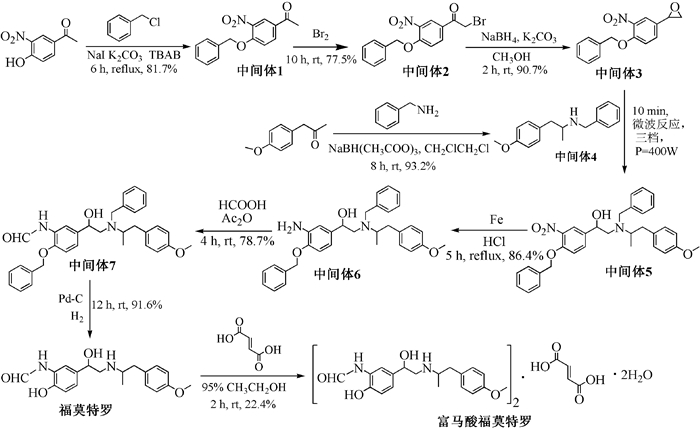
图式 1.
富马酸福莫特罗的合成路线
Scheme 1.
Synthetic route of formoterol fumarate

富马酸福莫特罗(Formoterol fumarate)是日本山之内制药株式会社中央研究所开发的第三代β2肾上腺素受体激动剂类平喘药物,1986年3月首次在日本上市,商品名为安通克(Atock)[1]。与其他平喘药物相比,富马酸福莫特罗具有高选择性,快速起效和良好的安全性和耐受性等特点[2~4]。在临床上主要使用富马酸盐水合物的一对构型为(R, R)和(S, S)的外消旋体[5]。
富马酸福莫特罗自上市以来,取得了良好的社会效益和销售业绩,被认为是控制和治疗哮喘的主要药物之一,因此对其合成工艺研究具有重要的现实意义。
富马酸福莫特罗合成的相关报道较多[6~16],基本上都是通过4-苄氧基-3-硝基苯基环乙烷与N-(4-甲氧基苯基-2-甲基乙基)苄胺两个中间体偶联、再经过硝基还原、甲酰化、去保护、与富马酸成盐最终得到富马酸福莫特罗。但是普遍存在产率不高、个别步骤条件苛刻等问题。在参考有关文献的基础上,本文设计了一条完整的合成路线(如图式 1所示),并对工艺进行了优化和改进,为富马酸福莫特罗的工业化生产提供实验基础。
Avance 400MHz型核磁共振波谱仪(瑞士Bruker公司);RY-1G型熔点仪(天津天光光学仪器有限公司);WBFY-201微电脑微波化学反应器(巩义市予华仪器有限责任公司)。
10%Pd/C、苄胺、氯苄购自阿拉丁试剂有限公司;氢气购自石家庄市西三教实用气体有限公司;4-羟基-3-硝基苯乙酮、硼氢化钠、对甲氧基苯基丙酮、乙酸酐、三乙酰氧基硼氢化钠购自北京偶合科技有限公司;其他试剂均购自天津市永大化学试剂有限公司。
在氮气保护下,向带有回流冷凝管的500mL四口瓶中依次加入50g(276.2mmol) 4-羟基-3-硝基苯乙酮、38.3g (303.9mmol)氯苄、4.15g(27.7mmol)碘化钠、38.05g(275.9mmol)碳酸钾、5.5g(17.1mmol)四丁基溴化铵、160mL水和80mL氯仿,溶液呈棕红色。回流反应6h,TLC监测原料反应完全。反应液冷却至室温,抽滤,滤液作为下次母液,滤饼先用水洗,再用二氯甲烷抽洗,干燥,得到61.2g黄色固体,收率81.7%,熔点126~128℃。1H NMR (500MHz,CDCl3)δ:8.43(d,J=2.0Hz,1H),8.11(d,J=6.5Hz,1H),7.45(d,J=7.5Hz,2H),7.40(t,J=7.5Hz,2H),7.35(t,J=7.0Hz,1H),7.19(d,J=9.0Hz,1H),5.32(s,2H),2.59(s,3H)。
向250mL四口瓶中加入10g(36.8mmol)中间体1和10mL氯仿,缓慢通入氮气,室温滴加6.4g溴的20mL氯仿溶液,滴毕,继续反应10h,在此过程中产物不断析出。抽滤,滤饼用环己烷冲洗,干燥得到10.0g白色固体,收率77.5%,熔点127~128℃。1H NMR (400MHz,CDCl3)δ:8.49(d,J=2.4Hz,1H),8.15(d,J=6.4Hz,1H),7.45(d,J=7.2Hz,2H),7.41(t,J=7.6Hz,2H),7.36(t,J=7.2Hz,1H),7.22(d,J=8.8Hz,1H),5.34(s,2H),4.38(s,2H)。
向100mL四口瓶中加入1.00g(2.87mmol)中间体2,0.074g(1.95mmol) NaBH4、0.74g(5.36mmol) K2CO3和30mL甲醇,室温反应2h,TLC监测原料完全反应,浓缩反应液,得到淡黄色固体2.18g,加入30mL二氯甲烷溶解,再加入20mL(1mol/L)盐酸,搅拌15min,静置,上层为淡黄色,下层为白色,分液,上层水相用20mL二氯甲烷萃取,有机相用水洗,合并有机相,无水硫酸镁干燥,过滤、浓缩滤液体积至约5mL,加入20mL石油醚,有少量白色固体析出,然后浓缩至干,得到0.712g白色粉末状固体,收率为90.7%,熔点为59~61℃。1H NMR(400MHz,CDCl3) δ:7.78(d,J=2.4Hz,1H),7.45(d,J=7.2Hz,2H),7.39(t,J=7.8Hz,3H),7.33(t,J=7.2Hz,1H),7.10(d,J=8.4Hz,1H),5.24(s,2H),3.89~3.79 (m,1H),3.21~3.11(m,1H),2.81~2.71(m,1H),1.54(s,1H)。
向250mL四口瓶中加入7.76g(36.6mmol)三乙酰氧基硼氢化钠、25mL 1, 2-二氯乙烷,冰浴-3~0 ℃下搅拌,滴加5.00g(30.5mmol)对甲氧基苯基丙酮、3.59g(33.5mmol)苄胺的25mL 1, 2-二氯乙烷溶液。滴毕,室温反应8h,TLC监测原料完全反应,将反应液浓缩,得到38.6g黄色胶状液体,加入100mL二氯甲烷溶解,然后加入150mL饱和碳酸钠水溶液,搅拌2h,静置,上层为无色,下层为黄色,分液,水相用10mL二氯甲烷萃取,用去离子水(150mL×3)洗有机相,分液,合并有机相,无水硫酸镁干燥,过滤,滤液减压蒸除溶剂,得到14.5g红棕色油状物,收率93.2%。1H NMR(400MHz,CDCl3) δ:7.32~7.26(m,2H),7.22(t,J=6.2Hz,3H),7.07(d,J=8.6Hz,2H),6.82(d,J=8.6Hz,2H),3.88~3.82(m,1H),3.79(s,3H),3.75~3.71(m,1H),2.95~2.84(m,1H),2.76~2.65 (m,1H),2.64~2.55 (m,1H),1.09(d,J=6.2Hz,3H)。
采用微波反应器向50mL烧瓶中加入0.446g(1.646mmol)中间体3、0.420g (1.646mmol)中间体4,设定功率为400W,反应时间为10min。TLC监测原料完全反应,反应液冷却至室温,得到0.865g红棕色胶状固态体粗品,直接用于下步反应。
向250mL四口瓶中加入6.10g(11.59mmol)中间体5、7.50g(134.10mmol)活化后的还原铁粉,然后加入15mL氯仿、50mL甲醇和15mL 1mol/L盐酸,搅拌,加热回流反应5h,TLC监测原料完全反应。冷却至室温,抽滤,用10mL二氯甲烷冲洗滤饼,将滤液倒入烧杯中,再依次加入70mL二氯甲烷、200mL 1mol/L氢氧化钠水溶液,搅拌1h,静置,水相为墨绿色,有机相为红棕色,抽滤,用去离子水(150mL×3)洗有机相,分液,无水硫酸镁干燥,过滤,浓缩,得到4.97g黑红色油状物,收率86.4%。1H NMR(400MHz,CDCl3)δ:7.47(d,J=7.2Hz,2H),7.42(t,J=7.2Hz,2H),7.36(t,J=7.2Hz,3H),7.32(d,J=6.0Hz,1H),7.29(s,1H),7.17(d,J=6.0Hz,1H),7.07~6.99(m,2H),6.88~6.82(m,3H),6.78~6.74(m,1H),6.66(t,J=7.2Hz,1H),5.10(s,2H),4.16(q,J=6.8Hz,1H),3.88~3.77 (m,6H),3.14~3.02(m,1H),2.82~2.61(m,2H),2.08(s,2H),1.30(t,J=7.2Hz,2H),1.10~1.01 (m,3H)。
向100mL四口瓶中加入15mL氯仿、0.65g(1.31mmol)中间体6、0.35g (7.61mmol)甲酸和0.54g(5.29mmol)醋酸酐。室温搅拌4h,待反应完毕后,将反应液浓缩得到1.51g红棕色液体;加入30mL甲醇、3.0g碳酸钠和2mL水的板结固体物,室温搅拌5h,反应液由红棕色变为黄色,TLC监测原料完全反应,过滤,得到黄色滤液。将滤液浓缩,向其中加入50mL水和50mL二氯甲烷后搅拌,静置,分液,用去离子水(50mL×3)洗有机相,无水硫酸镁干燥,过滤,浓缩,得到0.54g红棕色胶状物,收率78.7%。1H NMR (400MHz,CDCl3)δ:8.31(s,1H),7.76(s,1H),7.44~7.37 (m,5H),7.33(s,2H),7.13(d,J=6.2Hz,1H),7.06(d,J=8.3Hz,1H),7.01~6.95 (m,2H),6.91(d,J=8.5Hz,1H),6.83~6.77(m,2H),5.11(s,2H),4.63~4.37(m,1H),4.15~4.09 (m,1H),3.98~3.64(m,6H),3.11~2.97(m,1H),2.76~2.56(m,2H),2.04(s,2H),1.26(s,2H),1.00(t,J=6.1Hz,3H)。
向100mL高压反应釜中依次加入2.00g(3.81mmol)中间体7、0.316g 10%Pd/C和30mL无水甲醇。通入氮气置换釜内气体3次,确保排尽反应釜内的空气。通入氢气置换釜内气体3次,确保反应釜内充满氢气,通氢至0.2MPa。室温下开动搅拌12h,直至压力不变,保持30min。将反应液抽滤,浓缩得到淡黄色泡状固体1.20g,收率91.6%。ESI-MS(m/z):345[M]+;1H NMR (400MHz,CDCl3)δ:8.10(d,J=26.2Hz,1H),7.06~6.93(m,4H),6.88(d,J=8.0Hz,1H),6.84~6.80(m,2H),4.46~4.41(m,1H),3.78(d,J=1.4Hz,3H),2.90~2.76(m,2H),2.68~2.52(m,3H),2.23(s,2H),1.09~1.05(t,3H)。
向100mL单口瓶中加入0.13g(0.377mmol)福莫特罗、10mL 95%乙醇,然后加入0.02g(0.181mmol)富马酸的10mL异丙醇溶液,室温搅拌2h,反应液为淡黄色,TLC监测原料完全反应。将反应液浓缩,加入2mL乙醇溶解油状物,滴加约10mL石油醚,反应液变浑浊,逐渐有少量浅黄色固体析出,放入冰箱冷藏2h,抽滤,得0.042g淡黄色泡状固体,收率22.4%,熔点85~87 ℃。
中间体1是4-羟基-3-硝基苯乙酮与苄氯进行苄基化反应得到的产物,在已知的文献中后处理过程较为繁琐,通过水洗、干燥、浓缩、重结晶等过程得到产品。本文原料4-羟基-3-硝基苯乙酮质量(g)与水、氯仿(mL)比为5:16:8时,在反应过程中实现原料逐渐溶解,产品不断析出,从而通过抽滤、洗涤、干燥等简单的后处过程得到纯品。
在合成中间体2的α-羰基溴代反应中,以氯仿作溶剂,常温反应,反应结束产品在反应液中析出,通过抽滤、洗涤、干燥即可得到纯品,该方法操作简单,将反应用的溶剂氯仿回收后作下次反应的溶剂,反应速率急剧加快。
实验条件如1.2.2,考察液溴用量对反应收率的影响,结果如表 1所示。由表可知,随着液溴用量的增加,产物收率呈现先升高后下降趋势,这是因为当投料摩尔比低于1:1.1时,由于液溴易挥发导致有部分原料反应不完全,使收率偏低;投料比高于1:1.15时,液溴过量虽能使原料充分反应,但二溴取代副产物生成较多,影响收率。因而1:1.1为适宜的投料比。
 下载:
导出CSV
下载:
导出CSV
| 序号 | 中间体2:液溴 | 收率/% |
| 1 | 1:1 | 65.5 |
| 2 | 1:1.1 | 77.5 |
| 3 | 1:1.15 | 77.3 |
| 4 | 1:1.2 | 66.3 |
还原剂硼氢化钠分子中的每个氢都能进行反应。实验条件如1.2.3,考察中间体2与硼氢化钠摩尔配比对收率的影响,结果见表 2。随着硼氢化钠加入量的增加,收率先增加后基本不变。硼氢化钠量过少时,硼氢化钠水解导致还原反应不能充分进行,收率较低;硼氢化钠过量,收率基本不变。考虑到成本因素,中间体2与硼氢化钠摩尔比1:0.0275为适宜的投料比。
下载:
导出CSV
| 序号 | 中间体2:硼氢化钠 | 收率/% |
| 1 | 1:0.25 | 65.5 |
| 2 | 1:0.275 | 81.2 |
| 3 | 1:0.30 | 81.5 |
| 4 | 1:0.325 | 81.2 |
在发生环氧化反应时,同时还生成溴化氢,反应体系中如果没有碱作为缚酸剂,累积的溴化氢阻止了反应的继续进行,致使收率较低。为此,考察不同缚酸剂对中间体3收率的影响(实验条件见1.2.3),结果见表 3。由表可知,用碳酸钠、氢氧化钠、氢氧化钾作为缚酸剂时,产品收率较低,用碳酸钾作为缚酸剂时,有更为理想的收率。这是由于碱性较弱时不足以去除产生的溴化氢,而过于强的碱可能使卤代烷发生水解,导致收率下降;采用有机碱三乙胺时,虽然使得反应为均相体系,但三乙胺会生成季铵盐,消耗卤代烃,使收率下降,杂质增多。因此以碳酸钾作为缚酸剂较为合适。
下载:
导出CSV
| 序号 | 缚酸剂 | 收率/% |
| 1 | 碳酸钠 | 65.5 |
| 2 | 碳酸钾 | 81.2 |
| 3 | 氢氧化钠 | 75.9 |
| 4 | 氢氧化钾 | 46.3 |
考察对甲氧基苯基丙酮与三乙酰氧基硼氢化钠投料比对收率的影响(实验条件如1.2.4),结果见表 4。由表可知,还原剂三乙酰氧基硼氢化钠稍微过量有利于得到更高的收率,当投料比低于1:1.2收率较低,因此对甲氧基苯基丙酮与三乙酰氧基硼氢化钠摩尔比为1:1.2为优化的投料比。
下载:
导出CSV
| 序号 | 对甲氧基苯基丙酮:三乙酰氧基硼氢化钠 | 收率/% |
| 1 | 1:1 | 42.8 |
| 2 | 1:1.1 | 76.1 |
| 3 | 1:1.2 | 83.3 |
| 4 | 1:1.3 | 83.2 |
实验条件如1.2.4,分别选用乙腈、氯仿、四氢呋喃、二氯甲烷和1, 2二氯甲烷作为溶剂考察对收率的影响,结果见表 5。由表可见,以1, 2二氯乙烷作为溶剂时反应收率最高,因此1, 2二氯乙烷为合适的反应溶剂。
下载:
导出CSV
| 序号 | 溶剂 | 收率/% |
| 1 | 乙腈 | 76.5 |
| 2 | 氯仿 | 79.4 |
| 3 | 四氢呋喃 | 79.7 |
| 4 | 二氯甲烷 | 79.7 |
| 5 | 1, 2-二氯乙烷 | 83.3 |
关于中间体5的合成,文献[3]报道的方法中,多采用130℃高温无溶剂反应15h,此方法耗能较多耗时较长。本文利用微波反应器进行无溶剂反应,考察微波反应器不同功率对反应时间的影响(实验条件如1.2.5),结果见表 6。由表可知,微波功率为400W时,10min即可完成反应,与高温120℃无溶剂反应相比缩短了反应时间,降低了生产成本。功率太大会出现少许碳化,功率低时反应时间延长甚至不反应。
下载:
导出CSV
| 序号 | 档位/W | 结束时间/min |
| 1 | 800 | 3 |
| 2 | 640 | 5 |
| 3 | 400 | 10 |
| 4 | 240 | 90 |
| 5 | 80 | - |
本文对原有的路线进行了优化与改进。在制备中间体1的过程中,原料4-羟基-3-硝基苯乙酮质量(g)与溶剂水、氯仿体积(mL)比为5:16:8时,可以在反应过程中实现原料逐渐溶解,产品不断析出,使后处理简单易行;在制备中间体2时,中间体1质量(g)与溶剂体积(mL)比为1:3,反应结束产品即在反应液中析出,抽滤,环己烷冲洗并干燥后即可得到产品;制备中间体3时,采用一锅法,简化了反应步骤,在反应中加入了缚酸剂碳酸钾,使反应速率加快,收率提高;在制备中间体5时,采用微波合成,极大地缩短了反应时间。本文报道的富马酸福莫特罗合成工艺操作简单,适合工业化生产,总收率达7.46%。
金勇, 张少辉.药学进展, 1998, 22(3): 187~189. http://www.cnki.com.cn/Article/CJFD1998-YXJZ803.022.htm
R A Bartow, R N Brogden. Drugs, 1998, 55(2): 303~322. doi: 10.2165/00003495-199855020-00016
J Trofast, K Osterberg, B L Källsträm et al. Chirality, 1991, 3(6): 443~450. doi: 10.1002/chir.530030606
孔晓燕, 李华.西南国防医药, 2016, 26(7): 804~806. http://www.cnki.com.cn/Article/CJFDTotal-XNGF201607041.htm
F Campos, M P Bosch, A Guerrero. Tetrahed. Asymmetry, 2000, 11(13): 2705~2717. doi: 10.1016/S0957-4166(00)00238-X
赵丽琴, 赵冬梅, 张雅芳等.中国药物化学杂志, 2000, (4): 53~55. http://www.cnki.com.cn/Article/CJFDTotal-ZGYH200004015.htm
赵冬梅, 郭秋明, 张雅芳等.中国医药工业杂志, 2001, (7): 335~336. http://www.cnki.com.cn/Article/CJFDTotal-ZHOU200107026.htm
李公春, 吴长增, 牛亮峰等.山东化工, 2014, 43(12): 126~128. http://www.cnki.com.cn/Article/CJFDTotal-SDHG201412048.htm
P D de Koning, I R Gladwell, I B Moses et al. Org. Proc. Res. Dev., 2011, 15(6): 1247~1255. doi: 10.1021/op2001904
H Ling, S Wenjun, Z Qi et al. Bioorg. Med. Chem. Lett., 2014, 24(1): 249~253. https://www.sciencedirect.com/science/article/pii/S0960894X13013073
J W Trofast, E Jakupovic, K L Mansson. USP: 5434304, 1995.
I Navarro Muñoz, J Huguet Clotet. EPP: 2348013, 2011.
Y Gao, R Hett, K Q Fang et al. USP: 6268533, 2001.
S B Bhirud, S M Kadam, S B Gavhane et al. WOP: 2014184756, 2014.
C Li, Y Li, Y Zhang et al. Anal. Methods, 2018, 10: 548~553. doi: 10.1039/C7AY02562A
黎星术, 黄玲, 卢传君等. CN: 103664677, 2014.
表 1 投料比对中间体2收率的影响
Table 1. The influence of material ratio on the yield of intermediate 2
| 序号 | 中间体2:液溴 | 收率/% |
| 1 | 1:1 | 65.5 |
| 2 | 1:1.1 | 77.5 |
| 3 | 1:1.15 | 77.3 |
| 4 | 1:1.2 | 66.3 |
 下载: 导出CSV
下载: 导出CSV
表 2 投料比对中间体2收率的影响
Table 2. The influence of material ratio on the yield of intermediate 2
| 序号 | 中间体2:硼氢化钠 | 收率/% |
| 1 | 1:0.25 | 65.5 |
| 2 | 1:0.275 | 81.2 |
| 3 | 1:0.30 | 81.5 |
| 4 | 1:0.325 | 81.2 |
下载: 导出CSV
表 3 缚酸剂对中间体3反应结果的影响
Table 3. The influence of acid-binding agent on the reaction yield of intermediate 3
| 序号 | 缚酸剂 | 收率/% |
| 1 | 碳酸钠 | 65.5 |
| 2 | 碳酸钾 | 81.2 |
| 3 | 氢氧化钠 | 75.9 |
| 4 | 氢氧化钾 | 46.3 |
下载: 导出CSV
表 4 投料比对中间体4收率的影响
Table 4. The influence of material ratio on the yield of intermediate 4
| 序号 | 对甲氧基苯基丙酮:三乙酰氧基硼氢化钠 | 收率/% |
| 1 | 1:1 | 42.8 |
| 2 | 1:1.1 | 76.1 |
| 3 | 1:1.2 | 83.3 |
| 4 | 1:1.3 | 83.2 |
下载: 导出CSV
表 5 溶剂对中间体4收率的影响
Table 5. The influence of solvent on the yield of intermediate 4
| 序号 | 溶剂 | 收率/% |
| 1 | 乙腈 | 76.5 |
| 2 | 氯仿 | 79.4 |
| 3 | 四氢呋喃 | 79.7 |
| 4 | 二氯甲烷 | 79.7 |
| 5 | 1, 2-二氯乙烷 | 83.3 |
下载: 导出CSV
表 6 微波功率对反应时间影响
Table 6. The influence of microwave condition on the reaction process
| 序号 | 档位/W | 结束时间/min |
| 1 | 800 | 3 |
| 2 | 640 | 5 |
| 3 | 400 | 10 |
| 4 | 240 | 90 |
| 5 | 80 | - |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
