Received Date:
01 June 2019 Accepted Date:
08 October 2019 Available Online:
01 January 2020
Abstract:
Formic acid is the simplest carboxylic acid. It is colorless, low toxicity, and easy to transport and store at room temperature. Recently, formic acid is one of the most promising hydrogen storage materials, and the use of heterogeneous catalysts to decompose formic acid to produce hydrogen at room temperature has attracted wide attention of researchers. Compared to the other catalysts, Pd-based catalysts exhibit excellent performance for the decomposition of formic acid to yield hydrogen under mild condition. The properties and preparation methods of Pd-based catalysts and the applications in the fields of the production of hydrogen from formic acid decomposition are reviewed. The future research directions of Pd-based catalysts are also discussed.
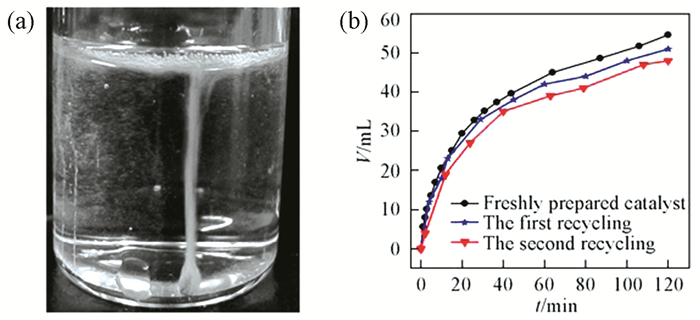
Figure 1.
(a) Photo for gas generation by the decomposition of formic acid in the presence of PdMg catalyst;
(b) Recycle tests of PdMg catalyst toward gas generation from formic acid at room temperature
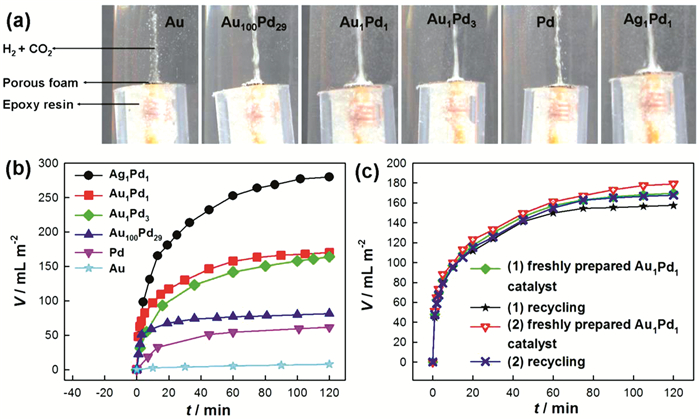
Figure 4.
(a) Photos for the hydrogen production at room temperature from the decomposition of formic acid on the AuPd and AgPd foam films; (b) The output volume of reforming gas on the foam films of AuPd and Ag1Pd1; (c) Cycling performances of the Au1Pd1 foam film[23]
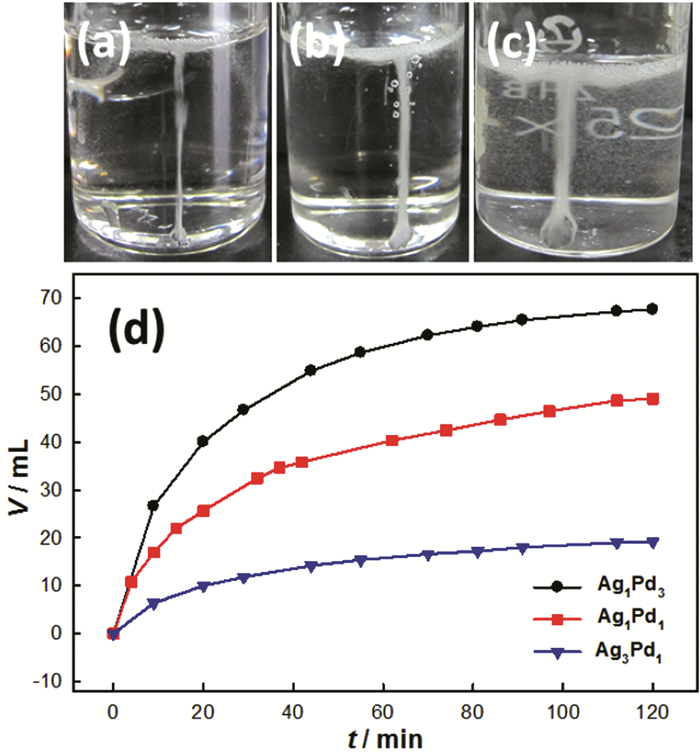
Figure 5.
(a~c) Photos for the hydrogen production at room temperature from the decomposition of formic acid on the AgPd catalysts; (d) The output volume of reforming gas on the foam films of AgPd catalysts[24]
Figure 1
(a) Photo for gas generation by the decomposition of formic acid in the presence of PdMg catalyst;
(b) Recycle tests of PdMg catalyst toward gas generation from formic acid at room temperature
Figure 4
(a) Photos for the hydrogen production at room temperature from the decomposition of formic acid on the AuPd and AgPd foam films; (b) The output volume of reforming gas on the foam films of AuPd and Ag1Pd1; (c) Cycling performances of the Au1Pd1 foam film[23]
Figure 5
(a~c) Photos for the hydrogen production at room temperature from the decomposition of formic acid on the AgPd catalysts; (d) The output volume of reforming gas on the foam films of AgPd catalysts[24]

 下载:
下载:

 下载:
下载:







