. Desalination, 2020, 491: 114569. doi: 10.1016/j.desal.2020.114569
Karaghouli A A, Kazmerski L L. Renew. Sustain. Energy Rev., 2013, 24: 343~356. doi: 10.1016/j.rser.2012.12.064
Mayor B. Desalination, 2019, 457: 75~84. doi: 10.1016/j.desal.2019.01.029
Semiat R. Environ. Sci. Technol., 2008, 42(22): 8193~8201. doi: 10.1021/es801330u
王凯军, 房阔, 宫徽, 等. 环境工程学报, 2018, 12(8): 2141~2152. https://www.cnki.com.cn/Article/CJFDTOTAL-ZGXH201902002.htm
Han D C, Zhang C M, Guan J, et al. Electrochim. Acta, 2020, 336: 135639. doi: 10.1016/j.electacta.2020.135639
Zhang H, Zhang W X, Shen J M, et al. Desalination, 2020, 473: 114173. doi: 10.1016/j.desal.2019.114173
黄宽, 唐浩, 刘丹阳, 等. 环境工程, 2016, 34(S1): 82~88. https://www.cnki.com.cn/Article/CJFDTOTAL-HJJZ201901030.htm
Vanysek P. CRC Hand Book of Chemistry and Physics, 1993: 5~92.
Li M, Park H G. ACS Appl. Mater. Interf., 2018, 10(3): 2442~2450. doi: 10.1021/acsami.7b14643
Porada S, Borchardt L, Oschatz M, et al. Energy Environ. Sci., 2013, 6(12): 3700~3712. doi: 10.1039/c3ee42209g
Chen Z L, Zhang H T, Wu C X, et al. Desalination, 2018, 433: 68~74. doi: 10.1016/j.desal.2017.11.036
Xu P, Drewes J E, Heil D, et al. Water Res., 2008, 42(10-11): 2605~2617. doi: 10.1016/j.watres.2008.01.011
Kang J, Kim T Y, Jo K, et al. Desalination, 2014, 352: 52~57. doi: 10.1016/j.desal.2014.08.009
Elisadiki J, Jande Y A C, Machunda R L, et al. Carbon, 2019, 147: 582~593. doi: 10.1016/j.carbon.2019.03.036
Korkmaz S, Kariper I A. J. Energy Storage, 2020, 27: 101038. doi: 10.1016/j.est.2019.101038
Shi P F, Wang C, Sun J Y, et al. Sep. Purif. Technol., 2020, 235: 116196. doi: 10.1016/j.seppur.2019.116196
Ding Z B, Xu X T, Li Y Q, et al. Desalination, 2019, 468: 114078. doi: 10.1016/j.desal.2019.114078
Pasta M, Wessells C D, Cui Y, et al. Nano Lett., 2012, 12(2): 839~843. doi: 10.1021/nl203889e
Lee J, Kim S, Kim C, et al. Energy Environ. Sci., 2014, 7(11): 3683~3689. doi: 10.1039/C4EE02378A
Chen F M, Huang Y X, Guo L, et al. Energy Environ. Sci., 2017, 10(10): 2081~2089. doi: 10.1039/C7EE00855D
Suss M E, Presser V. Joule, 2018, 2(1): 10~15. doi: 10.1016/j.joule.2017.12.010
Augustyn V, Simonbc P, Dunn B. Energy Environ. Sci., 2014, 7(5): 1597~1614. doi: 10.1039/c3ee44164d
Jaoude M A, Alhseinat E, Polychronopoulou K, et al. Electrochim. Acta, 2020, 330: 135202. doi: 10.1016/j.electacta.2019.135202
Hand S, Cusick R D. Environ. Sci. Technol., 2017, 51(20): 12027~12034. doi: 10.1021/acs.est.7b03060
Tang Y J, Zheng S S, Xu Y X, et al. Energy Storage Mater., 2018, 12: 284~309. doi: 10.1016/j.ensm.2018.02.010
Devaraj S, Munichandraiah N. J. Phys. Chem. C, 2008, 112: 4406~4417. doi: 10.1021/jp7108785
鞠广凯, 陶占良, 陈军. 物理化学学报, 2017, 33(7): 1421~1428. https://www.cnki.com.cn/Article/CJFDTOTAL-ZGZS201903001.htm
Lee H Y, Goodenough J B. J. Solid State Chem., 1999, 144(1): 220~223. doi: 10.1006/jssc.1998.8128
Toupin M, Brousse T, Bélanger D. Chem. Mater., 2004, 16(16): 3184~3190. doi: 10.1021/cm049649j
Tan G C, Lu S D, Xu N, et al. Environ. Sci. Technol., 2020, 54(9): 5843~5852. doi: 10.1021/acs.est.9b07182
Ma J, Cheng Y J, Wang L, et al. Chem. Eng. J., 2020, 384: 123329. doi: 10.1016/j.cej.2019.123329
Naguib M, Kurtoglu M, Presser V, et al. Adv. Mater., 2011, 23: 4248~4253. doi: 10.1002/adma.201102306
Malaki M, Maleki A, Varma R S. J. Mater. Chem. A, 2019, 7(18): 10843~10857. doi: 10.1039/C9TA01850F
Srimuk P, Kaasik F, Krüner B, et al. J. Mater. Chem. A, 2016, 4(47): 18265~18271. doi: 10.1039/C6TA07833H
Chen B B, Feng A H, Deng R X, et al. ACS Appl. Mater. Interf., 2020, 12(12): 13750~13758. doi: 10.1021/acsami.9b19684
Agartan L, Hantanasirisakul K, Buczek S, et al. Desalination, 2020, 477: 114267. doi: 10.1016/j.desal.2019.114267
Srimuk P, Halim J, Lee J H, et al. ACS Sustain. Chem. Eng., 2018, 6(3): 3739~3747. doi: 10.1021/acssuschemeng.7b04095
Srimuk P, Lee J H, Fleischmann S, et al. J. Mate. Chem. A, 2017, 5(30): 15640~15649. doi: 10.1039/C7TA03120C
Tang W J, Wang X L, Xie D, et al. J. Mater. Chem. A, 2018, 6(37): 18318~18324. doi: 10.1039/C8TA06905K
Han J L, Yan T T, Shen J J, et al. Environ. Sci. Technol., 2019, 53(21): 12668~12676. doi: 10.1021/acs.est.9b04274
Peng W J, Wang W, Han G H, et al. Desalination, 2020, 473: 114191. doi: 10.1016/j.desal.2019.114191
Jia F F, Sun K G, Yang B Q, et al. Desalination, 2018, 446: 21~30. doi: 10.1016/j.desal.2018.08.024
Yue Y F, Binder A J, Guo B K, et al. Angew. Chem. Int. Ed., 2014, 53: 3134~3137. doi: 10.1002/anie.201310679
You Y, Wu X L, Yin Y X, et al. Energy Environ. Sci., 2014, 7(5): 1643~1647. doi: 10.1039/C3EE44004D
Qian J F, Wu C, Cao Y L, et al. Adv. Energy Mater., 2018, 8: 1702619. doi: 10.1002/aenm.201702619
Wu XY, Wu C H, Wei C X, et al. ACS Appl. Mater. Interf., 2016, 8: 5393~5399. doi: 10.1021/acsami.5b12620
Lee J, Kim S, Yoon J. ACS Omega, 2017, 2(4): 1653~1659. doi: 10.1021/acsomega.6b00526
Guo L, Mo R W, Shi W H, et al. Nanoscale, 2017, 9(35): 13305~13312. doi: 10.1039/C7NR03579A
Vafakhah S, Guo L, Sriramulu D, et al. ACS Appl. Mater. Interf., 2019, 11: 5989~5998. doi: 10.1021/acsami.8b18746
Yu S C, Liu Z G, Tempel H, et al. J. Mater. Chem. A, 2018, 6(37): 18304~18317. doi: 10.1039/C8TA07313A
Qiu S, Wu X Y, Wang M Y, et al. Nano Energy, 2019, 64: 103941. doi: 10.1016/j.nanoen.2019.103941
Li X N, Zhu X B, Liang J W, et al. J. Electrochem. Soc., 2014, 161(6): A1181~A1187.
Liu Z X, An Y F, Pang G, et al. Chem. Eng. J., 2018, 353: 814~823. doi: 10.1016/j.cej.2018.07.159
Wang Q, Zhang M Y, Zhou C G, et al. J. Phys. Chem. C, 2018, 122(29): 16649~16654. doi: 10.1021/acs.jpcc.8b06120
Wang K, Liu Y, Ding Z B, et al. J. Mater. Chem. A, 2019, 7(19): 12126~12133. doi: 10.1039/C9TA01106D
Huang Y X, Chen F M, Guo L, et al. J. Mater. Chem. A, 2017, 5(34): 18157~18165. doi: 10.1039/C7TA03725B
Huang Y X, Chen F M, Guo L, et al. Desalination, 2019, 451: 241~247. doi: 10.1016/j.desal.2018.02.006
Cao J L, Wang Y, Wang L, et al. Nano Lett., 2019, 19(2): 823~828. doi: 10.1021/acs.nanolett.8b04006
Fang J Q, Wang S Q, Li Z T, et al. J. Mater. Chem. A, 2016, 4(4): 1180~1185. doi: 10.1039/C5TA08869K
Ko J S, Paul P P, Wan G, et al. Chem. Mater., 2020, 32(7): 3028~3035. doi: 10.1021/acs.chemmater.0c00004
Song W X, Ji X B, Wu Z P, et al. J. Mater. Chem. A, 2014, 2(15): 5358~5362. doi: 10.1039/c4ta00230j
Zhao W Y, Guo L, Ding M, et al. ACS Appl. Mater. Interf., 2018, 10(47): 40540~40548. doi: 10.1021/acsami.8b14014



表 1 常见脱盐技术能耗分析
Table 1. Energy consumption analysis of common desalination technologies
| 技术种类 | 热能消耗/(MJ/m3) | 盐水温度/℃ | 电能消耗/(kW·h)/m3 | 总体能耗/(kW·h)/m3 |
| 反渗透(RO) | 无 | 与进水温度一致 | 1.5~6 | 1.5~6 |
| 多级闪蒸(MSF) | 190~282 | 90~120 | 2.5~5 | 18.3~28.5 |
| 多效蒸馏(MED) | 145~230 | 50~90 | 2~2.5 | 14.2~21.6 |
| 电渗析(ED) | 无 | < 45 | 0.7~5.5 | 0.7~5.5 |
| 电容去离子(CDI) | 无 | 与进水温度一致 | 0.1~2.03 | 0.1~2.03 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
