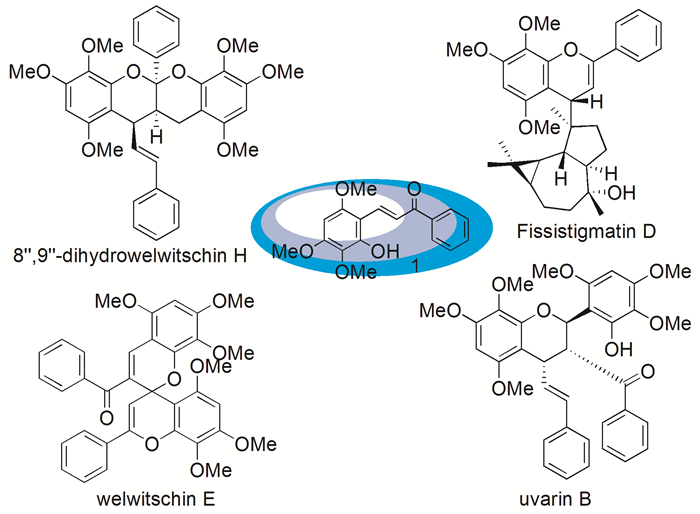
图式 1.
含2-羟基-3, 4, 6-三甲氧基查尔酮单元的天然产物
Scheme 1.
Natural products containing the 2-hydroxy-3, 4, 6-trimethoxychalcone unit

查尔酮及其衍生物广泛存在于蔬菜、水果、茶叶和其他植物当中,是生物体内合成黄酮和异黄酮类化合物的重要前体,具有抗肿瘤、抗炎、抗菌、抗病毒以及抑制和清除氧自由基等多种生物活性[1~7],是一类药物研发的重要先导化合物。截至目前,已经有部分查尔酮类化合物作为临床药物使用。例如,作为利胆药的美托查酮(metochalcone)和作为治疗胃溃疡及慢性胃炎药物的素法酮(sofalcone)[6, 7]。近来,基于其抗病毒、抗炎抗菌、免疫调节和抗纤维化等作用,方琦璐等[8]对其防治新型冠状病毒肺炎(COVID-2019)的潜在应用进行了探讨。此外,查尔酮类化合物在杀虫、杀螨和抗病毒等农药开发领域也展现出一定的潜力[9~11]。
天然产物2-羟基-3, 4, 6-三甲氧基查尔酮(1)于1992年由Colegate等[12]首次从番茄枝科植物Ellipeia cuneifolia的根中分离得到(图式 1)。随后,Nkunya等[13]在Uvaria dependens的根提取物中,Lien等[14]在Fissistigma bracteolatum的叶提取物中,Salae等[15]在Uvaria siamensis的根提取物中均分离到了该化合物。同时,在具有抗疟原虫活性的天然产物welwitschin E、uvarin B和8″, 9″-dihydrowelwitschin H中[15],以及香木兰烷型倍半萜杂黄酮分子Fissistigmatin D[16]中均含有2-羟基-3, 4, 6-三甲氧基查尔酮结构单元(图式 1)。
由于查尔酮及其衍生物多具有重要的生理活性,已经发展了Claisen-Schmidt缩合反应[17~19]、Suzuki偶联反应[20]、Heck反应[21, 22]、Wittig反应[23, 24]、Julia-Kocienski烯烃化反应[25]、Friedel-Crafts酰化反应[26]和光催化的Fries重排反应[27]等多种有效的合成查尔酮骨架的方法。尽管如此,尚未见关于查尔酮1的合成报道。
发展一种高效的合成化合物1的方法,对于该类查尔酮化合物的进一步生物活性研究以及对含有该片段的活性天然产物8″, 9″-dihydro-welwitschin H,Fissistigmatin D、Welwitschin E和Uvarin B的全合成研究具有重要的意义。本文介绍了一种以商业可得的1, 3, 5-三甲氧基苯为原料,在10g级规模高效合成化合物1的方法。
1, 3, 5-三甲氧基苯、高碘酸、三氧化铬、连二亚硫酸钠、氢化钠、碘甲烷、三氯氧磷、氢氧化钠、三氯化硼、2-溴苯乙酮、三苯基膦、乙腈、乙酸乙酯、N, N-二甲基甲酰胺(DMF)、四氢呋喃和二氯甲烷等均为市售分析纯级试剂。
上海顾村ZF-II型四用紫外分析仪;YRT-3型熔点仪;Bruker AMX-500型核磁共振谱仪;Finnigan MAT型质谱仪。
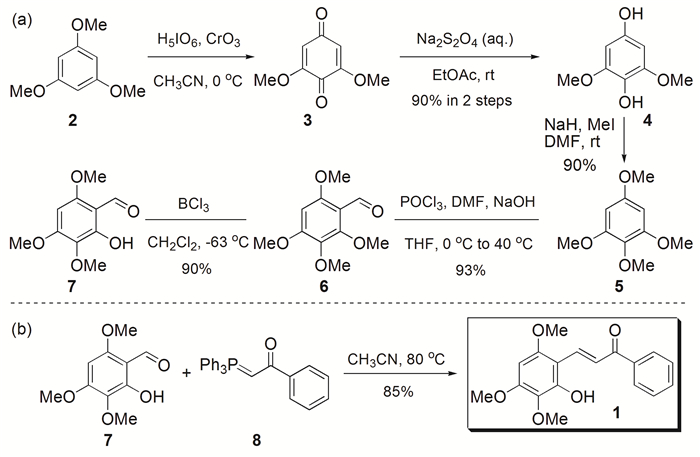
目标产物1的合成路线如图式 2所示。通过改进Maes等[28]合成2-羟基-3, 4, 6-三甲氧基苯甲醛(7)的甲基化和甲酰化条件,以商业可得的1, 3, 5-三甲氧基苯(2)为原料,经高碘酸氧化、保险粉还原、碘甲烷甲基化、Vilsmeier-Haack甲酰化和三氯化硼脱甲基共5步转化合成7(图式 2(a))。通过方法改良,将7的合成总收率从文献报道的22%[28]提高至68%。接着,由7和膦叶立德(8)经Wittig反应在50mmol规模以85%的产率合成了产物1(图式 2(b))。
2, 6-二苯甲醚-1, 4-二酚(4)的合成:向1L的圆底烧瓶中依次加入27.4g高碘酸和570mL乙腈,剧烈搅拌15min至高碘酸完全溶解;接着加入0.3g三氧化铬并将体系冷却至0℃;然后将5.05g的1, 3, 5-三甲氧基苯溶于30mL乙腈中并一次性加入体系,搅拌1h。TLC监测至原料2完全消耗后,旋蒸除去乙腈。所得棕色固体用200mL二氯甲烷和200mL水溶解后,用二氯甲烷萃取3次,饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后得中间体3, 直接用于下一步反应[29]。
向1L的圆底烧瓶中分别加入上述合成的中间体3和150mL乙酸乙酯,将20.88g连二亚硫酸钠溶于300mL水后逐滴加入体系中,室温下搅拌1h。TLC监测至原料3完全消耗后,用乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后,用乙酸乙酯打浆并过滤得4.59g淡黄色固体4[28],两步总产率90%,熔点158~161℃。1H NMR(500MHz,(CD3)2CO)δ:7.71(s,1H),6.44(s,1H),6.17(s,2H),3.77(s,6H);13C NMR (126MHz,(CD3)2CO)δ:150.04,148.20,129.18,93.54,55.56。
1, 2, 3, 5-四甲氧基苯(5)的合成:向500mL圆底烧瓶中加入3.02g氢化钠(60% in mineral oil)后用氩气进行保护,再加入80mL无水DMF;接着,将5.1g化合物4溶于100mL无水DMF后逐滴加入到上述溶液中,室温搅拌20min;最后,将17.03g碘甲烷缓慢滴加到体系中并搅拌1h。TLC监测至原料4完全消耗后,将反应液放入冰浴中冷却至0℃,缓慢滴入50mL水淬灭反应。减压旋蒸除去大部分DMF。浓缩液用100mL乙酸乙酯和100mL水稀释,并用乙酸乙酯萃取3次,有机相用饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后,经硅胶柱层析分离(洗脱剂∶乙酸乙酯/石油醚=1∶15)得5.5g无色油状物5[28],产率90%。1H NMR(500MHz,CDCl3)δ:6.15(s,2H),3.84(s,6H),3.79(s,3H),3.77(s,3H);13C NMR (126MHz,CDCl3)δ:156.63,154.11,132.71,92.08,61.40,56.47,55.93。
2, 3, 4, 6-四甲氧基苯甲醛(6)的合成:0℃氩气保护条件下,向50mL圆底烧瓶中依次加入1.54mL无水DMF和4mL无水四氢呋喃;接着,将1.86mL三氯氧磷逐滴加入到体系中并升至室温反应5min;最后,将1.98g化合物5溶于3mL无水四氢呋喃后滴加至体系中,升温至40℃搅拌2h。TLC监测至原料5完全消耗后,加入5.0mL 0.5mol/L的氢氧化钠溶液并搅拌10min。乙酸乙酯萃取,饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后,用硅胶柱层析分离(洗脱剂∶乙酸乙酯/石油醚=1∶3)得2.11g白色固体6[28],产率93%,熔点87~88℃。1H NMR(500MHz,CDCl3)δ:10.31(s,1H),6.28(s,1H),3.95(s,6H),3.90(s,3H),3.82(s,3H);13C NMR(126MHz,CDCl3)δ:187.91,159.19,158.89,156.64,135.94,112.59,91.68,62.07,61.17,56.20,56.09。
2-羟基-3, 4, 6-三甲氧基苯甲醛(7)的合成:向100mL的圆底烧瓶中加入2.6g化合物6,通氩气保护,再加入10mL无水二氯甲烷并将体系冷却至-63℃;接着,缓慢滴加30mL三氯化硼(1.0mol/L in CH2Cl2)溶液并搅拌1h。TLC监测至原料6完全消耗后,加入20mL水淬灭反应。二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后,用硅胶柱层析分离(洗脱剂:乙酸乙酯/石油醚=1∶5)得1.91g白色固体7[28],产率90%,熔点102~103℃。1H NMR(500MHz,CDCl3)δ:12.31(s,1H),10.18(s,1H),6.00(s,1H),4.00(s,3H),3.93(s,3H),3.87(s,3H);13C NMR(126MHz,CDCl3)δ:192.48,160.52,159.91,157.23,130.13,106.28,86.56,60.91,56.21,55.84。
向500mL的圆底烧瓶中依次加入26.22g三苯基膦、21.79g 2-溴苯乙酮和200mL甲苯,在80℃条件下搅拌反应4h。抽滤除去溶剂,滤饼用石油醚洗涤。接着,用120mL二氯甲烷/水(1∶1)的混合溶剂溶解滤饼,逐滴加入120mL 2.0mol/L的氢氧化钠溶液,室温搅拌3h后,用二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥。旋蒸除去溶剂后的膦叶立德试剂8直接用于下一步反应。
向250mL圆底烧瓶中依次加入10.6g中间体7、28.5g膦叶立德8和100mL乙腈,升温至80℃并搅拌反应6h。TLC监测至原料7完全消耗后,直接旋蒸除去溶剂并用硅胶柱层析分离(洗脱剂∶二氯甲烷/石油醚=1∶1分离除去杂质后,再用纯二氯甲烷洗脱)得13.3g黄色固体查尔酮1[12~14],产率85%,熔点135~137℃,1H NMR(500MHz,(CD3)2CO)δ:8.66(s,1H),8.27(d,J=15.7Hz,1H),8.07(d,J=15.9Hz,1H),8.03~7.99 (m,2H),7.62~7.57(m,1H),7.55~7.51(m,2H),6.37(s,1H),3.96(s,3H),3.95(s,3H),3.77(s,3H);13C NMR(126MHz,(CD3)2CO)δ:191.06,157.96,156.03,152.42,140.23,136.69,132.96,131.20,129.41,128.92,122.20,105.91,89.35,61.15,56.43,56.35;HRMS(ESI):理论值C18H19O5,m/z:315.1227 [M+H]+,实测值315.1230。
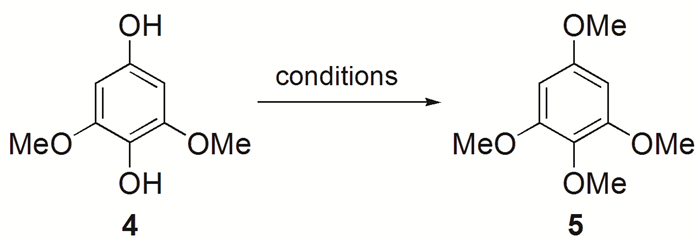
中间体5的合成中,Maes等[28]以硫酸二甲酯为甲基化试剂、碳酸钾为碱,在丙酮作溶剂、室温条件下反应24h以66%的产率获得甲基化产物(表 1,试验1)。为了缩短反应时间并提高反应产率,以碘甲烷作为甲基化试剂在与文献相同条件下进行反应,搅拌24h后以41%的产率获得产物5(表 1,试验2)。改用氢化钠作碱、DMF作溶剂时,在室温条件下搅拌2h后,以90%的产率获得甲基化产物5(表 1,试验3)。此步改良反应显著提高了收率并缩短了反应时间。
 下载:
导出CSV
下载:
导出CSV
 |
|||
| Entry | Conditions | Time/h | Yield/% |
| 1 | Me2SO4,K2CO3,acetone,rt | 24 | 66 |
| 2 | MeI,K2CO3,acetone,rt | 24 | 41 |
| 3 | MeI,NaH,DMF,rt | 2 | 90 |
中间体6的合成中,Maes等[28]运用Vilsmeier-Haack甲酰化反应,参考Iinum等[30]的方法,在室温条件下以61%的产率获得甲酰化产物(表 2,试验1)。为了提高反应收率,尝试了Katritzky等[31]的方法,先用正丁基锂攫氢,再与DMF发生加成消除反应,反应以38%的产率完成甲酰化(表 2,试验2)。最后,尝试了在40℃下进行Vilsmeier-Haack甲酰化反应(表 2,试验3),以93%的产率获得了甲酰化产物,明显提高了此步反应的产率。
下载:
导出CSV
 |
||
| Entry | Conditions | Yield/% |
| 1 | (a) POCl3,DMF,rt;(b) 2mol/L NaOH | 61 |
| 2 | (a) n-BuLi,DMF,THF,rt;(b) NH4Cl | 38 |
| 3 | (a) POCl3,DMF,THF,40 ℃;(b) 2mol/L NaOH | 93 |
目标产物查尔酮1的合成中,首先采用了Claisen-Schmidt缩合[17~19, 32]的方法(图式 3)。在室温条件下,40%的氢氧化钠溶液为碱、乙醇作溶剂,中间体7和苯乙酮9发生Claisen-Schmidt缩合反应,以63%的收率获得目标产物。但是,此反应较为缓慢,在0.1mmol规模尚需反应24h;而且产物难溶于乙醇,生成物在体系中呈粘稠状态,导致反应后处理困难,不利于放大量生产。
为了克服上述困难,鉴于课题组前期工作的积累[33],我们改用了Wittig反应[23, 24]的方法。用中间体7和膦叶立德8在乙腈作溶剂的条件下进行Wittig反应合成目标分子。当8为1.1倍量,反应温度为60℃时,有部分苯甲醛7未反应完全,产率为58%(表 3,试验1)。并且,原料7与查尔酮产物1极性相近,难以分离。为了提高反应转化率,降低分离难度,将8的用量提高至1.5倍量时,收率提高至73%,仍然有部分7剩余(表 3,试验2)。将反应温度升高至80℃时,原料7可以完全消耗,以85%的产率获得目标天然产物(表 3,试验3)。当反应温度升高至100℃时,反应收率不再有明显改善(表 3,试验4)。
下载:
导出CSV
 |
||||
| Entry | 7(equiv.) | 8(equiv.) | T/℃ | Yield/% |
| 1 | 1.0 | 1.1 | 60 | 58 |
| 2 | 1.0 | 1.5 | 60 | 73 |
| 3 | 1.0 | 1.5 | 80 | 85 |
| 4 | 1.0 | 1.5 | 100 | 83 |
本文通过对Maes等[28]合成2-羟基-3, 4, 6-三甲氧基苯甲醛(7)中甲基化和甲酰化条件的改进,以碘甲烷/氢化钠/DMF的室温条件为甲基化条件,以POCl3/DMF/THF的40℃加热为甲酰化条件,将7的合成总收率从文献报道的22%[28]提高至68%。并以7为原料,在乙腈为溶剂,80℃加热的条件下,通过Wittig反应在50mmol规模以85%的收率合成了天然产物2-羟基-3, 4, 6-三甲氧基查尔酮(1)。为该化合物的进一步活性研究以及对含有该片段的倍半萜杂黄酮分子Fissistigmatin D的全合成奠定了物质基础。
Mahapatra D K, Bharti S K, Asati V. Curr. Top. Med. Chem., 2017, 17(28): 3146~3169. doi: 10.2174/1568026617666170914160446
Dan W, Dai J. Eur. J. Med. Chem., 2020, 187: 111980~111999. doi: 10.1016/j.ejmech.2019.111980
Qin H, Zhang Z, Lekkala R, et al. Eur. J. Med. Chem., 2020, 193: 112215~112231. doi: 10.1016/j.ejmech.2020.112215
Mahapatra D K, Bharti S K, Asati V, et al. Eur. J. Med. Chem., 2019, 174: 142~158. doi: 10.1016/j.ejmech.2019.04.032
Zhuang C, Zhang W, Sheng C, et al. Chem. Rev., 2017, 117(12): 7762~7810. doi: 10.1021/acs.chemrev.7b00020
Sahu N K, Balbhadra S S, Choudhary J, et al. Curr. Med. Chem., 2012, 19(2): 209~225. doi: 10.2174/092986712803414132
Batovska D I, Todorova I T. Curr. Clin. Pharmacol., 2010, 5(1): 1~29. doi: 10.2174/157488410790410579
方琦璐, 辛文秀, 李清林, 等. 现代药物与临床, 2020, 35(4): 620~624. https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htm
严映坤, 徐侨, 高扬, 等. 有机化学, 2018, 38(07): 1763~1771. https://www.cnki.com.cn/Article/CJFDTOTAL-ZGXH201902002.htm
Koçyiǧit-Kaymakçıoǧlu B, Beyhan N, Tabanca N, et al. Med. Chem. Res., 2015, 24(10): 3632~3644. doi: 10.1007/s00044-015-1415-8
Kong W. B. Kor. Chem. Soc., 2013, 34(4): 1263~1265. doi: 10.5012/bkcs.2013.34.4.1263
Colegate S M, Din L B, Ghisalberti E L, et al. Phytochemistry, 1992, 31(6): 2123~2126. doi: 10.1016/0031-9422(92)80377-Q
Nkunya M H H, Waibel R, Achenbach H. Phytochemistry, 1993, 34(3): 853~856. doi: 10.1016/0031-9422(93)85372-X
Lien T P, Porzel A, Schmidt J, et al. Phytochemistry, 2000, 53(8): 991~995. doi: 10.1016/S0031-9422(99)00570-1
Salae A, Chairerk O, Sukkoet P, et al. Phytochemistry, 2017, 135: 135~143. doi: 10.1016/j.phytochem.2016.12.009
Porzel A, Phuong Lien T, Schmidt J, et al. Tetrahedron, 2000, 56(6): 865~872. doi: 10.1016/S0040-4020(99)01049-2
Song Q, Li X, Shen T, et al. Synth. Commun., 2003, 33(22): 3935~3941. doi: 10.1081/SCC-120026317
Hasaninejad A, Zare A, Balooty L, et al. Synth. Commun., 2010, 40(23): 3488~3495. doi: 10.1080/00397910903457282
Kulkarni P. Curr. Microwave Chem., 2015, 2(2): 144~149. doi: 10.2174/2213335601666141126220412
Buszek K R, Brown N. Org. Lett., 2007, 9(4): 707~710. doi: 10.1021/ol063027h
Wu X, Neumann H, Beller M. Angew. Chem. Int. Ed., 2010, 49(31): 5284~5288. doi: 10.1002/anie.201002155
Hermange P, Gøgsig T M, Lindhardt A T, et al. Org. Lett., 2011, 13(9): 2444~2447. doi: 10.1021/ol200686h
Barrios Antúnez D, Greenhalgh M D, Fallan C, et al. Org. Biomol. Chem., 2016, 14(30): 7268~7274. doi: 10.1039/C6OB01326K
Xu C, Chen G, Huang X. Org. Prep. Proced. Int., 1995, 27(5): 559~561. doi: 10.1080/00304949509458500
Kumar A, Sharma S, Tripathi V D, et al. Tetrahedron, 2010, 66(48): 9445~9449. doi: 10.1016/j.tet.2010.09.089
Shotter R G, Johnston K M, Jones J F. Tetrahedron, 1978, 34(6): 741~746. doi: 10.1016/0040-4020(78)88113-7
Ramakrishnan V T, Kagan J. J. Org. Chem., 1970, 35(9): 2901~2904. doi: 10.1021/jo00834a010
Maes D, Riveiro M E, Shayo C, et al. Tetrahedron, 2008, 64(19): 4438~4443. doi: 10.1016/j.tet.2008.02.059
Yamazaki S. Tetrahed. Lett., 2001, 42(19): 3355~3357. doi: 10.1016/S0040-4039(01)00432-4
Iinuma M, Matoba Y, Tanaka T, et al. Chem. Pharm. Bull., 1986, 34(4): 1656~1662. doi: 10.1248/cpb.34.1656
Katritzky A R, He H, Long Q, et al. Arkivoc, 2001, 2(3): U16~U24.
Yin G, Fan L, Ren T, et al. Org. Biomol. Chem., 2012, 10(44): 8877~8883. doi: 10.1039/c2ob26642c
Gao Y, Hou Y, Zhu L, et al. RSC Adv., 2019, 9(50): 29005~29009. doi: 10.1039/C9RA07198A

图式 1 含2-羟基-3, 4, 6-三甲氧基查尔酮单元的天然产物
Scheme 1 Natural products containing the 2-hydroxy-3, 4, 6-trimethoxychalcone unit

图式 2 2-羟基-3, 4, 6-三甲氧基查尔酮(1)的合成路线
Scheme 2 Synthetic route of 2-hydroxy-3, 4, 6-trimethoxychalcone(1)

图式 3 Claisen-Schmidt缩合法合成2-羟基-3, 4, 6-三甲氧基查尔酮(1)
Scheme 3 Synthesis of 2-hydroxy-3, 4, 6-trimethoxychalcone (1) via Claisen-Schmidt condensation
表 1 合成1, 2, 3, 5-四甲氧基苯5的条件优化
Table 1. Conditions optimization for the synthesis of 1, 2, 3, 5-tetramethoxybenzene (5)
|
|||
| Entry | Conditions | Time/h | Yield/% |
| 1 | Me2SO4,K2CO3,acetone,rt | 24 | 66 |
| 2 | MeI,K2CO3,acetone,rt | 24 | 41 |
| 3 | MeI,NaH,DMF,rt | 2 | 90 |
 下载: 导出CSV
下载: 导出CSV
表 2 合成2, 3, 4, 6-四甲氧基苯甲醛6的条件优化
Table 2. Conditions optimization for the synthesis of 2, 3, 4, 6-tetramethoxybenzaldehyde (6)
|
||
| Entry | Conditions | Yield/% |
| 1 | (a) POCl3,DMF,rt;(b) 2mol/L NaOH | 61 |
| 2 | (a) n-BuLi,DMF,THF,rt;(b) NH4Cl | 38 |
| 3 | (a) POCl3,DMF,THF,40 ℃;(b) 2mol/L NaOH | 93 |
下载: 导出CSV
表 3 合成2-羟基-3, 4, 6-三甲氧基查尔酮(1)的条件优化
Table 3. Conditions screening for the synthesis of 2-hydroxy-3, 4, 6-trimethoxychalcone (1)
|
||||
| Entry | 7(equiv.) | 8(equiv.) | T/℃ | Yield/% |
| 1 | 1.0 | 1.1 | 60 | 58 |
| 2 | 1.0 | 1.5 | 60 | 73 |
| 3 | 1.0 | 1.5 | 80 | 85 |
| 4 | 1.0 | 1.5 | 100 | 83 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们