金属有机化学是由多学科交叉渗透而发展起来的一门分支学科,其在催化反应中的应用是化学学科发展的重要环节之一,它为有机合成领域提供了众多高选择性、高产率且条件温和的方法[1 。CO2 由于其自然含量丰富、廉价、无毒性和固有的可再生性,是一种环境友好的C1资源[2 3 。20世纪70年代末首次发现金属配合物与CO2 和烯烃的反应[4a ,由于CO2 化学性质稳定且难以成键,往往只能通过使用高活性的金属试剂(例如格氏试剂或有机锂试剂)才可以克服CO2 的固有惰性[4 。因此开发能够高效活化CO2 的催化体系,将其化学转化为高附加值的精细化学品至关重要。过去几十年里在这个领域取得了一些较大的进步,其中最显著的就是以氮杂环卡宾(NHC)催化CO2 的羧化反应[5 。
NHC是一类具有碳烯结构的含氮杂环分子,其相邻两个氮原子上的孤对电子与卡宾碳原子空的p电子轨道存在给电子的共轭效应,这使得卡宾碳原子上电子云密度升高,亲核性增强。同时由于氮的电负性比碳强,可使卡宾碳原子上的孤对电子更趋于稳定状态,因此NHC相比于传统的膦配体具有更强的给电子能力和配位能力[1 。NHC已成为有机金属化学和无机配位化学中的常用配体,这不仅由于它们能与低氧化态或高氧化态的过渡金属结合,且由于它们特定的配位化学性质,在有机合成的关键步骤中能起到稳定和活化催化金属中心的作用[5 。本文结合近年来国内外NHC催化CO2 羧化反应的研究报道,分类归纳了催化体系中NHC-过渡金属配合物的类型,以期对该领域的发展起到一定的积极作用。
1.
Cu-NHCs配合物催化的羧化反应
与钯、镍等过渡金属相比,铜是一种廉价低毒性的金属,用铜来催化交叉偶联反应不仅可以节省贵金属的消耗,降低成本,而且可以减少对环境的污染,促进绿色化学的发展。氮杂环卡宾铜(Cu-NHCs)催化剂是一类价格低廉、毒性较低、易制备且结构种类丰富的NHC类催化剂[1 6 7 ,近年来,Cu-NHCs配合物已被证明是各种有机化合物与CO2 羧化得到相应羧酸或其衍生物的优良催化剂[8 10 ,现已广泛应用于各类有机催化反应中。
1.1
有机硼试剂参与的羧化反应
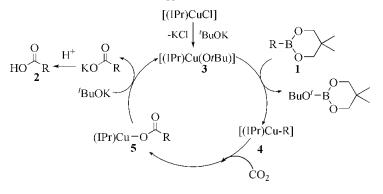
由于格氏试剂或有机锂试剂的活泼性太高,使得反应底物的官能团种类受限,而有机硼试剂作为较温和的有机金属试剂,则显示出较好的耐受性和良好的反应性。2008年,Hou等[11 12 报道了[(IPr)CuCl]催化烷基硼化合物(芳基硼酸酯和烷基链烯基硼酸酯)与CO2 的羧化反应。反应只需1(mol)%的[(IPr)CuCl]作催化剂,就可以高产率地获得各种官能化羧酸(式(1))。反应机理为:[(IPr)CuCl]和t -BuOK之间通过阴离子交换直接得到醇盐络合物3 ,醇盐络合物与有机硼酸酯反应,得到相应的有机铜络合物4 ;然后CO2 插入Cu-R产生羧酸盐5 ,最后该羧酸盐与t -BuOK再次复分解时可以再生醇盐络合物3 并释放出钾盐RCOOK,钾盐水解产生羧酸(图式 1
图式 1
在已有研究[11 14 的基础上,2011年Hou等[15 以[(IPr)CuCl]作催化剂,通过烯烃硼氢化原位产生的有机硼试剂与CO2 反应,高产率地得到相应的各种官能化羧酸(式(2))。
随后,Hou等[16 又首先报道了利用氮杂环卡宾铜[(SIMes)CuCl]作为催化剂,催化各类炔烃(例如二芳基炔烃、芳基/烷基炔烃和苯基乙炔)与乙硼烷化合物和CO2 的反应,并得到高区域和立体选择性的α, β-不饱和β-硼内酯衍生物9 (式(3))。反应的可能机理是:首先,催化剂[(SIMes)CuCl]与LiOt-Bu间发生阴离子交换生成10 ,然后与B2 (pin)2 反应生成硼铜络合物11 ,随后炔以顺式方式插入Cu-B键得到重要中间体β-硼烯基铜络合物12 ;其次,发生烯基铜络合物对CO2 的亲核加成,然后用硼原子取代生成的羧酸盐中的Cu部分,生成β-硼内酯衍生物13 ;最后,铜络合物13 与LiOt-Bu之间发生金属转移反应再生化合物10 并释放最终产物9 (图式 2
图式 2
2013年,Duong等[17 用Cu(I)/NHC作为催化剂,催化烯丙基硼酸频哪醇酯与CO2 反应,可得到各种各样的环状和非环状β, γ-不饱和羧酸化合物16 (式(4))。反应具有高度的区域选择性,有利于制备多种取代基团的羧酸,包括具有全碳四元中心的化合物。
2016年,Butcher等[18 报道了用ICyCuCl作催化剂进行区域选择性催化芳香族烯烃类化合物与双(频哪醇)合二硼和CO2 的羧化反应并能以中等至良好的产率获得产物(式(5))。通过该法可合成三类新化合物:硼-NSAID(非固醇类抗炎症药物)衍生物、氟代羧基化烯烃和二氟代内酯,它们具有较高的药物合成价值和药物化学应用潜力。
受之前研究工作[13 15 的启发,2017年Troels等[19 研究并开发了一种用二取代的烯烃和末端炔烃加氢羧化的方案(式(6))。这种合成方法适用于不同的环烯烃、苯乙烯和芪衍生物以及一些天然产物,提供了合成不同的仲羧酸和丙二酸衍生物的途径。该方法的优点在于反应不需要使用强碱性的醇盐,仅需氟盐即可。
与此同时,Kuge等[20 用IPrCuCl作催化剂,直接催化末端炔烃与CO2 的羧化反应,得到具有优异区域和立体选择性的α-烷基丙烯酸化合物23 (式(7))。反应的机理如图式 3 3 B反应生成炔基三乙基硼酸酯配合物24 ,该配合物随后在β-碳位置进行质子化,生成乙烯基阳离子中间体25 ;然后1, 2-乙基迁移到乙烯基碳正离子空的p轨道上,从而形成乙烯基二乙基硼烷中间体26 。随后,用卡宾-铜物种进行金属转移,得到乙烯基铜中间体27 ,该化合物再对CO2 进行亲核加成产生羧酸盐28 ,然后与MeOK反应再生卡宾-铜催化剂。
图式 3
1.2
有机铝试剂参与的羧化反应
2013年,Hou等[21 研究表明,由各种炔烃与氢化铝或甲基铝反应所得的烯基铝物质可以与CO2 进行羧化反应,得到各种α, β-不饱和羧酸化合物。作为羧化反应常用的高活性[CuCl(IPr)]配合物,其能够很好地与各种此类催化体系相匹配(式(8~11))。由于该反应具有高区域和立体选择性、简单的一锅反应操作以及使用CO2 作为起始原料,因此该方案是各种α, β-不饱和羧酸的一种实用且具有吸引力的绿色合成方法。
随后,他们[22 用混合的烷基氨基铝酸锂化合物i -Bu3 Al(TMP)Li对芳香族化合物进行去质子化,然后将所得的芳基铝物质与CO2 进行羧化反应,在催化量的NHC-Cu存在下,可以实现各种芳香族化合物与CO2 的邻位C-H键羧化,并得到相应的羧化产物(式(12))。该方案产物收率高,选择性好,适用底物范围广。除苯衍生物外,杂芳烃如苯并呋喃、苯并噻吩和吲哚衍生物也都可用来作底物。反应可能的机理为:铝酸盐试剂(i -Bu)3 Al(TMP)Li对芳族化合物进行质子化生成芳基铝40 ,然后经过金属转移、CO2 插入得到羧酸铜络合物42 ;该络合物与另一分子40再次进行金属转移反应得到羧酸铝物种43 ,随后水解即可得到化合物ArCOOH(图式 4
图式 4
1.3
有机锌试剂参与的羧化反应
2015年,Hou等[23 用卡宾铜[(IPr)CuCl]和二烷基锌试剂实现了炔胺的烷基化羧化反应(式(13))。这种三组分偶联反应可在温和条件下一锅法反应,并以良好至较高的产率生成β-β′-二取代的α, β-脱氢氨基酸衍生物47 。次年,他们[24 又首次开发了高效的联烯丙基胺与二烷基锌试剂和CO2 的烷基化羧化反应。其中烷基被引入受阻较少的γ-碳上,而羧基被引入烯丙酰胺的β-碳原子上,从而得到100%区域和立体选择性的各种(Z )-α, β-脱氢-β-氨基酸酯衍生物49 (式(14))。
1.4
有机锆试剂参与的羧化反应
2015年,Wang等[25 使用(IMes)CuCl作催化剂,在温和条件下能有效地催化各种链烯基二茂锆的羧化反应,以良好至较高的收率得到相应的α, β-不饱和羧酸(式(15))。反应先通过炔烃的顺序羰基化得到链烯基锆茂51 ,然后在室温下使用CO2 作为起始原料来进行羧化反应。该方法通常被用来制备各种三取代的α, β-不饱和羧酸。
1.5
C-H键与CO2 的直接羧化反应
研究开发C-H键直接官能化的高效催化体系是化学家们长期以来的目标。作为C-H键官能化的快速兴起领域之一,饱和脂肪烃以及芳烃、烯烃和炔烃的直接羧化和催化羧化对化学研究人员来说是相当有吸引力的,因为这些方案为C-C键和C-杂原子键的形成提供了环境友好的绿色替代方案[26 。在过去十年中,有机金属化学的显著进步为开发用于C-H键活化反应的金属催化剂奠定了基础。
1.5.1
Csp1 -H键和CO2 的羧化反应
2010年,Zhang等[27 报道了(IPr)CuCl催化端炔和烯丙基氯类化合物与CO2 的羧基耦合反应,并得到相应的炔酸丙烯酯类化合物54 (式(16))。该催化剂在实验中可以重复利用,且催化活性不受影响。
同年,Yu等[28 使用多齿N -杂环卡宾铜配合物(Poly-NHC)2 CuCl催化端炔与CO2 的羧化反应得到较高产率的芳基炔酸类化合物55 (式(17))。此反应条件温和、底物普适性广,且催化剂体系简单经济具有非常大的潜在应用价值。
异香豆素是一类重要的内酯,广泛存在于众多具有生物活性的天然产物中。人们也一直在开发新的合成策略,以用来方便地制备这些材料。2014年,Yoo等[29 报道了氮杂环卡宾铜配合物催化的芳炔、端炔和CO2 的多组分环化反应,合成异香豆素类化合物(式(18))。反应以氮杂环卡宾铜配合物作为催化剂,在乙腈和四氢呋喃的混合溶剂中反应,可高收率高选择性地得到6-endo-dig 型环化产物。
1.5.2
Csp2 -H键和CO2 的羧化反应
2010年,Boogaerts等[30 首次报道了利用铜络合物[Cu(IPr)X](X=OH, Cl)催化咪唑和多取代苯氟化合物的羧化反应(式(19))。该反应证明了氮杂环卡宾铜(I)氢氧化物配合物能够进行N-H键和C-H键的区域选择性羧化。
2012年,Hou等[31 9b 制备了基于1, 2, 3-三唑-5-亚烷基的铜配合物tzNHC-Cu,该配合物可有效催化杂芳族化合物(苯二噁唑和苯并噻唑衍生物)与CO2 的直接C-H羧化反应,并在用烷基碘化物处理后能以优异的收率得到相应的酯(式(20))。
1.5.3
Csp3 -H键和CO2 的羧化反应
2016年,Murakami等[32 通过紫外线照射,在亚化学计量酮和催化量铜络合物存在下,实现了光诱导的简单烯丙基的C-H键与CO2 的羧化反应(式(21))。
2.
2 Ag-NHCs配合物催化的羧化反应
2012年,Yu等[33 将Cu(I)-NHC的方法扩展到银催化剂,并制备了NHC聚合物负载的银纳米粒子催化剂体系(式(22))。独特的NHC聚合物和银纳米颗粒复合结构在催化活化末端炔烃和CO2 的偶联反应上表现出协同效应,使得反应产率大大提高。
2015年,吴兆轩等[34 用自己合成的两种金属双氮杂环卡宾催化剂催化一系列末端炔烃的羧化反应,并对催化反应的可行性及适宜反应条件进行了深入的探究(式(23))。实验证明,自由氮杂环卡宾可以作为极强的亲核试剂活化CO2 分子并形成加合物,对分子直接嵌入末端烯烃的羧化反应具有良好的催化作用。而且,反应条件温和,在1atm CO2 、室温、DMSO为溶剂、(1(mol)%) bis-(NHC)-Ag为催化剂、使用Cs2 CO3 提供碱性环境的条件下反应16h,产率可超过90%。
同年,Ikariya等[35 发现,NHC-Ag(I)羧酸盐配合物可用作高效催化剂,能够在相对温和的条件下催化烯丙基甲胺和CO2 的羧化反应生成5-烯基-1, 3-噁唑烷-2-酮衍生物,并且具有优异的选择性(式(24))。
此外,Yamada等[36 开发了三甲基(2-亚甲基-3-丁炔-1-基)硅烷衍生物和CO2 的反应,形成的C-C键可以再进一步羧化和环化,通过银催化剂和CsF的作用,最后得到相应的2-呋喃酮和2-吡喃酮衍生物(式(25))。结果还表明,当使用芳环取代的炔烃时,通过5-外-环化反应选择性地获得2-呋喃酮衍生物,而用烷基取代的炔烃的反应则以高选择性产生2-吡喃酮衍生物。
2016年,笔者课题组[37 通过使用合成的新型Ag-NHCs配合物实现了在常温条件下催化炔烃与CO2 的高活性和选择性羧化,反应能以良好的收率获得各种官能化的丙炔酸(式(26))。随后,进一步以1, 3-双(4-甲基苄基)咪唑-2-亚基银(I)氯化物为催化剂,成功地实现了末端炔烃、正丁基碘和CO2 的三组分羧化偶联反应,高产率地生成炔酸酯类化合物(式(27))[38 。
2017年,Yuan等[39 开发了一系列高活性的NHC/Ag原位催化体系,用来催化末端炔烃的直接羧化,并在温和条件下高效地得到相应的芳基和脂肪族羧酸(式(28))。反应过程中避免了光敏Ag络合物的合成,且该催化体系可以像无机银盐催化剂一样直接操作。
3.
Au-NHCs配合物催化的羧化反应
2010年,Boogaerts等[40 用新型的(IPr)AuOH催化剂来高产率和高选择性地催化芳烃和芳香族类杂环化合物C-H的直接羧化(式(29))。由于AuOH在活化过程的强碱性,(IPr)AuOH配合物可以在酸性的C-H键位置上来催化碳环和杂环的羧化,使得此类C-H键在不需要其他有机金属催化剂的情况下实现官能团化。
2013年,Hase等[41 使用IPr-Au(I)络合物在40℃条件下催化炔丙胺与CO2 羧化环化生成(Z )-5-亚烷基-2-噁唑烷酮类化合物77 (式(30))。该反应条件温和,且可以在无添加剂的情况下进行。
2016年,Fujita等[42 研究制备了新型树枝状NHC-Au(I)络合物,通过使用在外围层具有五(乙二醇)单元的两亲性树枝状NHC-Au(I)络合物作为催化剂,在室温条件下合成2-噁唑烷酮类化合物(式(31))。
4.
Ni-NHCs配合物催化的羧化反应
2017年,Diccianni等[43 研究了用氮杂环卡宾镍(Ni(IPr)2 )为催化剂、Et2 Zn为还原剂,通过催化1, 6-烯炔和1, 7-烯炔和CO2 反应来构建(杂)环α, β-不饱和羧酸类化合物79 和81 (式(32)和(33))。
5.
Rh-NHCs配合物催化的羧化反应
2018年,Cai等[44 报道了Rh(II)催化的2-吡啶酚的Csp2 -H羧化反应。反应条件优化过程中发现,以IMes为配体催化效果良好,而以ICy和IiPr为配体催化效远不如前者(式(34))。
6.
总结与展望
使用NHC-过渡金属络合物作为通用催化剂,可以高效、高选择性地实现使用CO2 作为可持续C1 源的各种转化。由于NHC配体独特的空间和电子特性,特别是它们相对于传统的膦配体而言具有的更高的σ-给电子能力,其能够稳定和活化催化作用的金属中心。另外,以NHC为配体的催化系统能较好地结合多种金属,具有普遍适用性。虽然科学家们已取得不小的成就,但是仍面临着许多困难。首先,与膦配体相比,市面上可用的NHC配体要相对少得多,如果可以量产并在较温和的条件下就可储存的话,就可以让所有研究者受益;其次,可利用优化的NHC配体类型很少,研究开发新型NHC配体可能会促进该领域的进一步进展。
综上所述,过渡金属-NHC配合物的使用极大地扩展了N-H键和C-H键的活化方法,这些研究可启发化学家们开发新的催化系统和设计新的催化转化以实现CO2 的高附加值化利用。相信随着研究的不断深入,NHC必将作为一类绿色高效的配体被越来越多的研究者使用。


 下载:
下载:





































 下载:
下载:





