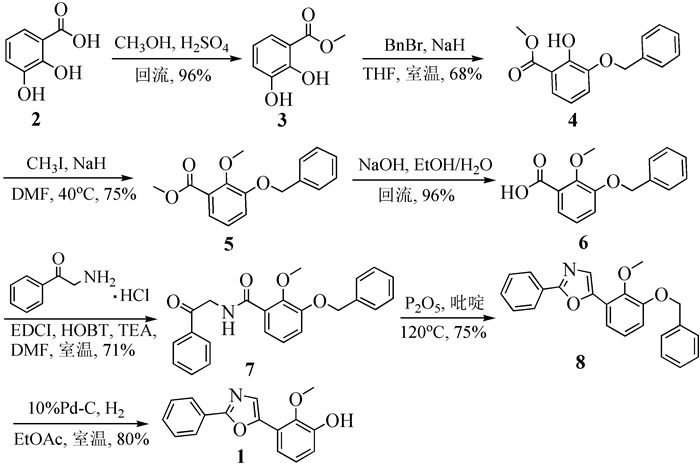
图 图式
化合物1的合成路线
Figure 图式.
Synthetic route of compound 1

中华裸蒴(Gymnotheca chinensis)作为我国植物的特有属之一[1],具有清热利湿等功效,是民间长期用作治疗肺痨咳嗽、腹胀水肿等疾病的传统草药[2]。2016年,肖世基等[3]首次从中华裸蒴中分离得到天然产物2-(3-羟基-2-甲氧基苯基)-5-苯基噁唑(Gymnothecaoxazoles A,1),并测试了该化合物对人肺癌细胞A549的抑制作用,无明显的生物活性(IC50>50μmol/L)。不过,文献报道一般含有2, 5-二苯基噁唑(PPO)结构的化合物均具有一定的杀菌活性,部分化合物还具有辐射防护和放射增敏作用,是国防工业和科学研究中极为常用的有机荧光闪烁剂,在医学、生物及化学化工等领域用于同位素的测定[4, 5],因此具有一定的研究价值。考虑到该化合物在自然界中的含量极少,全合成是获得此化合物的有效途径。为此,本文对Gymnothecaoxazoles A的合成方法进行研究,选择以2, 3-二羟基苯甲酸为起始原料,通过对羧基甲酯化,3-位羟基的选择性苄基保护,2-位羟基甲醚化,碱性条件下酯水解,再与2-氨基苯乙酮盐酸盐缩合生成重要中间体7,中间体7在P2O5作为脱水剂条件下环化得中间体8,最后Pd-C/H2脱苄基得到1,总收率为20%。反应路线见图式 1。
Mercury 400核磁共振谱仪(美国Varian公司);Xevo G2-S QTof四极杆飞行时间串联质谱仪(美国Waters公司);Tektronix X-4熔点仪(北京泰克仪器有限公司,温度未经校正);Tensor 37红外光谱仪(德国Bruker公司)。
2, 3-二羟基苯甲酸(99%)和2-氨基苯乙酮盐酸盐(98%)购自上海麦克林生化科技有限公司,其他所用试剂均为市售分析纯级试剂。
2, 3-二羟基苯甲酸甲酯(3)[6]的合成:将3.0g(19.5mmol)2, 3-二羟基苯甲酸2溶于30mL甲醇中,冰浴下逐滴加入2mL浓硫酸,氮气保护下回流4h。将反应液冷却至室温,减压浓缩蒸除部分甲醇,加入饱和碳酸氢钠至不再产生气泡,用乙酸乙酯(50mL×3)萃取,饱和食盐水洗涤,无水硫酸钠干燥,减压浓缩,得灰棕色固体3.15g,收率为96%,熔点75~76 ℃。1H NMR(400MHz,CDCl3)δ:10.90 (s,1H);7.38~7.35(m,1H);7.13~7.09(m,1H);6.82~6.77(m,1H);5.84~5.61(m,1H);3.96(s,3H);HRMS:理论值C8H7O4[M-H]-167.0344,实测值167.0347。
3-苄氧基-2-羟基苯甲酸甲酯(4)[7]的合成:冰浴下将1.06g(26.8mmol)60%的氢化钠加入到10mL干燥四氢呋喃中,缓慢滴加1.50g(8.9mmol)化合物3的2mL四氢呋喃溶液,冰浴下搅拌1h。加入1.83g(10.7mmol)苄溴,室温反应3h。将反应液倒入20mL冰水中,乙酸乙酯(30mL×3)萃取,无水硫酸钠干燥,减压浓缩后硅胶柱层析(淋洗剂为石油醚/乙酸乙酯,体积比20:1)分离纯化,得白色固体1.56g,收率为68%,熔点78~80℃。1H NMR (400MHz,DMSO-d6)δ:10.59(s,1H);7.45(d,2H,J=7.2Hz);7.41~7.30(m,3H);7.29(d,2H,J=8.0Hz);6.84(t,1H,J=8.0Hz);5.13(s,2H);3.88(s,3H)。HRMS:C15H14O4Na,理论值281.0790 [M+Na]+,实测值281.0790。
3-苄氧基-2-甲氧基苯甲酸甲酯(5):将1.27g(4.9mmol)化合物4溶于20mL无水DMF中,冰浴条件下加入0.22g(5.4mmol)60%的氢化钠和0.71g(5.0mmol)碘甲烷,氮气保护下40℃反应2h。将反应液倒入10mL冰水中,用乙酸乙酯(50mL×4)萃取,饱和食盐水洗涤(50mL×2),无水硫酸钠干燥后过滤,滤液减压蒸除溶剂,硅胶柱层析(淋洗剂为石油醚/乙酸乙酯,体积比40:1)分离纯化得无色油状物0.95g,收率为75%。1H NMR(400MHz, CDCl3)δ:7.48~7.42(m,2H);7.42~7.36(m,2H);7.36~7.31(m,2H);7.13~7.07(m,1H);7.07~7.02(m,1H);5.14(s,2H);3.95(s,3H);3.91(s,3H);13C NMR (101MHz,CDCl3)δ:166.8,152.6,149.7,136.7,128.7,128.1,127.3,126.3,123.8,122.8,118.1,71.1,61.6,52.3;HRMS:C16H16O4Na,理论值295.0946 [M+Na]+,实测值295.0947。
3-苄氧基-2-甲氧基苯甲酸(6)的合成:将0.95g(3.48mmol)化合物5加入到1.4g(34.8mmol)NaOH的20mL乙醇/水(体积比1:1)溶液中,回流反应3h,蒸除部分乙醇,用1mol HCl调节pH至2,乙酸乙酯(50mL×4)萃取,无水硫酸钠干燥,减压浓缩蒸干,得白色固体0.87g,收率为96%,熔点144~145 ℃。1H NMR (400MHz,CDCl3)δ:11.43(s,1H);7.77~7.74(m,1H);7.46~7.40(m,4H);7.39~7.35(m,1H);7.24~7.21(m,1H);7.20~7.15(m,1H);5.16(s,2H);4.11(s,3H)。HRMS:C15H13O4,理论值257.0814 [M-H]-,实测值257.0824。
3-苄氧基-2-甲氧基-N-(2-氧代-2-苯乙基)苯甲酰胺(7)[8]的合成:取0.76g(2.94mmol)化合物6和2-氨基苯乙酮盐酸盐0.48g(3.54mmol)溶于20mL DMF,加入0.62g(3.24mmol)1-乙基-(3-二甲基氨基丙基)碳化二亚胺盐酸盐(EDCI)、0.44g(3.24mmol)1-羟基苯并三唑(HOBT)和0.40g(3.98mmol)三乙胺,室温反应24h。反应完全后将反应液倒入20mL蒸馏水中,二氯甲烷(50mL×4)萃取,饱和氯化钠(50mL×2)洗涤,无水硫酸钠干燥,减压浓缩后硅胶柱层析(淋洗剂为石油醚/乙酸乙酯,体积比5:1)分离纯化得白色固体0.82g,收率为71%,熔点110~112 ℃。1H NMR (400MHz,CDCl3)δ:9.26(s,1H);8.06~8.02(m,2H);7.81~7.77(m,1H);7.66~7.60(m,1H);7.55~7.50(m,2H);7.49~7.46(m,2H);7.44~7.39(m,2H);7.38~7.34(m,1H);7.15~7.11(m,2H);5.16(s,2H);5.05~4.98(m,2H);4.10(s,3H);13C NMR (101MHz,CDCl3)δ:194.2,165.1,151.8,148.6,136.5,134.6,133.9,128.9,128.6,128.1,127.9,127.3,126.0,124.1,123.3,117.7,71.1,61.5,47.1;HRMS:C23H22NO4,理论值376.1549 [M+H]+,实测值376.1551。
2-(3-苄氧基-2-甲氧基苯基)-5-苯基噁唑(8)的合成:取0.26g(0.69mmol)化合物7加入到48mL封管中,再加入5mL吡啶和0.11g(0.76mmol)五氧化二磷,拧紧封盖,120℃下反应4h。反应液用5%HCl调节pH=3,二氯甲烷(20mL×3)萃取,无水硫酸钠干燥,减压浓缩后硅胶柱层析(淋洗剂为石油醚/乙酸乙酯,体积比1:2)分离纯化得白色固体0.19g,收率75%,熔点129~130℃。1H NMR(400MHz,CDCl3)δ:7.76~7.73(m,2H);7.67(dd,J=7.6,1.6Hz,1H);7.51~7.47(m,3H);7.47~7.38(m,4H);7.37~7.32(m,2H);7.15~7.05(m,2H);5.19(s,2H);4.07(s,3H);13C NMR (101MHz,CDCl3)δ:159.4,152.9,151.3,148.3,136.9,129.0,128.7,128.4,128.1,127.4,124.3,123.4,122.3,121.9,116.6,71.1,61.4。HRMS:C23H20NO3,理论值358.1443[M+H]+,实测值358.1439。
2-(3-羟基-2-甲氧基苯基)-5-苯基噁唑(gymnothecaoxazoles A)(1)的合成:取0.10g(0.28mmol)化合物8加入10mL反应瓶中,加入4mL乙酸乙酯和0.03g(0.28mmol) 10%钯碳,氢气环境下剧烈搅拌,室温反应6h,将反应液抽滤,滤液减压浓缩,石油醚-甲醇重结晶,得白色固体0.06g,收率为80%。1H NMR (400MHz,(CD3)2CO)δ:8.35(s,1H);7.72(d,J=7.6Hz, 2H);7.58(s,1H);7.42(dd,J=7.6、1.6Hz,1H);7.37(t,J=7.6Hz,2H);7.26(t,J=7.6Hz,1H),7.01~6.92(m,2H);3.84(s,3H);13C NMR (101MHz,(CD3)2CO)δ:160.1,152.3,152.0,147.0,129.9,129.3,129.1,125.5,124.9,124.5,122.6,121.2,119.3,61.6。HRMS:C16H14NO3,理论值268.0974 [M+H]+,实测值268.0978。
在目标化合物gymnothecaoxazoles A的合成过程中,最初采用的合成路线是直接将起始原料2, 3-二羟基苯甲酸的羧基和3-位羟基用苄溴保护后再对2-位羟基甲基化,但由于空间位阻过大而收率较低。于是根据需要对路线进行了优化,选择先将羧基甲酯化,再选择性地对3-位羟基进行苄基保护,最后对2-位羟基进行甲基化合成重要中间体5,收率明显提高。接下来化合物5经过水解、胺化得到α-酰胺基取代酮7,再在P2O5作为脱水剂的条件下经过Robinson-Gabriel法关环合成了噁唑环,此步反应需要在封管中无水条件下进行。最后一步是Pd-C/H2催化下的还原脱苄基保护得到天然产物gymnothecaoxazoles A,总收率为20%。本文的合成路线原料廉价易得、反应条件温和、操作简单,能够方便快捷地制备gymnothecaoxazoles A。
应俊生, 张玉龙. 中国种子植物特有属. 北京: 科学出版社, 1994: 540.
马林, 吴丰, 陈若芸. 中国中药杂志, 2003, 28(3): 196~198. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=zgzy200303001&dbname=CJFD&dbcode=CJFQ
S J Xiao, D L Guo, M S Zhang et al. Chin. Chem. Lett., 2016, 27(2016): 1064~1066. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=fxkb201607015&dbname=CJFD&dbcode=CJFQ
L N Shishkina, M A Smotriaeva, M V Kozlov et al. Radiat. Biol. Radioecol., 2004, 45(2004): 610~615. http://europepmc.org/abstract/MED/16304778
J T Ahokas, C Davies, N Jacobsen et al. Pharmacol. Toxicol., 1987, 61(1987): 184~190. http://europepmc.org/abstract/med/3684951
M A Dean, S R Hitchcock. Tetrahed. Asymm., 2010, 21(20): 2471~2478. https://www.sciencedirect.com/science/article/abs/pii/S0957416610006853
R S Coleman, E B Grant. J. Org. Chem., 1991, 56(4): 1357~1359. doi: 10.1021/jo00004a006
A Moreau, A Couture, E Deniau et al. Synthesis, 2004, 10: 1664~1670. doi: 10.1055/s-2004-829114

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载: