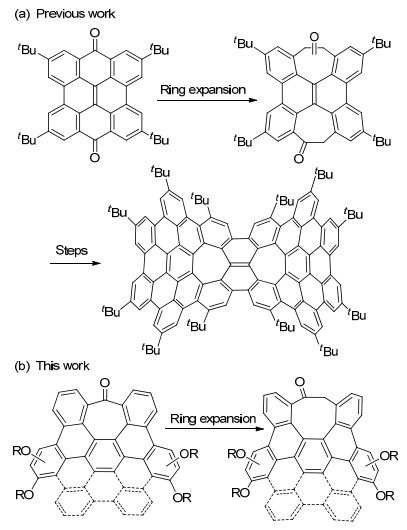
Scheme 1.
(a) Reported introduction of seven-membered rings into polycyclic arenes by ring expansion, and (b) introduction of an eight-membered ring to polycyclic arenes by ring expansion

Understanding how the topological defects define the geometries and properties of curved nanocarbons is of great importance in the research of carbon nanoscience.[1] One common topological defect that cause the polycyclic framework to contort is non-hexagonal rings. Embedding four-[2] or five-membered rings[3] into an otherwise planar polycyclic arene leads to positive curvature, which is the topological charater of aromatic bowls; while embedding seven-[4-6] or eight-membered rings[7] into an otherwise planar polycyclic arene leads to negative curvature, which is the topological charater of aromatic saddles. Negatively curved polycyclic arenes (also known as aromatic saddles) represent fragments in carbon Schwarzites (also known as Mackay crystals), [8] which are theoretical negatively curved carbon allotropes of aesthetic structures and interesting properties but have not been synthesized unambiguously yet.[9-10] Negatively curved polycyclic arenes contain structual infromation of carbon Schwarzites and can in principle be used as building blocks in a bottom-up approch to the synthesis of carbon Schwarzites. Moreover, negatively curved polycyclic arenes present unique stereochemistry and properties that are not available to planar or positively curved polycycylic arenes. For these reasons, negatively curved polycyclic arenes have recently received more and more attentions.[11]
Difficulty in the synthesis of negatively curved polycyclic arenes is mainly associated with their considerable strain, which is more significant in polycyclic arenes containing eight-membered rings.[12] So far, only a handful of methods are availabe for constructing eight-membered rings in polycyclic arenes.[7, 13] Moreover, already-formed eight-membered rings may suffer skeletal rearrangement during the oxidative cyclodehydrogenation reaction, [14] which predominates in the synthesis of large polycyclic arenes. As a result, new synthetic strategies need to be explored for introduction of eight-membered rings into polycyclic arenes. Recently, our group successfully utilized a ring expansion strategy to construct the seven-membered rings in negatively curved polycyclic arenes as shown in Scheme 1a.[6] Herein, we demonstate that this strategy can be extended for introduction of an eight-membered ring into polycyclic aromatic frameworks as shown in Scheme 1b.
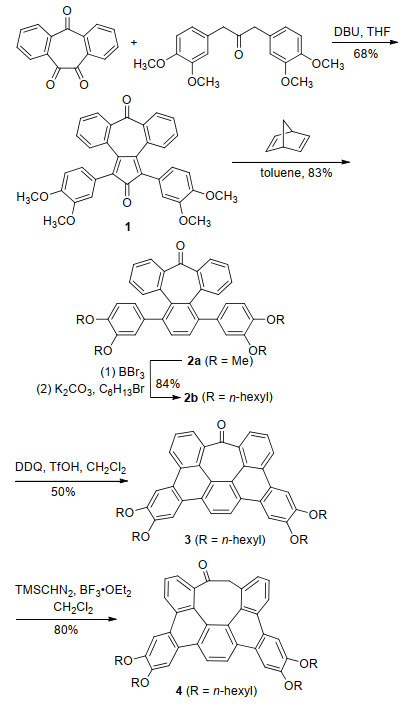
The ring expansion strategy in the synthesis of two polycyclic arenes containing an eight-membered ring, namely 4 (Scheme 2) and 9 (Scheme 3) was tested. As shown in Scheme 2, the synthesis of 4 started with 2, 3:6, 7-diben- zocycloheptadiene-1, 4, 5-trione, which can be easily obtained by a one-step oxidation of commercially available 5-dibenzosuberenone[15]. Double Aldol condensation of the trione with 1, 3-bis(3, 4-dimethoxyphenyl)-2-propanone[16] afforded cyclopentadiene-one 1 in a yield of 68%. Diels-Alder reaction of 1 and norbornadiene, followed by retro-Diels-Alder reaction afforded 2a in a yield of 83%. After the methyl groups in 2a were replaced by longer hexyl chains to provide sufficient solubility, the Scholl reaction[17-18] of 2b with 2, 3-dichloro-5, 6-dicyano-1, 4-ben- zoquinone (DDQ) and triflic acid (TfOH)[19] resulted 3 with the formation of two carbon-carbon bonds in a moderate yield. Here, the oxidative cyclodehydrogenation is facilitated by the alkoxyl groups that are positioned meta and para to the reaction site.[20] Treatment of 3 with trimethylsilyldiazomethane (TMSCHN2)[21-22] at the presence of boron trifluoride etherate enabled ring expansion and thus afforded 4 in a yield of 80%.
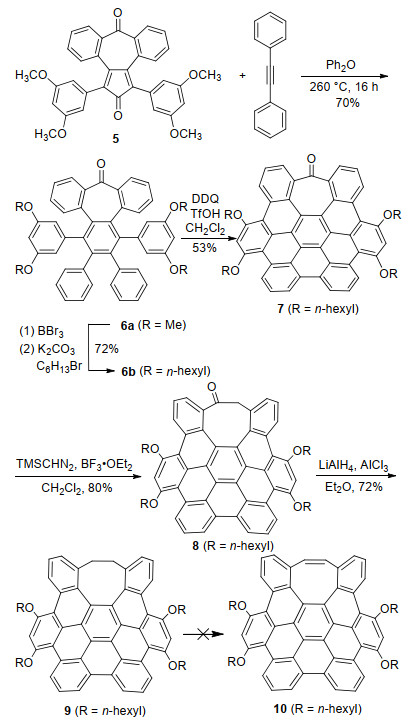
Scheme 3 shows the synthesis of 9, an octagon embedded hexabenzocoronene (HBC) from 5, which, similar to 1, was prepared from 2, 3:6, 7-dibenzocycloheptadiene-1, 4, 5- trione following the reported procedure.[15] Diels-Alder reaction of 5 with diphenyl acetylene followed by decarbonylation gave hexaphenylbenzene derivative 6a. After the methyl groups in 6a were replaced by longer hexyl chains to increase solubility, the Scholl reaction of 6b with DDQ and TfOH led to 7 with the formation of five carbon-carbon bonds in a yield of 53%. The four alkoxy groups in 6b are important to activate the desired positions for oxidative cyclodehydrogenation[23] as noted earlier with the synthesis of the heptagon-embedded HBC.[4] Treatment of 7 with TMSCHN2 at the presence of boron trifluoride etherate enabled ring expansion and thus afforded 8 in a yield of 80%. This indicates that the ring expansion strategy is applicable to curved and extended π-backbones. Decarbonylation of 8 by LiAlH4/AlCl3 led to 9 in a yield of 72%. Attempted dehydrogenation of 9 with a variety of reagents, such as DDQ or Pd/C for direct dehydrogenation, and bromination followed by elimination, all failed to yield 10 presumably because the reactivity of four benzylic methylene groups is changed by the contorted eight-membered ring.
Single crystals of 9 suitable for X-ray crystallography were obtained from solutions in dichloromethane and ethyl acetate by slow evaporation of solvents.[24] In the single crystals, 9 exists as a pair of enantiomers, which have helical chirality due to the embedded [5]helicene subunit. However, this chiral structure can be racemized through the eight-membered ring flipping as suggested by the broad peaks at δ 3.10~3.24 in 1H NMR at room temperature for the methylene groups in the eight-membered ring. As shown in Figure 1a, the π-backbone of 9 (even with two sp3 carbon atoms in the eight-membered ring excluded) is curved like a saddle and thus presents negative curvature. As shown in Figure 1b, the C—C bonds shown in magenta, which exhibit bond lengths of 0.147~0.149 nm, are significantly longer than a typical C—C aromatic bond (0.138~0.140 nm) but resemble C—C single bonds between sp2-sp2 carbons, which have a typical bond length of 0.145~0.148 nm.[25] As shown in Figure 1c, molecules of 9 stack with π-π interactions, which exhibit two different π-to-π distances between neighboring molecules. The π-to-π distance of 0.348 nm is associated with overlap of roughly three benzenoid rings, while the π-to-π distance of 0.357 nm is associated with overlap of roughly one benzenoid ring.
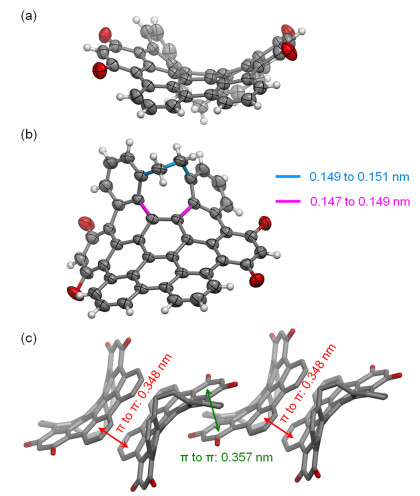
(a) Side view showing the saddle-like shape; (b) top view with C—C single bonds in the eight-membered ring highlighted; (c) molecular packing with π-π stacking. Hexyl groups removed for clarity. Carbon and oxygen atoms in (a) and (b) are shown as grey and red ellipsoids, respectively, at 50% probability level
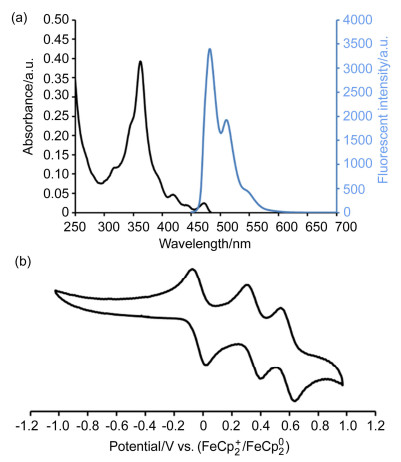
Compound 9 is soluble in common organic solvents resulting in yellow solutions and appears green fluorescence upon excited with UV light. As shown in Figure 2a, the UV-vis absorption of 9 exhibits an intense peak at 363 nm and weak absorption band at 472 nm. The fluorescent spectrum of 9 exhibits a small Stoke shift of 10 nm, which suggests that the polycyclic aromatic backbone of 9 is as rigid as that of the earlier reported heptagon-embedded HBC.[4] As estimated from the absorption edge, the HOMO-LUMO gap of 9 is 2.58 eV. As shown in Figure 2b, the cyclic voltammogram of 9 in CH2Cl2 exhibits two reversible oxidation waves with a half-wave oxidation potential of 0.38 and 0.62 V vs. ferrocenium/ferrocene (Fc+/Fc), respectively. From the half-wave oxidation, the HOMO energy level of 9 is estimated as -5.48 eV.[26]
In summary, the successful syntheses of 4 and 9 indicate that the ring expansion is a useful strategy to introduce eight-membered rings into polycyclic aromatic frameworks. The single crystal X-ray crystallography reveals that 9 has a negatively curved π-backbone, which is π-isoelectronic to hexabenzocoronene (HBC), an well- known planar polycyclic arene.[27] The study on the electronic structure of 4 sheds light on the effect of an eight-mem- bered ring defect on a planar polycyclic aromatic system. This ring expansion strategy opens a new avenue to synthesis of negatively curved polycyclic arenes. Further study on the π-expansion of 4 is in progress in our laboratory.
The reagents and starting materials employed were commercially available and used without any further purification or made following reported methods as indicated. Unless otherwise noted, all reactions were performed with dry solvents under an atmosphere of nitrogen in dried glassware with standard vacuum-line techniques. Anhydrous and oxygen-free dichloromethane, tetrahydrofuran, and toluene were purified by an Advanced Technology Pure-Solv PS-MD-4 system. NMR spectra were recorded on a Brucker ADVANCE Ⅲ 400 MHz spectrometer (1H NMR: 400 MHz, 13C NMR: 100 MHz). Chemical shift values (δ) were expressed using residual solvent protons (1H NMR, δH=7.26 for CDCl3; 13C NMR, δC=77.16 for CDCl3) as internal standard. Mass spectra were recorded on a Bruker Autoflex speed MALDI-TOF spectrometer. X-ray crystallography data were collected on a Bruker AXS Kappa ApexII Duo Diffractometer. UV-vis absorption spectra were recorded on a Varian CARY 1E UV-vis spectrophotometer. Fluorescence spectra were taken on a Hitachi F-4500 spectrofluorometer. Melting points, without correction, were measured using a Nikon Polarized Light Microscope ECLIPSE 50i POL equipped with an INTEC HCS302 heating stage. The cyclic voltammetry was performed in a solution of CH2Cl2 with 0.1 mol/L Bu4NPF6 as the supporting electrolyte, at a scan rate of 50 mV•s-1. Ferrocene/ferrocenium was used as the internal standard. Potentials were referenced to ferrocenium/ferrocene (Fc+/ Fc).
1, 3-Bis(3, 4-dimethoxyphenyl)dibenzo[e, h]azulene-2, 8- dione (1): To a solution of 2, 3:6, 7-dibenzocyclohepta- diene-1, 4, 5-trione (2.70 g, 9.4 mmol) and 1, 3-bis(3, 4-dime- thoxyphenyl)-2-propanone (3.12 g, 9.4 mmol, 1 equiv.) in 90 mL of anhydrous tetrahydrofuran (THF) was added 1, 8-diazabicyclo(5.4.0)undec-7-ene (DBU) (0.7 mL, 4.7 mmol, 0.5 equiv.). The reaction mixture was stirred at room temperature for 5 h and then quenched with 1 mol/L HCl aqueous solution. The resulting mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried with anhydrous MgSO4 and concentrated under reduced pressure. The crude mixture was triturated with cold methanol/diethyl ether (V:V=20:1) and dried under vacuum to afford 3.40 g (6.4 mmol) of 1 as a dark green solid in a yield of 68%. m.p. 214~216 ℃; 1H NMR (CDCl3) δ: 7.62 (d, J=7.6 Hz, 2H), 7.39~7.25 (m, 6H), 6.89~6.81 (m, 4H), 6.61 (d, J=1.6 Hz, 2H), 3.89 (s, 6H), 3.65 (s, 6H); 13C NMR (CDCl3) δ: 200.9, 199.8, 149.8, 149.2, 148.7, 141.5, 131.8, 131.5, 130.2, 130.1, 128.3, 126.7, 123.4, 123.0, 112.6, 111.2, 55.9, 55.7; HRMS (MALDI-TOF) calcd for C34H26O6 530.1724, found 530.1724.
1, 4-Bis(3, 4-dimethoxyphenyl)tribenzo[a, c, e]cyclohep- tatriene-9-one (2a): To a solution of 1 (2.71 g, 5.1 mmol) in 17 mL of toluene was added bicyclo[2.2.1]hepta-2, 5-diene (1.0 mL, 9.8 mmol, 1.9 equiv.) under atmosphere of nitrogen. The reaction mixture was refluxed for 72 h. The crude mixture was concentrated under reduced pressure and purified by column chromatography on silica gel with hexane/CH2Cl2/diethyl ether (V:V:V=1:1:1) as eluent to afford 2.23 g (0.43 mmol) of 2a as a white solid in a yield of 83%. m.p. 282~287 ℃; 1H NMR (CDCl3) δ: 7.54 (s, 2H), 7.51, (d, J=7.6 Hz, 2H), 7.23~7.19 (m, 2H), 7.00~6.97 (m, 4H), 6.81 (br s, 4H), 6.25 (br s, 2H), 3.88 (s, 6H), 3.53 (s, 6H); 13C NMR (CDCl3) δ: 199.7, 148.3, 147.9, 145.9, 141.6, 135.6, 135.0, 134.6, 133.7, 131.3, 129.5, 127.6, 124.9, 121.8, 113.2, 111.0, 55.9, 55.7; HRMS (MALDI- TOF) calcd for C35H28O5 528.1931, found 528.1939.
1, 4-Bis(3, 4-dihexoxyphenyl)tribenzo[a, c, e]cyclohepta- triene-9-one (2b): To a suspension of 2a (164 mg, 0.31 mmol) in 20 mL of anhydrous CH2Cl2 was added solution of BBr3 (5.0 mL, 1 mol/L in CH2Cl2, 5.0 mmol, 16 equiv.) at 0 ℃ under atmosphere of nitrogen. The reaction mixture was warmed to room temperature and heated to reflux for 4 h, and then was cooled with ice bath and quenched with H2O. The mixture was extracted with diethyl ether, washed with water and brine, dried with anhydrous MgSO4, and concentrated under reduced pressure. The resulting tetraol was used in the next step without further purification.
To a suspension of the crude tetraol and K2CO3 (858 mg, 6.2 mmol, 20 equiv.) in 3 mL of N, N-dimethylformamide (DMF) was added 1-bromohexane (0.67 mL, 6.2 mmol, 20 equiv.) under atmosphere of nitrogen. The reaction mixture was heated at 80 ℃ overnight, cooled to room temperature, and then quenched with water. The resulting mixture was extracted with diethyl ether, washed with water and brine, dried with anhydrous MgSO4. The resulting solution was concentrated under reduced pressure and the crude product was purified by column chromatography on silica gel with hexane/CH2Cl2 (V:V=1:1~1:2) as eluent to afford 213 mg (0.26 mmol) of 2b as pale-yellow oil in a yield of 84%. 1H NMR (CDCl3) δ: 7.52 (s, 2H), 7.50 (d, J=7.2 Hz, 2H), 7.20 (t, J=8.0 Hz, 2H), 7.02~6.94 (m, 4H), 6.78 (br s, 6H), 4.02~3.93 (m, 4H), 3.73~3.47 (m, 4H), 1.84~1.71 (m, 8H) 1.48~1.35 (m, 24H), 0.95~0.90 (m, 12H); 13C NMR (CDCl3) δ: 199.7, 148.6, 147.9, 145.8, 141.7, 135.5, 135.2, 134.6, 133.7, 131.2, 129.3, 127.4, 124.8, 121.9, 115.4, 113.5, 31.7, 31.6, 29.3, 29.1, 25.7, 25.8, 22.7, 22.7, 14.1, 14.1; HRMS (MALDI-TOF) calcd for C55H68O5 808.5061, found 808.5053.
3: To a solution of 2b (213 mg, 0.26 mmol) and 2, 3-dichloro-5, 6-dicyanobenzoquinone (131 mg, 0.58 mmol, 2.2 equiv.) in 80 mL of anhydrous CH2Cl2 was added trifluoromethanesulfonic acid (1.4 mL, 15.8 mmol, 61 equiv.) under atmosphere of nitrogen. The reaction mixture was kept stirred at 0 ℃ for 30 min and then quenched with 10% NaHCO3 aqueous solution. The resulting mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried with anhydrous MgSO4 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel with hexane/CH2Cl2 (V:V=1:1~1:2) as eluent to afford 104 mg (0.13 mmol) of 3 as a pale-yellow solid in a yield of 50%. m.p. 145~147 ℃; 1H NMR (CDCl3) δ: 8.64 (s, 2H), 8.61 (d, J=7.2 Hz, 2H), 7.87 (m, 4H), 7.81 (2H), 7.78 (t, J=8.0 Hz, 2H), 4.23~4.19 (m, 8H), 1.98~1.93 (m, 8H), 1.61~1.57 (m, 8H), 1.45~1.40 (m, 16H), 0.98~0.92 (m, 12H); 13C NMR (CDCl3) δ: 202.7, 150.1, 150.1, 141.4, 130.5, 129.3, 127.4, 126.9, 126.3, 125.8, 124.9, 123.7, 123.5, 122.1, 107.2, 106.0, 31.8, 29.4, 29.4, 25.9, 22.8, 22.8, 14.2, 14.2; HRMS (MALDI-TOF) calcd for C55H64O5 804.4748, found 804.4735.
4: To a solution of 3 (55 mg, 0.07 mmol) in 6 mL of anhydrous CH2Cl2 was added boron trifluoride diethyl etherate (0.1 mL, 0.81 mmol, 12 equiv.) at 0 ℃ under atmosphere of nitrogen. The reaction mixture was kept stirred at 0 ℃ for 10 min. Then, a solution of (trimethylsilyl)diazomethane (0.05 mL, 2.0 mol/L in hexane, 0.1 mmol, 1.4 equiv.) was added into the mixture. The resulting mixture was kept stirred at 0 ℃ for 2.5 h. The reaction mixture was quenched with water. The resulting mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried with anhydrous MgSO4 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel with hexane/CH2Cl2 (V:V=1:2) as eluent to afford 46 mg (0.056 mmol) of 4 as yellow solid in a yield of 80%. m.p. 191~194 ℃; 1H NMR (CDCl3) δ: 8.577~8.582 (m, 2H), 8.39 (d, J=7.2 Hz, 1H), 8.29~8.27 (m, 2H), 7.93 (s, 1H), 7.78 (s, 1H), 7.74 (s, 1H), 7.73 (t, J=7.6 Hz, 1H), 7.58~7.56 (m, 2H), 7.42 (t, J=7.6 Hz, 1H), 4.60 (d, J=10.8 Hz, 1H), 4.32~4.14 (m, 8H), 3.86 (d, J=10.8 Hz, 1H), 2.00~1.86 (m, 8H), 1.55~1.51 (m, 8H), 1.40~1.37 (m, 16H), 0.97~0.89 (m, 12H); 13C NMR (CDCl3) δ: 194.4, 150.5, 150.0, 150.0, 149.9, 136.0, 135.1, 132.2, 132.0, 131.5, 130.3, 129.9, 129.8, 128.5, 128.1, 127.8, 127.1, 125.7, 125.4, 125.2, 124.9, 124.7, 124.6, 124.5, 122.7, 122.2, 121.3, 107.5, 107.3, 106.5, 106.2, 69.6, 69.5, 69.3, 47.7, 31, 8, 31.7, 29.4, 29.3, 29.3, 25.9, 25.8, 22.8, 22.7, 14.2, 14.2; HRMS (MALDI- TOF) calcd for C56H66O5 818.4905, found 818.4913.
1, 4-Bis(3, 5-dimethoxyphenyl)-2, 3-diphenyl-tribenzo- [a, c, e]cycloheptatriene-9-one (6a): To a solution of 5 (3.0 g, 5.7 mmol) and diphenylacetylene (1.02 g, 5.7 mmol, 1 equiv.) in 4 mL of diphenyl ether was refluxed for 16 h under atmosphere of nitrogen. The mixture was cooled to room temperature, filtered and triturated with cold methanol to afford 2.72 g (4.0 mmol) of 6a as beige solid in a yield of 70%. m.p. not melt when heated to 300 ℃. 1H NMR (CDCl3) δ: 7.44 (d, J=7.6 Hz, 2H), 7.39 (d, J=7.6 Hz, 2H), 7.17 (t, J=7.6 Hz, 2H), 7.11 (d, J=8.0 Hz, 2H), 7.06 (t, J=7.2 Hz, 2H), 6.95 (t, J=7.6 Hz, 2H), 6.85 (t, J=7.2 Hz, 2H), 6.76 (t, J=7.6 Hz, 2H), 6.46 (d, J=7.6 Hz, 2H), 6.32 (s, 2H), 6.00 (t, J=2.4 Hz, 2H), 5.63 (s, 2H), 3.54 (s, 6H), 3.36 (s, 6H); 13C NMR (CDCl3) δ: 200.3, 159.8, 159.3, 146.1, 142.3, 141.3, 140.2, 135.0, 132.8, 131.7, 130.1, 129.1, 127.3, 127.3, 126.0, 125.8, 124.4, 111.8, 108.3, 99.3, 55.4, 55.3; HRMS (MALDI-TOF) calcd for C47H36O5 680.2557, found 680.2563.
1, 4-Bis(3, 5-dihexoxyphenyl)-2, 3-diphenyl-tribenzo- [a, c, e]cycloheptatriene-9-one (6b): To a suspension of 6a (1.0 g, 1.5 mmol) in 120 mL of anhydrous CH2Cl2 was added a solution of BBr3 (30.0 mL, 1 mol/L in CH2Cl2, 30 mmol, 20 equiv.) at 0 ℃ under atmosphere of nitrogen. The reaction mixture was warmed to room temperature and heated to reflux for 4 h, and then was cooled with ice bath and quenched with H2O. The mixture was extracted with diethyl ether, washed with water and brine, dried with anhydrous MgSO4, and concentrated under reduced pressure. The resulting tetraol was used in the next step without further purification.
To a suspension of the crude tetraol and K2CO3 (4.1 g, 30 mmol, 20 equiv.) in 15 mL of DMF was added 1-bromohexane (4.2 mL, 30 mmol, 20 equiv.) under atmosphere of nitrogen. The reaction mixture was heated at 80 ℃ overnight, cooled to room temperature, and then quenched with water. The resulting mixture was extracted with diethyl ether, washed with water and brine, dried with anhydrous MgSO4. The resulting solution was concentrated under reduced pressure and the crude product was purified by column chromatography on silica gel with hexane/CH2Cl2 (V:V=1:1~1:2) as eluent to afford 1.04 g (1.08 mmol) of 6b as white solid in a yield of 72%. m.p. 109~112 ℃; 1H NMR (CDCl3) δ: 7.42 (d, J=7.6 Hz, 2H), 7.36 (d, J=7.6 Hz, 2H), 7.14~7.11 (m, 4H), 7.02 (t, J=7.6 Hz, 2H), 6.95 (t, J=8.4 Hz, 2H), 6.81 (t, J=7.2 Hz, 2H), 6.75 (t, J=7.6 Hz, 2H), 6.45 (d, J=7.6 Hz, 2H), 6.30 (s, 2H), 6.00 (t, J=2.4 Hz, 2H), 5.61 (s, 2H), 3.74~3.70 (m, 2H), 3.61~3.55 (m, 4H), 3.37~3.33 (m, 2H), 1.62~1.54 (m, 4H), 1.52~1.51 (m, 4H), 1.34~1.30 (m, 24H), 0.93~0.90 (m, 12H); 13C NMR (CDCl3) δ: 200.4, 159.3, 158.8, 146.1, 142.6, 142.1, 141.4, 140.3, 135.4, 135.1, 132.8, 131.8, 130.2, 129.0, 127.3, 127.2, 126.0, 125.7, 124.3, 112.6, 109.3, 100.8, 68.3, 68.1, 31.7, 29.2, 29.0, 22.8, 14.2; HRMS (MALDI-TOF) calcd for C67H76O5 960.5687, found 960.5681.
7: To a solution of 6b (777 mg, 0.81 mmol) and 2, 3-di- chloro-5, 6-dicyanobenzoquinone (1.01 g, 4.44 mmol, 5.5 equiv.) in 400 mL of anhydrous CH2Cl2 was added trifluoromethanesulfonic acid (18 mL, 202 mmol, 250 equiv.) under atmosphere of nitrogen. The reaction mixture was stirred at room temperature for 18 h and then carefully quenched with 10% NaHCO3 solution. The resulting mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried with anhydrous MgSO4 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel with hexane/CH2Cl2 (V:V=1:1~1:2) as eluent and subsequently purified by preparative thin layer chromatography with hexane/CH2Cl2 (V:V=1:1) as eluent to afford 392 mg (0.43 mmol) of 7 as yellow solid in a yield of 53%. m.p. 194~196 ℃; 1H NMR (CDCl3) δ: 9.71 (d, J=8.4 Hz, 2H), 9.40 (d, J=8.0 Hz, 2H), 8.99 (d, J=8.0 Hz, 2H), 8.04 (t, J=8.0 Hz, 2H), 7.84 (d, J=7.2 Hz, 2H), 7.77 (t, J=8.0 Hz, 2H), 7.16 (s, 2H), 4.32~4.24 (m, 8H), 2.10~2.03 (m, 4H), 1.98~1.91 (m, 4H), 1.67~1.63 (m, 8H), 1.46~1.35 (m, 16H), 0.97~0.91 (m, 12H); 13C NMR (CDCl3) δ: 199.6, 156.8, 156.2, 140.4, 130.7, 130.5, 130.1, 129.5, 128.9, 128.1, 127.4, 126.9, 126.6, 126.5, 124.9, 124.9, 124.5, 112.2, 111.3, 99.3, 70.0, 69.7, 31.7, 31.6, 29.5, 26.2, 26.1, 22.8, 22.7, 14.2, 14.1; HRMS (MALDI-TOF) calcd for C67H66O5 950.4905, found 950.4901.
8: To a solution of 7 (392 mg, 0.41 mmol) in 28 mL of anhydrous CH2Cl2 was added boron trifluoride diethyl etherate (0.70 mL, 5.67 mmol, 14 equiv.) at 0 ℃ under atmosphere of nitrogen. The reaction mixture was kept stirred at 0 ℃ for 10 min. Then, a solution of (trimethylsilyl)diazomethane (0.3 mL, 2.0 mol/L in hexane, 0.6 mmol, 1.5 equiv.) was added into the mixture slowly. The resulting mixture was kept stirred at 0 ℃ for 2.5 h. The reaction mixture was quenched with water. The resulting mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried with anhydrous MgSO4 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel with hexane/CH2Cl2 (V:V=1:1~1:2) as eluent to afford 318 mg (0.33 mmol) of 8 as yellow solid in a yield of 80%. m.p. 258~262 ℃; 1H NMR (CDCl3) δ: 9.59 (d, J=8.0 Hz, 1H), 9.46 (d, J=8.0 Hz, 1H), 9.25 (t, J=8.0 Hz, 2H), 9.00 (d, J=7.6 Hz, 2H), 8.34 (d, J=7.6 Hz, 1H), 8.08~8.05 (m, 2H), 7.65 (t, J=7.6 Hz, 1H), 7.47~7.40 (m, 2H), 7.14 (d, J=6.8 Hz, 2H), 4.40~4.20 (m, 4H), 4.18~4.05 (m, 4H), 4.08 (d, J=11.2 Hz, 2H), 3.66 (d, J=10.8 Hz, 2H), 2.12~1.98 (m, 8H), 1.63~1.45 (m, 8H), 1.42~1.25 (m, 16H), 1.00~0.92 (m, 12H); 13C NMR (CDCl3) δ: 193.3, 157.9, 156.4, 155.9, 155.8, 136.0, 135.1, 133.1, 132.6, 131.4, 131.3, 130.4, 130.3, 129.8, 128.2, 128.0, 127.9, 127.8, 127.3, 127.1, 126.9, 126.7, 126.5, 126.3, 126.1, 125.5, 125.4, 125.2, 124.7, 124.2, 124.1, 124.0, 123.1, 112.4, 112.1, 111.7, 108.8, 98.9, 98.1, 70.0, 69.8, 69.6, 69.4, 48.1, 31.8, 31.7, 31.7, 29.5, 29.5, 29.4, 26.2, 26.2, 26.1, 22.8, 22.7, 22.7, 22.7, 14.2, 14.2, 14.2; HRMS (MALDI-TOF) calcd for C68H68O5 964.5061, found 964.5070.
9: To a mixture of 8 (40 mg, 0.04 mmol), lithium aluminum hydride (39 mg, 1.0 mmol, 25 equiv.), and aluminum chloride (48 mg, 0.34 mmol, 9 equiv.) was added 5 mL of anhydrous THF at room temperature under atmosphere of nitrogen. The reaction mixture was kept stirred at room temperature for 16 h. The reaction mixture was quenched with water. The resulting mixture was extracted with CH2Cl2. The organic layer was washed with brine, dried with anhydrous MgSO4 and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel with hexane/CH2Cl2 (V:V= 3:1) as eluent to afford 27 mg (0.029 mmol) of 9 as yellow solid in a yield of 72%. m.p. 173~178 ℃; 1H NMR (CDCl3) δ: 9.43 (d, J=8 Hz, 2H), 9.15 (d, J=8.0 Hz, 2H), 8.99 (d, J=8.0 Hz, 2H), 8.05 (t, J=8.0 Hz, 2H), 7.44 (t, J=8.0 Hz, 2H), 7.29 (d, J=6.8 Hz, 2H), 7.03 (s, 2H), 4.37~4.15 (m, 8H), 3.24~3.10 (m, 4H), 2.07~1.96 (m, 8H), 1.64~1.60 (m, 8H), 1.45~1.38 (m, 8H), 1.26 (s, 16H), 0.97~0.87 (m, 12H); 13C NMR (CDCl3) δ: 156.3, 155.2, 139.7, 132.5, 129.5, 129.4, 129.1, 128.7, 126.4, 126.1, 126.1, 125.9, 125.5, 124.4, 122.8, 122.3, 120.0, 119.1, 112.1, 111.4, 98.5, 69.8, 69.6, 37.6, 31.8, 31.8, 29.9, 29.6, 26.2, 26.2, 22.8, 22.7, 14.3, 14.2, 14.2; HRMS MALDI-TOF calcd for C68H70O4 950.5269, found 950.5273.
Supporting Information X-ray crystallographic infor-mation files for compounds 9 and NMR spectra. The Supporting Information is available free of charge via the Internet at http://siocjournal.cn/.
(a) Narita, A.; Wang, X.-Y.; Feng, X.; Müllen, K. Chem. Soc. Rev. 2015, 44, 6616.
(b) Segawa, Y.; Ito, H.; Itami, K. Nat. Rev. Mater. 2016, 1, 15002.
(c) Rickhaus, M.; Mayor, M.; Juríček, M. Chem. Soc. Rev. 2017, 46, 1643.
(d) Cruz, C. M.; Castro-Fernández, S.; Maçôas, E.; Millán, A.; Campaña, A. G. Synlett 2019, 30, 997.
Bharat; Bhola, R.; Bally, T.; Valente, A.; Cyrański, M. K.; Dobrzycki, Ł.; Spain, S. M.; Rempała, P.; Chin, M. R.; King, B. T. Angew. Chem., Int. Ed. 2010, 49, 399. doi: 10.1002/anie.200905633
Barth, W. E.; Lawton, R. G. J. Am. Chem. Soc. 1966, 88, 380. doi: 10.1021/ja00954a049
Luo, J.; Xu, X.; Mao, R.; Miao, Q. J. Am. Chem. Soc. 2012, 134, 13796. doi: 10.1021/ja3054354
(a) Kawasumi, K.; Zhang, Q.; Segawa, Y.; Scott, L. T.; Itami, K. Nat. Chem. 2013, 5, 739.
(b) Márquez, I. R.; Fuentes, N.; Cruz, C. M.; Puente-Muñoz, V.; Sotorrios, L.; Marcos, M. L.; Choquesillo-Lazarte, D.; Biel, B.; Crovetto, L.; Gómez-Bengoa, E.; González, M. T.; Martin, R.; Cuerva, J. M.; Campaña, A. G. Chem. Sci. 2017, 8, 1068.
(c) Fukui, N.; Kim, T.; Kim, D.; Osuka, A. J. Am. Chem. Soc. 2017, 139, 9075.
(d) Gu, X.; Li, H.; Shan, B.; Liu, Z.; Miao, Q. Org. Lett. 2017, 19, 2246.
(e) Oki, K.; Takase, M.; Mori, S.; Shiotari, A.; Sugimoto, Y.; Ohara, K.; Okujima, T.; Uno, H. J. Am. Chem. Soc. 2018, 140, 10430.
(f) Fernández-García, J. M.; Evans, P. J.; Medina Rivero, S.; Fernández, I.; García-Fresnadillo, D.; Perles, J.; Casado, J.; Martín, N. J. Am. Chem. Soc. 2018, 140, 17188.
(g) Farrell, J. M.; Grande, V.; Schmidt, D.; Würthner, F. Angew. Chem. 2019, 131, 16656.
Pun, S. H.; Chan, C. K.; Luo, J.; Liu, Z.; Miao, Q. Angew. Chem., Int. Ed. 2018, 57, 1581. doi: 10.1002/anie.201711437
(a) Feng, C.-N.; Kuo, M.-Y.; Wu, Y.-T. Angew. Chem., Int. Ed. 2013, 52, 7791.
(b) Sakamoto, Y.; Suzuki, T. J. Am. Chem. Soc. 2013, 135, 14074.
(c) Miller, R. W.; Duncan, A. K.; Schneebeli, S. T.; Gray, D. L.; Whalley, A. C. Chem. Eur. J. 2014, 20, 3705.
(d) Chen, F.; Hong, Y. S.; Shimizu, S.; Kim, D.; Tanaka, T.; Osuka, A. Angew. Chem., Int. Ed. 2015, 54, 10639.
(e) Miller, R. W.; Averill, S. E.; Van Wyck, S. J.; Whalley, A. C. J. Org. Chem. 2016, 81, 12001.
(f) Cheung, K. Y.; Chan, C. K.; Liu, Z.; Miao, Q. Angew. Chem., Int. Ed. 2017, 56, 9003.
(g) Pun, S. H.; Wang, Y.; Chu, M.; Chan, C. K.; Li, Y.; Liu, Z.; Miao, Q. J. Am. Chem. Soc. 2019, 141, 9680.
Mackay, A. L.; Terrones, H. Nature 1991, 352, 762.
Kim, K.; Lee, T.; Kwon, Y.; Seo, Y.; Song, J.; Park, J.; Lee, H.; Park, J. Y.; Ihee, H.; Cho, S. J.; Ryoo, R. Nature 2016, 535, 131. doi: 10.1038/nature18284
Braun, E.; Lee, Y.; Moosavi, S. M.; Barthel, S.; Mercado, R.; Baburin, I. A.; Proserpio, D. M.; Smit, B. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E8116. doi: 10.1073/pnas.1805062115
(a) Pun, S. H.; Miao, Q. Acc. Chem. Res. 2018, 51, 1630.
(b) Márquez, I. R.; Castro-Fernández, S.; Millán, A.; Campaña, A. G. Chem. Commun. 2018, 54, 6705.
(c) Stępień, M.; Majewski, M. A. Angew. Chem., Int. Ed. 2019, 58, 86.
Christoph, H.; Grunenberg, J.; Hopf, H.; Dix, I.; Jones, P. G.; Scholtissek, M.; Maier, G. Chem. Eur. J. 2008, 14, 5604. doi: 10.1002/chem.200701837
(a) Wong, H. N. C.; Sondheimer, F. Tetrahedron 1981, 37, 99.
(b) Wong, H. N. C. Acc. Chem. Res. 1989, 22, 145.
(a) Müller, M.; Iyer, V. S.; Kübel, C.; Enkelmann, V.; Müllen, K. Angew. Chem., Int. Ed. Engl. 1997, 36, 1607.
(b) Nobusue, S.; Fujita, K.; Tobe, Y. Org. Lett. 2017, 19, 3227.
(c) Tamoto, A.; Aratani, N.; Yamada, H. Chem. Eur. J. 2017, 23, 16388.
Cheung, K. Y.; Xu, X.; Miao, Q. J. Am. Chem. Soc. 2015, 137, 3910. doi: 10.1021/jacs.5b00403
Resendiz, M. J. E.; Garcia-Garibay, M. A. Org. Lett. 2005, 7, 371. doi: 10.1021/ol0480527
(a) Grzybowski, M.; Skonieczny, K.; Butenschön, H.; Gryko, D. T. Angew. Chem. Int. Ed. 2013, 52, 9900.
(b) Grzybowski, M.; Sadowski, B.; Butenschön, H.; Gryko, D. Angew. Chem., Int. Ed. 2019, 59, 2998.
(a) Scholl, R.; Mansfeld, J. Ber. Dtsch. Chem. Ges. 1910, 43, 1734.
(b) Scholl, R.; Seer, C.; Weitzenböck, R. Ber. Dtsch. Chem. Ges. 1910, 43, 2202.
Zhai, L.; Shukla, R.; Rathore, R. Org. Lett. 2009, 11, 3474. doi: 10.1021/ol901331p
(a) Plunkett, K. N.; Godula, K.; Nuckolls, C.; Tremblay, N.; Whalley, A. C.; Xiao, S. Org. Lett. 2009, 11, 2225.
(b) Zhai, L.; Shukla, R.; Wadumethrige, S. H.; Rathore, R. J. Org. Chem. 2010, 75, 4748.
(c) Ip, H.-W.; Ng, C.-F.; Chow, H.-F.; Kuck, D. J. Am. Chem. Soc. 2016, 138, 13778.
(a) Hennig, R.; Metz, P. Angew. Chem. Int. Ed. 2009, 48, 1157.
(b) Sakai, T.; Ito, S.; Furuta, H.; Kawahara, Y.; Mori, Y. Org. Lett. 2012, 14, 4564.
(a) Chaffins, S.; Brettreich, M.; Wudl, F. Synthesis 2002, 9, 1191.
(b) Sletten, E. M.; Nakamura, H.; Jewett, J. C.; Bertozzi, C. R. J. Am. Chem. Soc. 2010, 132, 11799.
King, B. T.; Kroulik, J.; Robertson, C. R.; Rempala, P.; Hilton, C. L.; Korinek, J. D.; Gortari, L. M. J. Org. Chem. 2007, 72, 2279. doi: 10.1021/jo061515x
CCDC2003773 contains the crystallographic data for 9. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Anslyn, E. V.; Dougherty, D. A. Modern Physical Organic Chemistry, University Science Books, Sausalito, 2004, Chapter 1, p. 22.
The commonly used formal potential of the redox couple of ferrocenium/ferrocene (Fc+/Fc) in the Fermi scale is -5.1 eV, which is calculated on the basis of an approximation neglecting solvent effects using a work function of 4.46 eV for the normal hydrogen electrode (NHE) and an electrochemical potential of 0.64 V for (Fc+/Fc) versus NHE. See: Cardona, C. M.; Li, W.; Kaifer, A. E.; Stockdale, D.; Bazan, G. C. Adv. Mater. 2011, 23, 2367.
(a) Wu, J.; Pisula, W.; Müllen, K. Chem. Rev. 2007, 107, 718.
(b) Zhi, L.; Müllen, K. J. Mater. Chem. 2008, 18, 1472.

Scheme 1 (a) Reported introduction of seven-membered rings into polycyclic arenes by ring expansion, and (b) introduction of an eight-membered ring to polycyclic arenes by ring expansion

Figure 1 Crystal structure of 9
(a) Side view showing the saddle-like shape; (b) top view with C—C single bonds in the eight-membered ring highlighted; (c) molecular packing with π-π stacking. Hexyl groups removed for clarity. Carbon and oxygen atoms in (a) and (b) are shown as grey and red ellipsoids, respectively, at 50% probability level

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:


