表 1
CSF-1R抑制剂的结构和生物活性数据
Table 1.
Structure and activity data of CSF-1R inhibitors

集落刺激因子-1受体激酶(CSF-1R)是由人类c-fms原癌基因编码的细胞表面糖蛋白[1, 2]。它是Ⅲ型受体酪氨酸激酶家族成员之一,类似的激酶还有KIT、Fit3和PDGFR[3]。CSF-1R可以调控单核巨噬细胞谱系细胞的存活、增殖、分化和功能[4]。CSF-1R及其配体在胞内过度表达,可引起这一蛋白激酶诱导的信号通路异常激活,从而参与多种疾病过程。由于CSF-1在许多肿瘤组织和炎症部位存在过度表达,因此抑制CSF-1R及其配体CSF-1的表达为治疗乳腺癌、肾细胞癌、前列腺癌、肺癌、骨疾病和炎症提供了一个有吸引力的策略[5]。许多具有不同结构骨架的抑制剂已经被开发出来并呈现出显著的生物活性,例如,氰基咪唑甲酰胺类、氨基嘧啶类、吡啶类、喹啉类、噌啉类和4-芳酰胺基-3-甲基异噁唑类[6, 7],其中Pexidartinib已于2019年完成Ⅲ期临床试验,有望用于治疗罕见病腱鞘巨细胞瘤[8]。然而,这些化合物对蛋白激酶靶标选择性不强,而且还观察到存在某些副作用,例如心脏毒性、呕吐、眼睛肿胀和腹泻[9]。因此,寻找新型小分子CSF-1R激酶抑制剂具有重要意义。
嘧啶以及含嘧啶单元的并环化合物具有多种药理活性,是公认的药物分子设计中的特权结构。2018年,Ding等[11]报道了一系列2-氧代-3, 4-二氢嘧啶并[4, 5-d]嘧啶衍生物作为CSF-1R激酶抑制剂。在此之前,他们曾将此类结构作为不可逆表皮生长因子受体(EGFR)抑制剂[10]。化合物1就是先前设计的不可逆EGFR抑制剂,近来研究发现其对CSF-1R有较强的抑制作用(IC50=30nmol/L)。随后他们通过对化合物1的结构进行结构修饰,得到了一系列新的CSF-1R抑制剂,且生物活性较好。因此,本研究拟采用分子对接和三维定量构效关系(3D-QSAR)来研究2-氧代-3, 4-二氢嘧啶并[4, 5-d]嘧啶衍生物结构与活性之间的关系,以此为研究开发靶向CSF-1R新型骨架的先导化合物提供参考。
本研究中54个2-氧代-3, 4-二氢嘧啶并[4, 5-d]嘧啶类的CSF-1R小分子抑制剂均来源文献[11]。这一系列化合物来自于同一个实验活性测试小组,并且测试条件相同。根据分子结构多样性和活性范围,以近似4:1的比例随机选择其中40个化合物作为训练集,其余14个化合物作为测试集。所有化合物的结构和生物活性见表 1。
 下载:
导出CSV
下载:
导出CSV
| No | R1 | R2 | R3 | R4 | pIC50 | Predicted Value | |
| CoMFA | CoMSIA | ||||||
 |
 |
||||||
| 1* |  |
- | - | - | 7.523 | 8.063 | 8.050 |
| 2* |  |
- | - | - | 8.301 | 8.012 | 7.979 |
| 3* |  |
- | - | - | 8.260 | 8.073 | 7.945 |
| 4 |  |
- | - | - | 8.174 | 8.232 | 8.214 |
| 5 |  |
- | - | - | 7.996 | 8.015 | 8.089 |
| 6 |  |
- | - | - | 7.987 | 7.912 | 7.944 |
| 7 |  |
- | - | - | 7.697 | 7.628 | 7.662 |
| 8 |  |
- | - | - | 7.684 | 7.620 | 7.684 |
| 9 |  |
- | - | - | 7.547 | 7.558 | 7.517 |
| 10 |  |
- | - | - | 7.514 | 7.534 | 7.521 |
| 11* |  |
- | - | - | 7.699 | 8.042 | 7.756 |
| 12 |  |
- | - | - | 7.644 | 7.644 | 7.658 |
| 13* |  |
- | - | - | 6.743 | 7.408 | 7.408 |
| 14 |  |
- | - | - | 7.680 | 7.655 | 7.655 |
 |
|||||||
| 15* | - |  |
- | - | 8.284 | 7.967 | 7.971 |
| 16 | - |  |
- | - | 8.252 | 8.255 | 8.352 |
| 17 | - |  |
- | - | 7.818 | 7.809 | 7.827 |
| 18 | - |  |
- | - | 7.742 | 7.762 | 7.762 |
| 19 | - |  |
- | - | 7.879 | 7.916 | 7.879 |
| 20 | - |  |
- | - | 7.830 | 7.759 | 7.803 |
| 21 | - |  |
- | - | 7.785 | 7.835 | 7.769 |
| 22 | - |  |
- | - | 7.686 | 7.765 | 7.716 |
| 23 | - |  |
- | - | 8.292 | 8.332 | 8.238 |
| 24 | - |  |
- | - | 8.347 | 8.244 | 8.321 |
| 25* | - |  |
- | - | 8.237 | 7.987 | 7.755 |
| 26* | - |  |
- | - | 7.553 | 8.216 | 7.813 |
| 27 | - |  |
- | - | 6.821 | 6.806 | 6.78 |
| 28* | - |  |
- | - | 8.347 | 7.991 | 7.927 |
| 29 | - |  |
- | - | 8.699 | 8.400 | 8.313 |
| 30* | - |  |
- | - | 8.357 | 8.047 | 7.881 |
| 31 | - |  |
- | - | 8.678 | 8.738 | 8.678 |
 |
 |
||||||
| 32 | - | - | H | 4-甲基-哌嗪-1-基 | 7.866 | 7.824 | 7.892 |
| 33 | - | - | 2′-CH3 | 4-甲基-哌嗪-1-基 | 7.695 | 7.717 | 7.646 |
| 34 | - | - | 2′-OCH3 | 4-甲基-哌嗪-1-基 | 7.517 | 7.452 | 7.589 |
| 35 | - | - | 3′-OCH3 | 4-甲基-哌嗪-1-基 | 8.237 | 8.200 | 8.187 |
| 36 | - | - | 3′-F | 4-甲基-哌嗪-1-基 | 7.767 | 7.846 | 7.826 |
| 37 | - | - | 3′-Cl | 4-甲基-哌嗪-1-基 | 7.830 | 7.947 | 7.855 |
| 38* | - | - | 3′-CF3 | 4-甲基-哌嗪-1-基 | 7.496 | 7.893 | 7.380 |
| 39 | - | - | 3′-ethyl | 4-甲基-哌嗪-1-基 | 8.000 | 8.164 | 8.148 |
| 40* | - | - | 3′-isopropyl | 4-甲基-哌嗪-1-基 | 7.588 | 8.134 | 8.189 |
| 41 | - | - | 3′-CH3 |  |
7.385 | 7.481 | 7.376 |
| 42 | - | - | 3′-CH3 |  |
7.599 | 7.540 | 7.617 |
| 43* | - | - | 3′-CH3 |  |
7.587 | 8.312 | 8.428 |
| 44 | - | - | 3′-CH3 |  |
8.310 | 8.315 | 8.316 |
| 45 | - | - | 3′-CH3 |  |
8.328 | 8.323 | 8.314 |
| 46 | - | - | 3′-CH3 |  |
8.167 | 8.228 | 8.210 |
| 47 | - | - | 3′-CH3 |  |
8.208 | 8.202 | 8.255 |
| 48 | - | - | 3′-CH3 |  |
8.187 | 8.264 | 8.282 |
| 49 | - | - | 3′-CH3 |  |
8.155 | 8.112 | 8.140 |
| 50 | - | - | 3′-CH3 |  |
7.987 | 7.992 | 7.964 |
| 51 | - | - | 3′-CH3 |  |
8.143 | 8.177 | 8.156 |
| 52* | - | - | 3′-CH3 |  |
8.108 | 8.309 | 8.229 |
| 53 | - | - | 3′-CH3 |  |
8.469 | 8.412 | 8.466 |
| 54 | - | - | - |  |
8.528 | 8.510 | 8.504 |
| *代表测试集 | |||||||
所有化合物的分子结构均由Chem Bio Draw 11.0构建[12],并通过SYBYL-X 2.0中“Minimize”功能模块,将所有分子结构进行分子力学优化,从而得到低能稳定构象。优化过程:采用Powell法,在Tripos力场条件下,加载Gasteiger-Hückel电荷,最大迭代次数为1000次,收敛阈值为0.005kcal·(mol·Å)-1,其他参数均采用系统缺省值[13]。
运用Molegro Virtual Docker 5.5(MVD)进行分子对接研究。CSF-1R的靶蛋白晶体结构(PDB ID:3DPK)[14]来自PDB数据库。在对接之前,提取原配体8C5、除去水及辅酶,部分氨基酸添加质子化状态。以该晶体复合物中的原配体作为参照[15],先利用原配体8C5进行自身对接验证对接方法的可靠性;再将54个抑制剂分子与CSF-1R受体蛋白对接。所有对接计算均使用MolDock Simplex Evolution算法和基于网格的MolDock Score(GRID)函数进行[16],对接参数设置如下:网格分辨率为0.03Å,最大迭代次数为500次,输出30个构象。
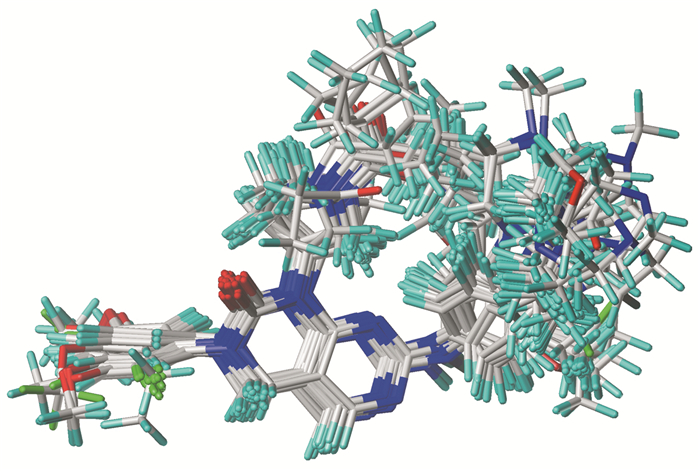
分子叠合是构建可靠模型的关键步骤,3D-QSAR模型的预测能力和统计质量与其密切相关[17]。在本研究中,采用基于分子对接的方法进行叠合,将每个分子对接后所输出的30个构象根据MolDock Score和Hbond选取优势构象进行叠合(图 1)。
本文通过SYBYL-X 2.0软件对叠合后的训练集化合物构建3D-QSAR模型。CoMFA和CoMSIA是目前使用最广泛的两种3D-QSAR方法。为了得到CoMFA和CoMSIA描述符,叠合好的化合物位于3D立方晶格中,栅格间距为2Å。在CoMFA分析中,以范德华半径为1.52Å且电荷为+1.0的sp3杂化碳为探针原子,应用Lennard-Jones和库仑势来计算每个格点的立体场能量和静电场能量,其能量截断值为30kcal·mol-1。对于CoMSIA模型,还计算了疏水场、氢键供体场和氢键受体场,作为立体场和静电场的延伸。CoMSIA建模使用一个+1.0电荷、范德华半径为1.0Å、疏水性为+1.0、氢键供体和受体为+1.0的sp3杂化碳作为探针来计算。另外,衰减系数α设定为0.3。在CoMSIA中,引入Gaussian函数来评估探针原子和每个分子原子之间的距离。
偏最小二乘(PLS)回归方法用于处理分子场和生物活性之间的线性相关性[18]。使用留一法(LOO)进行交叉验证分析得到交叉验证相关系数(q2)和最佳组分数(N)。q2可以通过式(1)[19, 20]计算。
|
$ q^{2}=1-\frac{\sum\left(Y_{\text {pred }}-Y_{\text {actual }}\right)^{2}}{\sum\left(Y_{\text {actual }}-Y_{\text {mean }}\right)^{2}} $ |
(1) |
其中,Ymean、Ypred和Yactual分别是训练集化合物pIC50的平均值、预测值和实验值。接着基于N执行非交叉验证分析,可获得相关系数r2、标准误差估计(SEE)、F值及每个场的贡献值。
CoMFA和CoMSIA模型的统计参数列于表 2。当模型的r2>0.6,且q2>0.5,则所构建的3D-QSAR模型具有可靠性和良好的预测能力[21]。
下载:
导出CSV
| No. | q2 | N | SEE | r2 | F | Field contribution | ||||
| S | E | H | D | A | ||||||
| CoMFA | 0.725 | 9 | 0.086 | 0.960 | 79.542 | 0.760 | 0.240 | - | - | - |
| CoMSIA-SE | 0.574 | 6 | 0.184 | 0.799 | 21.888 | 0.905 | 0.095 | - | - | - |
| CoMSIA-SH | 0.588 | 8 | 0.093 | 0.953 | 76.679 | 0.272 | - | 0.728 | - | - |
| CoMSIA-ED | 0.176 | 4 | 0.321 | 0.352 | 4.749 | - | 0.509 | - | 0.491 | - |
| CoMSIA-HA | 0.519 | 8 | 0.114 | 0.927 | 49.529 | - | - | 0.683 | - | 0.317 |
| CoMSIA-SEH | 0.610 | 10 | 0.097 | 0.951 | 56.354 | 0.233 | 0.082 | 0.685 | - | - |
| CoMSIA-SED | 0.618 | 8 | 0.171 | 0.837 | 19.898 | 0.612 | 0.101 | - | 0.286 | - |
| CoMSIA-SHD | 0.636 | 9 | 0.088 | 0.958 | 76.184 | 0.228 | - | 0.653 | 0.119 | - |
| CoMSIA-SHA | 0.547 | 8 | 0.110 | 0.933 | 53.551 | 0.179 | - | 0.555 | - | 0.266 |
| CoMSIA-EDA | 0.345 | 7 | 0.229 | 0.698 | 10.548 | - | 0.207 | - | 0.156 | 0.638 |
| CoMSIA-EHA | 0.520 | 10 | 0.117 | 0.929 | 38.009 | - | 0.057 | 0.672 | - | 0.270 |
| CoMSIA-EHD | 0.545 | 9 | 0.121 | 0.921 | 38.992 | - | 0.125 | 0.759 | 0.116 | - |
| CoMSIA-HDA | 0.528 | 10 | 0.097 | 0.951 | 56.089 | - | - | 0.632 | 0.108 | 0.260 |
| CoMSIA-SEHD | 0.604 | 9 | 0.113 | 0.931 | 45.031 | 0.194 | 0.124 | 0.581 | 0.100 | - |
| CoMSIA-SEHA | 0.541 | 10 | 0.112 | 0.935 | 41.428 | 0.161 | 0.089 | 0.531 | - | 0.220 |
| CoMSIA-SHDA | 0.564 | 10 | 0.093 | 0.955 | 61.921 | 0.166 | - | 0.522 | 0.095 | 0.218 |
| CoMSIA-SEDA | 0.583 | 10 | 0.161 | 0.866 | 18.705 | 0.353 | 0.108 | - | 0.146 | 0.393 |
| CoMSIA-EHDA | 0.528 | 10 | 0.126 | 0.918 | 32.368 | - | 0.107 | 0.561 | 0.094 | 0.238 |
| CoMSIA-SEHDA | 0.552 | 10 | 0.115 | 0.931 | 39.213 | 0.138 | 0.102 | 0.471 | 0.088 | 0.202 |
由表 2可知,CoMFA模型的q2为0.725,最佳组分数N为9,r2、SEE和F分别为0.960、0.086和79.542。这些统计数据表明CoMFA模型具有较好的预测能力。立体场与静电场的贡献分别为0.760和0.240,说明化合物的生物活性受立体场影响更大。
构建了不同的CoMSIA模型组合。从表 2可知,CoMSIA-SHD是最佳模型。该模型的q2及N为0.636和9,r2、SEE和F分别为0.958、0.088和76.184。模型中立体场、疏水场和氢键供体场的贡献分别为0.228、0.653和0.119。
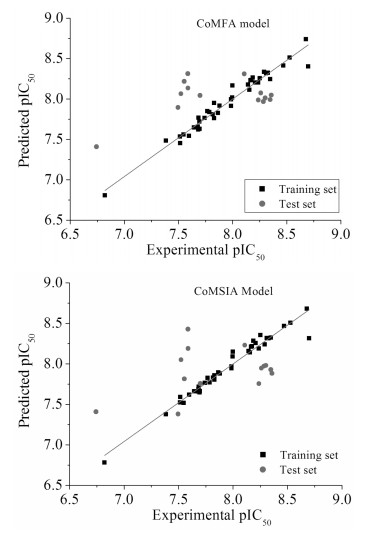
根据所构建的CoMFA、CoMSIA模型对训练集和测试集生物活性的实验值及预测值作散点图进行线性回归分析。散点图可以反映数据集中各点离散程度以及其生物活性的实验值和预测值的差异[22],从图 2可见,实验值和预测值基本分布在线性回归趋势线两侧,说明两者具有较好的相关性。
三维等势图能够直观地阐明化合物的生物活性及其分子结构之间的关系。以活性最好的化合物29作为参比分子进行CoMFA和CoMSIA模型的三维等势图分析(图 3、图 4)。
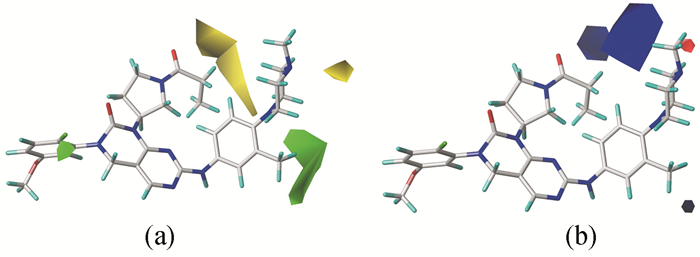
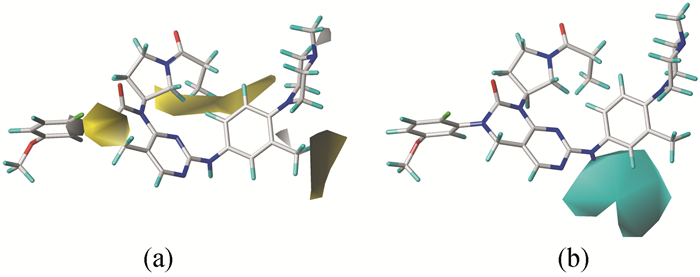
图 3(a)为CoMFA模型立体场等势图,绿色区域表示增大基团体积有利于化合物生物活性的提高,黄色区域则相反。从图 3(a)可见,与亚氨基相连的苯环上甲基取代基附近有一块绿色区域,表明在该位置引入大体积基团有利于提高化合物活性。例如,引入甲氧基后的化合物35(pIC50=8.237),其生物活性明显高于化合物32(pIC50=7.866)。此外,有两块黄色区域分布在R1取代基附近,表明体积较大基团在这个位置对化合物活性起抑制作用。化合物5(pIC50=7.996)、9(pIC50=7.547)和10(pIC50=7.514)均引入较大体积基团,故表现出比化合物2(pIC50=8.301)更低的活性。在哌嗪基的N-4右上方有一块黄色区域,表明引入较大体积基团会降低化合物的生物活性。例如,在N-4位置引入-COCH3的化合物43(pIC50=7.587),与引入甲基的化合物29(pIC50=8.699)相比,其活性明显降低。在R2取代基的氟原子上方有一块绿色区域,说明此处有较大基团时可以提高活性,例如,化合物23(pIC50=8.292)的活性比化合物16(pIC50=8.252)的高一点。
CoMFA模型的静电场等势图如图 3(b)所示,红色区域代表负电性基团有利于活性的提高,而蓝色区域代表正电性基团有利于提高活性。可以观察到在哌嗪基的C-5附近有一块红色区域,表明引入正电性基团会影响活性降低,化合物52(pIC50=8.108)由于在此处引入甲基,其活性比化合物29(pIC50=8.699)低。与亚氨基相连的苯环上3′-CH3附近有一小块蓝色区域,说明在此处有正电性基团对生物活性提高有利,因此,化合物29(pIC50=8.699)的活性比化合物37(pIC50=7.830)的高可以得到解释。在R1取代基的亚甲基附近有一块蓝色区域,表明引入正电性基团有利于提高生物活性,如经加氢还原,化合物2(pIC50=8.301)活性明显高于化合物1(pIC50=7.523);又或者通过加成反应引入甲基,化合物4(pIC50=8.174)、化合物7(pIC50=7.697)的活性比化合物1的高。此外,有一大块蓝色区域分布在哌嗪基的N-CH3左侧,表明这一区域引入正电性基团有利于活性提高,可以尝试在这个位置引入正电性基团。
CoMSIA模型的立体场与CoMFA模型类似,在这里不再阐述。图 4(a)是CoMSIA模型的疏水场等势图,黄色区域表示增加疏水基团可以化合物的活性增加,白色区域则表示引入亲水基团有利于活性提高。可以看到,R2取代基上的氟原子上方有一块黄色区域,表明在这位置引入疏水基团有利于活性的增加。同时有一小块白色区域覆盖在R2取代基的C-F上,故疏水基团和亲水基团在此处对活性的影响不大,如化合物2(pIC50=8.301)与化合物23(pIC50=8.292)就是这种情况。与亚氨基相连的苯环上3′-CH3也出现类似R2取代基的情况,区别是那小块白色区域位于C-C旁边,在此处疏水基团及亲水基团对活性影响不大,化合物37(pIC50=7.830)与化合物32(pIC50=7.866)的活性相近。当苯环的3′位均为疏水基团如-OCH3、-F、-Cl等时,对活性的影响起主要作用的是立体场。还有一大块黄色区域覆盖在R1取代基的末端甲基,表明增加疏水基团有利于活性提高。例如,在此处为羟基取代的化合物11(pIC50=7.699)的活性要比化合物2(pIC50=8.301)、5(pIC50=7.996)的低。另外在哌嗪基的N-4上有小块白色区域,说明引入亲水基团有利于活性。
图 4(b)为CoMSIA模型的氢键供体场等势图,青色区域表示氢键供体基团对活性有利,紫色则相反。由图可见,一大块青色区域覆盖了亚氨基上的氮原子,这说明该基团也许是氢键供体,可能与CSF-1R靶蛋白在结合位点形成氢键相互作用。从对接结果可知,亚氨基上的氮原子与关键氨基酸Cys666形成氢键相互作用,验证了前面的推测。
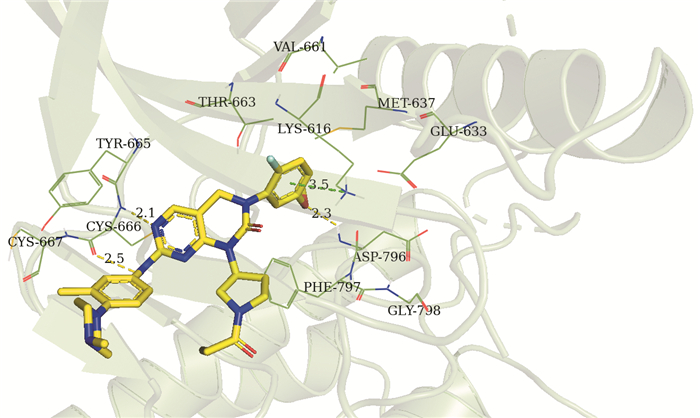
对54个化合物进行分子对接研究,结果发现,所有化合物在对接后均与CSF-1R靶蛋白对接口袋的氨基酸残基形成1~3个氢键,其中Cys666是关键氨基酸残基,均能与所有化合物形成氢键相互作用。为了进一步说明化合物与CSF-1R靶蛋白的结合特征,选取活性最好的化合物29的对接结果进行分析。图 5是化合物29与CSF-1R氨基酸残基的结合图,从图可知,29所在的结合口袋由氨基酸残基Lys616、Glu633、Met637、Val661、Thr663、Tyr665、Cys666、Cys667、Asp796、Phe797和Gly798形成。Cys666与29中嘧啶环上的N原子及亚氨基的氢原子形成氢键相互作作用,Asp796与R2取代基中的氧原子形成氢键相互作用。此外,二氢嘧啶环上的苯环与Lys616形成π-阳离子作用。
为了寻找高选择性的CSF-1R小分子抑制剂,本研究对54个2-氧代-3, 4-二氢嘧啶并[4, 5-d]嘧啶类化合物进行分子对接和3D-QSAR研究。基于分子对接叠合构建了预测能力较好的CoMFA模型(q2=0.725,r2=0.960)和CoMSIA模型(q2=0.636,r2=0.958),根据两模型的三维等势图可进一步指导此类化合物的优化设计。分子对接研究表明,这些化合物可与Cys666、Asp796等氨基酸残基形成氢键,其中Cys666是关键氨基酸残基,与之结合可增强化合物与CSF-1R靶蛋白的结合稳定性。通过对分子对接和CoMFA、CoMSIA的结果的综合分析,为设计改造该类化合物提供了理论依据。
L Donner, L A Fedele, C F Garon et al. J. Virol., 1982, 41(2):489~500.
C J Sherr, C W Rettenmier, R Sacca et al. Cell, 1985, 41(3):665~676. doi: 10.1016/S0092-8674(85)80047-7
M A Lemmon, J Schlessinger. Cell, 2010, 141(7):1117~1134. doi: 10.1016/j.cell.2010.06.011
F J Pixley, E R Stanley. Trends Cell Biol., 2004, 14(11):628~638. doi: 10.1016/j.tcb.2004.09.016
S Patel, M R Player. Curr. Top. Med. Chem., 2009, 9(7):599~610. doi: 10.2174/156802609789007327
M I El-Gamal, H S Anbar, K H Yoo et al. Med. Res. Rev., 2013, 33(3):599~636. doi: 10.1002/med.21258
M I El-Gamal, S K Al-Ameen, D M Al-Koumi et al. J. Med. Chem., 2018, 61(13):5450~5466. doi: 10.1021/acs.jmedchem.7b00873
W D Tap, H Gelderblom, E Palmerini et al. Lancet, 2019, S0140~6736(19):30764.
A Kumari, O Silakari, R K Singh. Biemed. Pharmacother., 2018, 103:662~679. doi: 10.1016/j.biopha.2018.04.046
S Xu, T Xu, L Zhang et al. J. Med. Chem., 2013, 56(21):8803~8813. doi: 10.1021/jm4012388
Q Xun, Z Zhang, J Luo et al. J. Med. Chem., 2018, 61(6):2353~2371. doi: 10.1021/acs.jmedchem.7b01612
G W A Milne. J. Chem. Inf. Model., 2010, 50(11):2053. doi: 10.1021/ci100385n
Y Fang, Y Lu, X Zang et al. Sci. Rep., 2016, 6:23634. doi: 10.1038/srep23634
H Huang, D A Hutta, J M Rinker et al. J. Med. Chem., 2009, 52(4):1081~1099. doi: 10.1021/jm801406h
张淑贞, 郑超, 朱长进.物理化学学报, 2015, 31(12):2395~2404. doi: 10.3866/PKU.WHXB201510142
吴倩, 李先国, 李燕等.化学通报, 2016, 79(6):509~515. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20151018001&flag=1
Y F Li, Y Q Chang, J Deng et al. Sci. Rep., 2016, 6:34387. doi: 10.1038/srep34387
W Zhang, Z Wei, C Lin et al. J. Mol. Struct., 2019, 1186:11~22. doi: 10.1016/j.molstruc.2019.02.107
张玲, 刘鹰翔, 赵钟祥等.化学通报, 2018, 81(2):148~157. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20170929002&flag=1
G Yan, X Yang, B Albijanic et al. Sol. Energy, 2019, 184:187~194. doi: 10.1016/j.solener.2019.03.092
A Cherkasov, E N Muratov, D Fourches et al. J. Med. Chem., 2014, 57(12):4977~5010. doi: 10.1021/jm4004285
宋昱, 任聪, 杨彭真等.化学通报, 2019, 82(5):446~451. http://www.hxtb.org/ch/reader/view_abstract.aspx?file_no=20180927001&flag=1

图 2 CoMFA和CoMSIA模型pIC50实验值及预测值的线性回归图
Figure 2 Linear regression between experimental and predicted pIC50 for the CoMFA and CoMSIA models

图 3 CoMFA模型的立体场等势图(a)和静电场等势图(b)
Figure 3 Steric contour maps (a) and electrostetic contour maps (b) for CoMFA model

图 4 CoMSIA模型的疏水场等势图(a)和氢键供体场等势图(b)
Figure 4 Hydrophobic (a) and H-bond donor (b) contour maps for CoMSIA model

图 5 化合物29与CSF-1R氨基酸残基结合图
Figure 5 Binding mode of compound 29 and CSF-1R amino acid residues
表 1 CSF-1R抑制剂的结构和生物活性数据
Table 1. Structure and activity data of CSF-1R inhibitors
| No | R1 | R2 | R3 | R4 | pIC50 | Predicted Value | |
| CoMFA | CoMSIA | ||||||
|
|
||||||
| 1* | |
- | - | - | 7.523 | 8.063 | 8.050 |
| 2* | |
- | - | - | 8.301 | 8.012 | 7.979 |
| 3* | |
- | - | - | 8.260 | 8.073 | 7.945 |
| 4 | |
- | - | - | 8.174 | 8.232 | 8.214 |
| 5 | |
- | - | - | 7.996 | 8.015 | 8.089 |
| 6 | |
- | - | - | 7.987 | 7.912 | 7.944 |
| 7 | |
- | - | - | 7.697 | 7.628 | 7.662 |
| 8 | |
- | - | - | 7.684 | 7.620 | 7.684 |
| 9 | |
- | - | - | 7.547 | 7.558 | 7.517 |
| 10 | |
- | - | - | 7.514 | 7.534 | 7.521 |
| 11* | |
- | - | - | 7.699 | 8.042 | 7.756 |
| 12 | |
- | - | - | 7.644 | 7.644 | 7.658 |
| 13* | |
- | - | - | 6.743 | 7.408 | 7.408 |
| 14 | |
- | - | - | 7.680 | 7.655 | 7.655 |
|
|||||||
| 15* | - | |
- | - | 8.284 | 7.967 | 7.971 |
| 16 | - | |
- | - | 8.252 | 8.255 | 8.352 |
| 17 | - | |
- | - | 7.818 | 7.809 | 7.827 |
| 18 | - | |
- | - | 7.742 | 7.762 | 7.762 |
| 19 | - | |
- | - | 7.879 | 7.916 | 7.879 |
| 20 | - | |
- | - | 7.830 | 7.759 | 7.803 |
| 21 | - | |
- | - | 7.785 | 7.835 | 7.769 |
| 22 | - | |
- | - | 7.686 | 7.765 | 7.716 |
| 23 | - | |
- | - | 8.292 | 8.332 | 8.238 |
| 24 | - | |
- | - | 8.347 | 8.244 | 8.321 |
| 25* | - | |
- | - | 8.237 | 7.987 | 7.755 |
| 26* | - | |
- | - | 7.553 | 8.216 | 7.813 |
| 27 | - | |
- | - | 6.821 | 6.806 | 6.78 |
| 28* | - | |
- | - | 8.347 | 7.991 | 7.927 |
| 29 | - | |
- | - | 8.699 | 8.400 | 8.313 |
| 30* | - | |
- | - | 8.357 | 8.047 | 7.881 |
| 31 | - | |
- | - | 8.678 | 8.738 | 8.678 |
|
|
||||||
| 32 | - | - | H | 4-甲基-哌嗪-1-基 | 7.866 | 7.824 | 7.892 |
| 33 | - | - | 2′-CH3 | 4-甲基-哌嗪-1-基 | 7.695 | 7.717 | 7.646 |
| 34 | - | - | 2′-OCH3 | 4-甲基-哌嗪-1-基 | 7.517 | 7.452 | 7.589 |
| 35 | - | - | 3′-OCH3 | 4-甲基-哌嗪-1-基 | 8.237 | 8.200 | 8.187 |
| 36 | - | - | 3′-F | 4-甲基-哌嗪-1-基 | 7.767 | 7.846 | 7.826 |
| 37 | - | - | 3′-Cl | 4-甲基-哌嗪-1-基 | 7.830 | 7.947 | 7.855 |
| 38* | - | - | 3′-CF3 | 4-甲基-哌嗪-1-基 | 7.496 | 7.893 | 7.380 |
| 39 | - | - | 3′-ethyl | 4-甲基-哌嗪-1-基 | 8.000 | 8.164 | 8.148 |
| 40* | - | - | 3′-isopropyl | 4-甲基-哌嗪-1-基 | 7.588 | 8.134 | 8.189 |
| 41 | - | - | 3′-CH3 | |
7.385 | 7.481 | 7.376 |
| 42 | - | - | 3′-CH3 | |
7.599 | 7.540 | 7.617 |
| 43* | - | - | 3′-CH3 | |
7.587 | 8.312 | 8.428 |
| 44 | - | - | 3′-CH3 | |
8.310 | 8.315 | 8.316 |
| 45 | - | - | 3′-CH3 | |
8.328 | 8.323 | 8.314 |
| 46 | - | - | 3′-CH3 | |
8.167 | 8.228 | 8.210 |
| 47 | - | - | 3′-CH3 | |
8.208 | 8.202 | 8.255 |
| 48 | - | - | 3′-CH3 | |
8.187 | 8.264 | 8.282 |
| 49 | - | - | 3′-CH3 | |
8.155 | 8.112 | 8.140 |
| 50 | - | - | 3′-CH3 | |
7.987 | 7.992 | 7.964 |
| 51 | - | - | 3′-CH3 | |
8.143 | 8.177 | 8.156 |
| 52* | - | - | 3′-CH3 | |
8.108 | 8.309 | 8.229 |
| 53 | - | - | 3′-CH3 | |
8.469 | 8.412 | 8.466 |
| 54 | - | - | - | |
8.528 | 8.510 | 8.504 |
| *代表测试集 | |||||||
 下载: 导出CSV
下载: 导出CSV
表 2 CoMFA和CoMSIA模型的统计参数
Table 2. The statistical parameters of CoMFA and CoMSIA models
| No. | q2 | N | SEE | r2 | F | Field contribution | ||||
| S | E | H | D | A | ||||||
| CoMFA | 0.725 | 9 | 0.086 | 0.960 | 79.542 | 0.760 | 0.240 | - | - | - |
| CoMSIA-SE | 0.574 | 6 | 0.184 | 0.799 | 21.888 | 0.905 | 0.095 | - | - | - |
| CoMSIA-SH | 0.588 | 8 | 0.093 | 0.953 | 76.679 | 0.272 | - | 0.728 | - | - |
| CoMSIA-ED | 0.176 | 4 | 0.321 | 0.352 | 4.749 | - | 0.509 | - | 0.491 | - |
| CoMSIA-HA | 0.519 | 8 | 0.114 | 0.927 | 49.529 | - | - | 0.683 | - | 0.317 |
| CoMSIA-SEH | 0.610 | 10 | 0.097 | 0.951 | 56.354 | 0.233 | 0.082 | 0.685 | - | - |
| CoMSIA-SED | 0.618 | 8 | 0.171 | 0.837 | 19.898 | 0.612 | 0.101 | - | 0.286 | - |
| CoMSIA-SHD | 0.636 | 9 | 0.088 | 0.958 | 76.184 | 0.228 | - | 0.653 | 0.119 | - |
| CoMSIA-SHA | 0.547 | 8 | 0.110 | 0.933 | 53.551 | 0.179 | - | 0.555 | - | 0.266 |
| CoMSIA-EDA | 0.345 | 7 | 0.229 | 0.698 | 10.548 | - | 0.207 | - | 0.156 | 0.638 |
| CoMSIA-EHA | 0.520 | 10 | 0.117 | 0.929 | 38.009 | - | 0.057 | 0.672 | - | 0.270 |
| CoMSIA-EHD | 0.545 | 9 | 0.121 | 0.921 | 38.992 | - | 0.125 | 0.759 | 0.116 | - |
| CoMSIA-HDA | 0.528 | 10 | 0.097 | 0.951 | 56.089 | - | - | 0.632 | 0.108 | 0.260 |
| CoMSIA-SEHD | 0.604 | 9 | 0.113 | 0.931 | 45.031 | 0.194 | 0.124 | 0.581 | 0.100 | - |
| CoMSIA-SEHA | 0.541 | 10 | 0.112 | 0.935 | 41.428 | 0.161 | 0.089 | 0.531 | - | 0.220 |
| CoMSIA-SHDA | 0.564 | 10 | 0.093 | 0.955 | 61.921 | 0.166 | - | 0.522 | 0.095 | 0.218 |
| CoMSIA-SEDA | 0.583 | 10 | 0.161 | 0.866 | 18.705 | 0.353 | 0.108 | - | 0.146 | 0.393 |
| CoMSIA-EHDA | 0.528 | 10 | 0.126 | 0.918 | 32.368 | - | 0.107 | 0.561 | 0.094 | 0.238 |
| CoMSIA-SEHDA | 0.552 | 10 | 0.115 | 0.931 | 39.213 | 0.138 | 0.102 | 0.471 | 0.088 | 0.202 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们