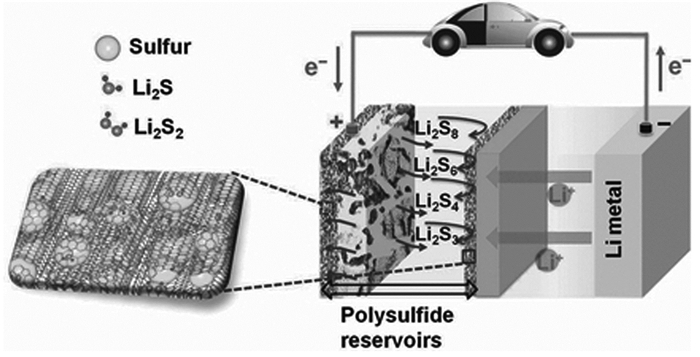
图 1
Li-S电池的工作原理示意图
Figure 1.
Schematic operation proposed for the rechargeable Li-S battery

随着科技的不断发展,人类社会对能源的需求量日益增加,高容量电池也越来越成为关注的焦点,目前锂离子电池的能量密度虽已达150~200Wh/kg[1, 2],但是传统的LiCoO2[3]、LiFeO4[4, 5]、LiNiO2[6, 7]的理论容量有限,很难进一步提升其电池容量[8~11]。锂硫(Li-S)电池具有高的理论比容量(1675mAh/g)和高的理论比能量(2600Wh/kg)[12~14];而且硫储量丰富,价格低廉,不会对环境造成污染[15]。因此,Li-S电池的发展对减少化石燃料使用及环境保护有重要的意义,在电动储能方面也有很好的应用前景。
单质硫理论比容量虽然高达1675mAh/g,与锂组装成电池,理论比能量可达2600Wh/kg,在二次电池中具有很明显的优势,但是Li-S电池目前能够实现的电池容量和理论值还有一定的差距,容量衰减快、循环寿命短等因素限制了Li-S电池的实用化进程[16~18]。这源于Li-S电池中长链多硫化物Li2Sn(n>2)在电解液中的溶解[19]。锂硫电池放电过程主要按方程(1)~(6)逐步进行[20],其工作原理如图 1所示。
正极放电开始时,固态的硫单质会在正极表面放电形成液态硫单质,然后按照式(3)~(6)放电过程进行,最终形成Li2S沉积在正极表面[21]。
|
$ {\rm S_8+16Li}\to 8{\rm Li_2S(s)} $ |
|
$ {\rm S_8(s)}\to {\rm S_8(l)} $ |
|
$ {\rm S_8+2Li}\to {\rm Li_2S_8} $ |
|
$ {\rm Li_2S_8+2Li}\to2{\rm Li_2S_4} $ |
|
$ {\rm Li_2S_4+2Li}\to2{\rm Li_2S_2(s)} $ |
|
$ {\rm Li_2S_2(s)+2Li}\to2{\rm Li_2S(s)} $ |
但是长链Li2Sn(n>2)易于溶解到电解液中,受浓度梯度影响,长链Li2Sn穿梭至锂负极表面发生腐蚀反应(式(7)~(10))[22],形成Li2S2/Li2S及链长较短的Li2Sn,Li2S2/Li2S会继续与穿梭至负极表面的长链Li2Sn发生歧化反应形成链长较短的Li2Sn。这种短链的Li2Sn会穿梭到正极表面被氧化形成长链Li2Sn,引起快速的电池容量衰减。这种Li2Sn在正负极间穿梭的现象称为穿梭效应[23, 24]。穿梭效应会造成活性物质的损失,降低电池的可存储性能,使绝缘的Li2S2/Li2S覆盖在负极,降低Li电极的导电性等。它是导致电池容量衰减快和循环寿命短等问题的重要因素[14]。
|
$ {\rm Li_2S}_n+2{\rm Li}\to{\rm Li_2S(s)}+{\rm Li_2S}_{(n-1)} $ |
|
$ {\rm Li_2S}_n+2{\rm Li}\to{\rm Li_2S_2(s)}+{\rm Li_2S}_{(n-2)} $ |
|
$ {\rm Li_2S}{\rm(s)+ Li_2S}_n\to{\rm Li_2S}_k+{\rm Li_2S}_{(n-k+1)} $ |
|
$ {\rm Li_2S}_2{\rm(s)+ Li_2S}_n\to{\rm Li_2S}_k+{\rm Li_2S}_{(n-k+2)} $ |
为了解决锂硫电池中长链Li2Sn的溶解问题,目前国内外研究人员对正极材料的改良主要从长链Li2Sn的包覆、吸附、催化反应三个思路开展。本文将从这几个方面出发分析总结近年来提高锂硫电池性能研究的现状。
目前,在锂硫电池正极材料的合成过程中,为了抑制锂硫电池的穿梭效应、提高正极材料的循环利用率,研究人员主要从Li2Sn的包覆、Li2Sn的吸附及最新提出的催化Li2Sn快速分解方面开展研究。
Li2Sn包覆是解决锂硫电池穿梭效应十分有效的思路,包括碳骨架、导电聚合物、金属氧化物的包覆等。研究人员倾向于制备具有巨大表面积和丰富孔结构的纳米包覆结构材料以提高硫的负载率,但当硫正极电导率较差或硫颗粒较大时,有效物质硫不能得到充分利用。另外当正极材料孔径较大时,吸附能力较弱的多孔材料(如多孔碳)不能有效限制Li2Sn的溶解;当孔径较小时,硫的负载率将明显减少。一些包覆结构的正极材料(如金属氧化物)能对Li2Sn提供良好的吸附作用,但通常需要再添加sp2杂化碳等材料改善其导电性能,却因此也缩减了锂硫电池的能量密度。因此,研究人员致力于从调控正极材料孔径大小、增强对Li2Sn的吸附作用、提高硫的负载率及导电性等方面设计和筛选锂硫电池正极包覆材料。
锂硫电池正极中硫本身导电性很差(电导率为5×10-30S·m-1(25℃)),在放电反应过程中长链Li2Sn溶解造成穿梭效应,另外在锂化反应期间正极材料存在较大的体积膨胀[25~34]。因此,引入碳骨架材料需要从以下几个方面考量:(1)高导电性,确保硫阴极中有效的导电网络,提高硫有效物质的利用率,确保在氧化还原过程中快速运输Li离子;(2)提高孔体积容量,提高硫有效物质的负载率;(3)适度增加包覆厚度和减小包覆碳孔径尺寸,抑制多硫化物溶解到本体电解液中;(4)增强正极材料的机械性能。研究人员设计了多种碳质材料来改善锂硫电池的性能,如表 1所示。
 下载:
导出CSV
下载:
导出CSV
| 正极材料 | 合成方法 | 形态 | 硫:载体质量比/硫负载率/(wt)% | 电压窗口/V (vs.Li+) |
初次放电容/(mAh/g) (容量保持%,循环次数/倍率) |
渗硫方法 | 尺寸/nm | 文献 |
| 3D超支化中空碳纳米棒封装硫(CNR-S)纳米复合阴极 | 化学气相传输/冷凝法和模板法 | 超支化中空碳纳米棒 | -/71.5 | 1.7~2.6 | 1378(84.8%,C275/0.1C); 990(94.4%,C500/1C)) | 熔体扩散 | - | [25] |
| 高度有序的中-微孔核壳碳结构(MMCS)负载硫复合阴极 | 模板法和纳米喷涂表面涂覆法 | 高度有序的中孔/微孔碳核壳结构 | 3:1/72 | 3.0~1.0 | 1037/C2(80%,C200/C2,/0.5C) | 熔体扩散 | 微孔、介孔 | [39] |
| 中空多孔CNT封装MWNT-硫(TTCN-S)复合阴极 | 模板法 | CNT包覆CNT结构 | 4:1/71 | 1.95~2.7 | 1274(72%,C50/500mAg-1); 750(72.2%,C200/2Ag-1) | 熔体扩散 | 微孔、介孔 | [40] |
| 分层多孔碳-硫(HPC-S)复合阴极 | 超声波喷雾热解 | 微孔包覆中孔和大孔分层多孔碳结构 | 1:1/46 | 1.7~2.6 | 1412(77%,C500/2.4C) | 熔体扩散 | 微孔、介孔、大孔 | [41] |
| 3D高效导电MWCNT多孔微球-硫(MS-C/S)复合阴极 | 喷雾干燥 | MWCNT缠结构建三维多孔微球 | 7:3/70 | 1.8~2.6 | 983(87.2%,C100/0.2C); 921(87.5%,C100/0.5C) | 熔体扩散 | - | [42] |
| 纳米结构石墨烯/SWNT@活化多孔碳-硫(GSH@APC-S)复合阴极 | 化学气相渗透化学气相沉积 | 多孔碳原位生长在导电石墨烯/CNT混合支架上的全碳纳米结构 | 1.5:1、5:1/50、77 | 1.5~3.0 | 50(wt)%,1010 (86.8%,C150/1C);77wt%,914(72%,C150/1C) | 熔体扩散 | 0.5~5 | [43] |
| 微孔碳纳米片组装的自支撑整体碳-硫复合阴极 | - | 微孔碳纳米片组装的自支撑整体碳 | -/70 | 1.7~2.8 | 1068(70.8%,C100/0.5C) | 电解法 | 微孔 | [44] |
| 中空碳纳米纤维-硫(HCNF-S)复合阴极 | 混合溶剂法制备 | 中空碳纳米纤维 | 1.5:1/60.8 | 1.5~3.0 | 1090(55.1%,C100/1C) | 熔体扩散 | - | [27] |
| 中空碳纳米纤维-硫复合阴极 | 模板法 | 中空碳纳米纤维 | -/75 | 1.7~2.6 | 900,(81.1% C30/C150/0.2C) | 熔体扩散 | - | [26] |
| 高度有序介孔碳-硫(CMK-3-S)复合阴极 | 模板法和纳米浇注法 | 高度有序介孔碳 | 7:3/70 | - | 1320,(83.3%,C20) | 熔体扩散 | 3~4 | [33] |
| 高孔隙率球形有序介孔碳纳米粒子-硫(S-BMC/S)复合阴极 | 模板法 | 球形有序介孔碳 | -/49.7、61.4、70.0 | 1.5~3.0 | 49.7(wt)%:1200(60.8%,C100/1C); 61.4&70.0(wt)%:1070(65.4%,C100/1C) | 熔体扩散 | 3.1~6 | [29] |
| 石墨烯层状多孔碳-硫(L-GPCS)复合阴极 | 水热法原位聚合 | 层状石墨烯多孔碳 | -/68 | 1.7~2.6 | 885.5(70%,C100/0.5) | 熔体扩散 | 1~10 | [30] |
| 空心碳胶囊/硫(C@S)复合阴极 | 模板法 | 空心碳球 | 7:3/70 | 1.7~3.1 | 1071(91%,C100/0.5C) | 熔体扩散 | 3 | [34] |
从表 1分析可知,锂硫电池正极性能的改善很难通过单一的碳结构材料来实现。sp2杂化纳米碳,如碳纳米管(CNT)和石墨烯具有非凡的机械强度和导电性,但是倾向于粘连在一起,具有很少的孔结构以储存硫有效物质。纳米结构的多孔碳提供巨大的表面积和丰富的孔结构,但电导率较差,降低了硫有效物质的利用率。另外当孔径较大时虽可以储存更大量的硫有效物质,但不能有效限制Li2Sn的穿梭效应。考虑到环-S8分子(0.76nm)之间的大小兼容性,当孔尺寸小于2nm时可以有效包覆Li2Sn[35~38],但其有限的孔体积减少了硫有效物质的负载量。
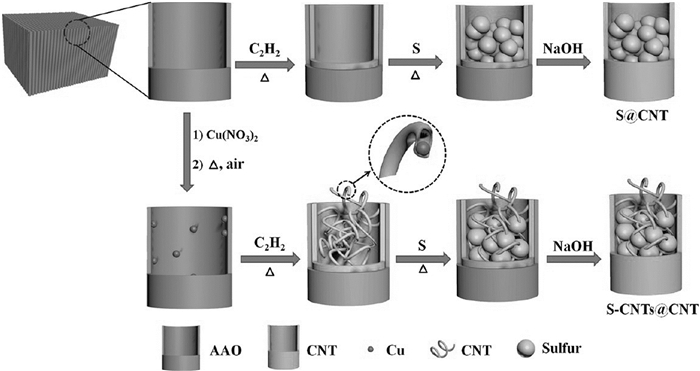
因此,为了有效改善锂硫电池正极的性能,可以将不同尺寸和类型的碳纳米材料复合到正极材料中[39~43],或引用更优的渗硫方法[44]。近年来,研究人员为提高锂硫电池的电化学性能设计了多种新型碳材料[45, 46]。Jin等[47]以氧化铝膜(AAO)为模板,经过硝酸铜处理后和C2H2/N2混合气体反应,形成CNTs@CNT@AAO复合材料,与升华硫按质量比1:5混合后渗硫,然后除去模板,制备出一种200nm大尺寸CNT包覆20nm小尺寸CNT和硫的复合结构(见图 2)。大CNT提高了硫的负载量,小尺寸CNT提高了硫正极材料的利用率,这种结构有效地通过物理作用限制长链Li2Sn的溶解。该CNTs@CNT@AAO复合材料作正极可以实现85.2(wt)%的硫负载率。在0.1C倍率下首次放电容量为1633mAh/g,而且体现了良好的倍率性能和循环稳定性。在5C倍率下150次循环后仍保持954mAh/g的放电容量。
Xiong等[48]通过水热法合成碳质层包覆碲线的均质Te/C复合纳米线,经过高温煅烧和化学刻蚀形成分层微孔-介孔碳纳米管(HMMCNT),这种材料中介孔的中空孔道能提高硫的负载率,较厚的微孔碳纳米管壁能有效限制Li2Sn溶解,良好的导电性有利于促进电子和离子转移,提高了硫有效物质的利用率,进而提高了电化学性能。在1.6A/g (0.95C)倍率下经过150次循环可以保持80.2%的放电容量。
Lou等[49]以一锅法无模板合成SnO2作为硬质模板,在SnO2内外表面均匀涂覆葡萄糖衍生的多糖,在还原气氛下碳化并将SnO2还原成金属锡,除去金属锡后,得到双壳中空碳球(DHCS,其透射电镜(TEM)照片见图 3),这种材料具有很高的比表面积(748m2/g)和大的总孔体积(1.685cm3/g),提高了硫的负载量,其本身的双层结构也能进一步限制多硫分子的渗出。通过熔体扩散渗硫,可以获得70(wt)%硫负载率的双壳中空碳球/硫(DHCS/S)复合结构。这种材料应用于锂硫电池正极,能有效限制Li2Sn的溶解,体现出优良的电化学性能,在0.1C倍率下初次放电容量为1020mAh/g,100次循环后电池容量保持在690mAh/g。
导电聚合物与硫和有机电解质有较好的相容性,聚合物包覆的硫正极复合材料通常在较低温度下使用原位一锅法合成,工艺较简单[50~52]。在缓解正极材料体积变化的同时,导电聚合物因其独特的官能团和链结构,可以通过链间或链内键合限制硫和多硫化物的移动[1, 53, 54]。
Wen等[55]通过原位聚合将聚吡咯(PPy)引入多壁碳纳米管(MWCNT)的表面,成功构建了PPy/MWCNT核/壳结构复合材料。复合材料中PPy的质量分数分别10%、25%、40%、60%,统一与硫以3:7质量比混合,在155℃渗硫得到硫质量分数为70%的S/PPy/MWCNT复合正极材料。研究发现,PPy质量分数为25%时电池体现出最好的电化学性能,初次放电容量为1275mAh/g,100次循环后放电容量仍为725.8mAh/g。
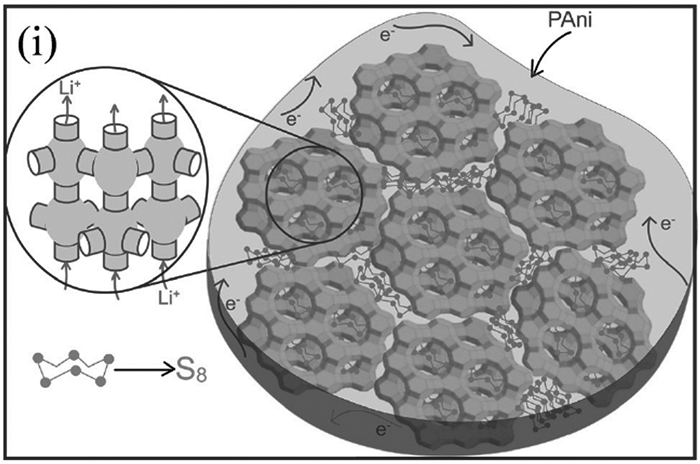
Bhattacharyya等[56]将硫和多硫化物有效封闭包裹在导电聚苯胺(PANI)鞘中的非碳质离子导电沸石(NaY)主体内,这种NaY-PANI材料在电池运行时为电子(通过PANI)和离子(通过NaY通道和穿过PANI鞘)提供不同的传输路径(见图 4)。首先,将升华硫渗入脱空气的沸石(NaY)内,形成NaY-S复合材料,使其浸泡在合成的氯化苯胺溶液中并使其干燥;然后加入H2O2,通过形成自由基引发聚合反应,涂覆在NaY-S材料表面,形成(S-NaY-PANI)复合材料。分别以不同硫负载率(30%~75%)的复合材料作正极,发现硫质量分数为65%的电池的电化学性能最优,200次循环后仍可保持600mAh/g的电池容量。
Luo等[57]提出用管状PPy包裹生物质衍生的碳纳米球(BCSs)结构作为锂硫电池的正极材料。首先,高温碳化处理澄清的西瓜汁获得碳纳米球,再通过吡咯原位聚合合成PPy包裹碳纳米球的复合材料(BCS@T-PPy),进一步与硫以质量比3:7混合,使用熔体扩散法将硫渗入复合材料中得到BCS@T-PPy/S。这种复合锂硫电池正极材料在0.5C倍率下500次循环后可保持685.8mAh/g的电池容量。
Gao等[58]使用一锅法原位合成三元还原氧化石墨烯/PPy/硫(rGO/PPy/S)分层纳米结构。在rGO/PPy/S结构中rGO/PPy和S之间形成C—S、S=C=S和S=O化学键,能有效降低Li2Sn的溶解。这种结构可以达到69.4(wt)%硫负载率,具有良好的倍率性能和循环性能。将其作为锂硫电池正极材料,在1C倍率下初次放电容量可达991.5mAh/g,400次循环后电池容量能保持63.2%。Jung等[59]在155℃下将氧化石墨烯(GO)和硫以质量比1:3混合渗硫合成GO-S,所制备的GO-S与导电碳和粘合剂藻酸钠按一定比例混合制成电极,再通过转印法将通过氧化聚合法制备的均匀PANI膜可控复合在硫阴极上,限制Li2Sn的穿梭。具有PANI层的硫阴极显示出更好的循环性能。在1C的倍率下初次放电容量为935mAh/g,200次循环后电池容量仍可以保持初始放电容量的96.4%。
Zhang等[60]通过编织双生物高分子(N-GG-XG)瓜尔胶(GG)和黄原胶(XG)来构建机械坚固的网络型粘合剂,和硫复合后得到高硫负载电极S@N-GG-XG,其具有26.4mAh/cm2的超高面积比容量和很好的倍率性能,在5C倍率下可以实现737mAh/g的电池容量。Chen等[61]将MWCNT和硫以质量比1:4复合,再通过原位聚合涂覆PANI制备MWCNT-S@PANI正极材料。MWCNT良好的导电性和PANI对Li2Sn的有效吸附不仅提高了正极的导电性能,而且能减少有效物质的流失。其首次放电容量可以达到1225mAh/g,400次循环后仍可保持760mAh/g的电池容量。
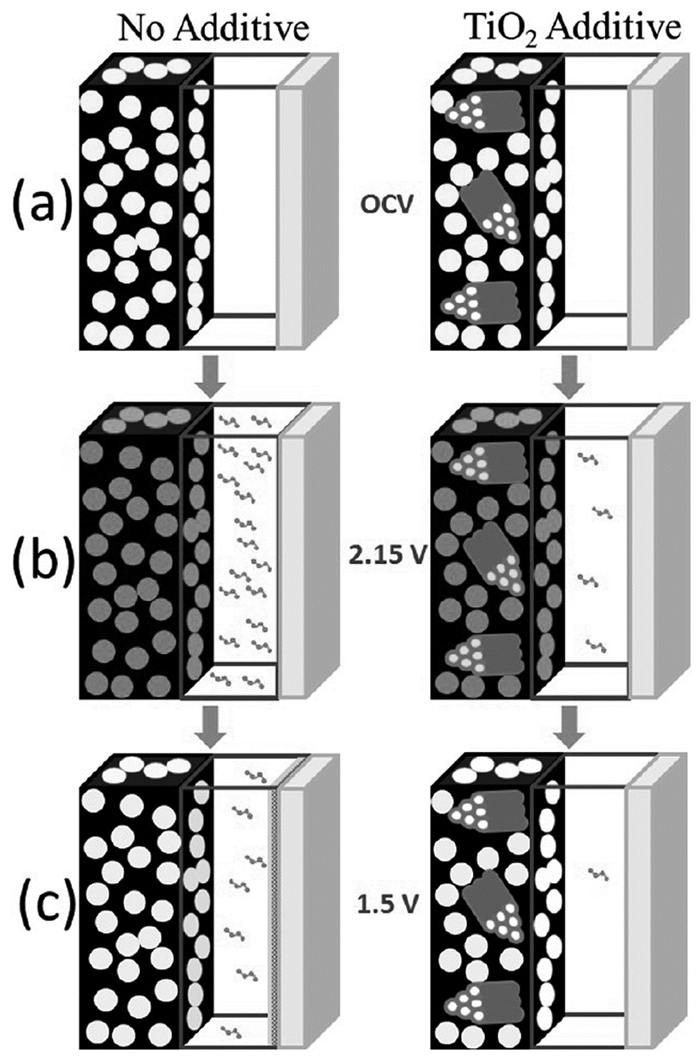
为了抑制锂硫电池的穿梭效应,许多研究人员从金属氧化物包覆的思路出发,制备多孔金属氧化物对Li2Sn进行包覆[62~64]。孔状结构为循环过程正极体积膨胀提供了足够的空间,保持了正极材料结构稳定性;另外,Li2Sn由于表面物理包覆和表面化学吸附而被有效地限制在金属氧化物的孔中,从而有效缓解了它的溶解。Cui等[65]通过聚苯乙烯(PS)蛋白石模板法制备3D反蛋白石TiO2。首先将PS分散在水性溶液中,在干燥的过程中PS会自聚形成六边形密堆结构,进一步在这种结构上沉积无定形TiO2,最后在氢环境下除去PS模板,同时形成氢还原3D反蛋白石TiO2-x结构,再将升华硫渗入孔隙中形成TiO2-x/S复合结构(见图 5)。这种复合材料作为正极时具有高电导性、高的硫负载率且限制了Li2Sn的溶解。在C/5倍率下200次循环后可维持890mAh/g的电池放电容量。

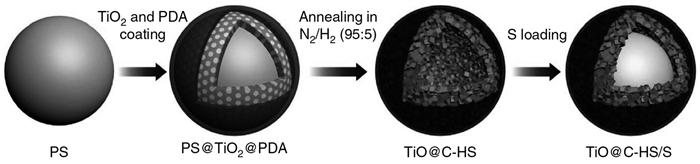
Lou等[66]先将PS和聚多巴胺(PDA)与TiO2复合形成PS@TiO2@PDA复合结构,再在N2/H2混合气体中处理形成TiO@中空碳微球(TiO@C-HS),将质量比为7:3的升华硫和TiO@C-HS的混合物在155℃渗硫形成TiO@C-HS/S电池正极材料,其合成示意图见图 6。其中TiO@C-HS具有很好的金属导电性和吸附Li2Sn的能力。所制备的正极材料具有优良的循环稳定性,在0.2C倍率下500次循环后仍能保持750mAh/g的电池容量。
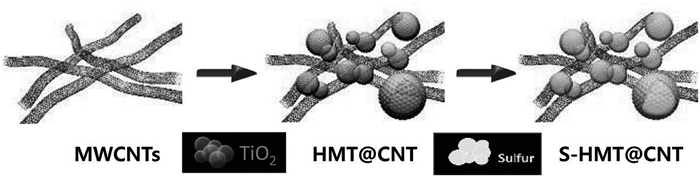
Sun等[67]将水热合成法制得的空心TiO2(HMT)球嵌入CNT间形成HMT@CNT复合结构,再和硫以质量比1:1.5混合渗硫得到56(wt)%硫含量的S-HMT@CNT复合正极材料(见图 7)。空心的TiO2及CNT良好的机械性能缓解了正极材料的体积膨胀,CNT可以有效提高正极材料的导电性能。这种正极材料在1C倍率下首次放电容量可达1113mAh/g,100次循环后电池容量仍可以保存93.5%。
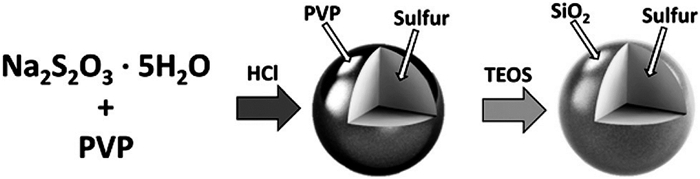
Ozkan等[68]用两步湿化学合成法合成SiO2并涂覆在硫颗粒表面,通过物理和化学吸附作用将硫有效成分保留在正极材料中,减少正极有效物质的流失,提高了电化学性能(其合成路线见图 8)。并且通过在这种复合结构中加入rGO可提高正极材料的导电性。在50次循环后电池容量保持为763.2mAh/g。
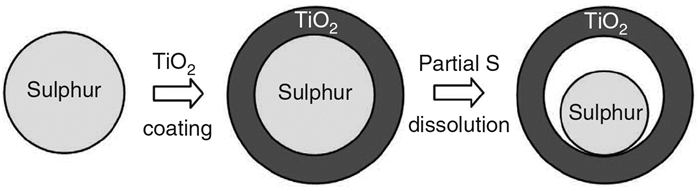
Cui等[69]合成了一种具有71(wt)%硫负载率的TiO2/S蛋壳结构复合正极材料(制备过程如图 9所示)。其能有效缓解充放电过程中的体积变化,而且TiO2对Li2Sn提供了有效的吸附作用。在0.5C的倍率下首次放电容量可达1030mAh/g,1000次循环的每循环容量衰减率仅0.033%。
锂硫电池穿梭效应的另一个解决思路是通过正极材料表面对Li2Sn的物理化学吸附作用阻止Li2Sn溶解到电解液中,从而有效解决Li2Sn的穿梭问题[70~72]。从吸附结构界面角度分析,长链或短链的Li2Sn是本征极性物质,末端硫含有大部分负电荷[73, 74],极性Li2Sn和极性基底通过形成强的化学作用力吸附在正极材料表面[75~77]。由于与非极性石墨烯表面微弱的分子间相互作用,Li2Sn会从电极正极溶解到电解液中,最终腐蚀锂电极。通常含有O2-的金属氧化物具有很强的极性表面[77~82],纳米尺寸的Mg0.6Ni0.4O是首个在锂硫电池阴极中作为添加剂用于吸附Li2Sn的金属氧化物[83]。另外,通过N、S、B等对GO或rGO[84~86]进行掺杂改性,可使碳质材料具有较强的极性。因此,研究人员向正极添加金属氧化物、GO/杂原子掺杂的rGO体系等,将长链Li2Sn通过强化学作用固定在锂硫电池的正极,减弱锂硫电池穿梭效应,提高锂硫电池的性能。
金属氧化物中O2-氧化态氧表面极性较强,所以对Li2Sn具有比较强的吸附作用,因此,研究人员通过向锂硫电池正极材料中添加金属氧化物以减弱多硫分子的溶解[79, 80]。Nazar等[87]向含有硫的介孔碳和介孔二氧化硅的复合材料中添加不同形态的介孔TiO2作正极材料,进一步提高正极对Li2Sn的吸附作用(如图 10所示)。在放电过程中,Li2Sn更倾向于吸附在TiO2孔道内。研究发现,比表面积最高、孔径为5nm的α-TiO2作为正极材料添加剂的电极表现出最好的循环性能。首次放电容量为1201mAh/g,在高倍率下循环200次后电池容量仍保持在750mAh/g。Cui等[88]用水热法合成TiO2纳米管,进一步在TiO2纳米管上涂覆SiO2,制得SiO2涂覆的TinO2n-1,再除去SiO2得到Magneéli相TinO2n-1。将Magneéli相TinO2n-1和升华硫以质量比1:3混合形成TinO2n-1-S复合材料作正极,相比于TiO2-S正极材料具有更好的电池性能。他们应用第一性原理和实验分析得出,Magneéli相TinO2n-1中低配位的Ti位点能更有效的吸附Li2Sn并提高电池的循环性能。电池在0.1C倍率下首次放电容量为1044mAh/g,100次循环后电池容量能保持99%。
Han等[89]用原子层沉积方法在rGO表面上沉积0.5nm厚度的Al2O3作为化学吸附基底以减少多硫分子的溶解。涂有超薄Al2O3原子沉积层的复合材料作正极,初次放电容量可达1136mAh/g,相比于无Al2O3涂覆正极材料提高了25%。Wang等[90]将尖晶石结构的NiFe2O4生长在CNT上,这种材料不仅为Li2Sn提供O2-、Ni2+、Fe3+等强吸附位点,而且CNT也能有效提高材料的导电性能。与硫复合后制备一种76(wt)%硫负载率的CNT/NiFe2O4-S三元复合材料阴极,在0.1C倍率下可实现1350mAh/g的可逆放电容量,在1C倍率下经过500次循环的每循环容量衰减率仅为0.009%。
为了抑制锂硫电池的穿梭效应,提高正极材料的循环利用率,研究人员发现杂原子(N、S、B等)掺杂的碳材料作基底能够有效吸附Li2Sn,甚至可以产生较金属氧化物更有效的物理化学吸附作用[91~96],在锂硫电池领域有很好的应用前景。Tan等[95]从第一性原理计算角度对比石墨烯和杂原子掺杂的石墨烯对Li2Sn的吸附作用,后者可有效提高材料对Li2Sn的吸附作用(见图 11)。
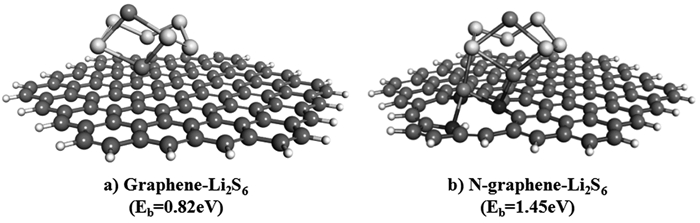
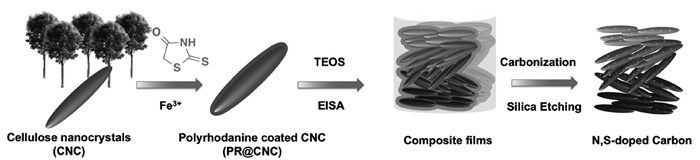
Zhou等[97]分别以尿素、硫化钠、硫脲作为N、S掺杂的石墨烯的N源和S源,合成出一种N、S共同掺杂的海绵体结构石墨烯。能够实现在0.2C倍率下1200mAh/g的放电容量,具有良好的循环稳定性,500次循环的每循环容量衰减率为0.078%。Nazar等[98]使用生物源可持续和低成本的纤维素纳米晶体,基于液晶驱动自组装形成纳米杆状介孔碳材料结构(CNC),在CNC结构上均匀涂覆聚草氨酸(PR),加入正硅酸乙酯(TEOS)提供间隔物以保持碳结构,形成PR@CNC/TEOS复合材料;进一步热解刻蚀硅形成N、S双掺杂的碳材料结构(图 12为合成原理示意图),渗硫后可实现70(wt)%硫负载率。以这种复合材料作锂硫电池正极材料可增强对Li2Sn的吸附作用,在2C倍率下初次放电容量为830mAh/g,1100次充放电循环后电池容量每循环衰减率仅为0.052%。
Yuan等[99]将一种由N掺杂石墨烯和富硼碳层结合的复合材料在155℃渗硫后作为锂硫电池的正极材料,正极中的带负价的N和带正电的B分别与正极表面长链Li2Sn的Li+、S2-n提供化学吸附作用,并且可以实现正极70(wt)%硫负载率,将其应用于锂硫电池正极,在2C倍率下1500次循环后电池容量每循环衰减率为0.035%。Wang等[100]将乙二胺和GO在溶液状态下75℃共热,获得氨基处理的氧化石墨烯(EFG),在155℃渗硫制备硫负载率为60(wt)%的(EFG-S)复合材料。以这种复合材料作正极,表现出良好的倍率性能和循环稳定性,在4C高倍率下循环350次后仍能够保持80%的电池容量。第一性原理计算也表明杂原子掺杂石墨烯对Li2Sn具有良好的物理化学吸附作用,能有效限制Li2Sn的穿梭效应[71]。
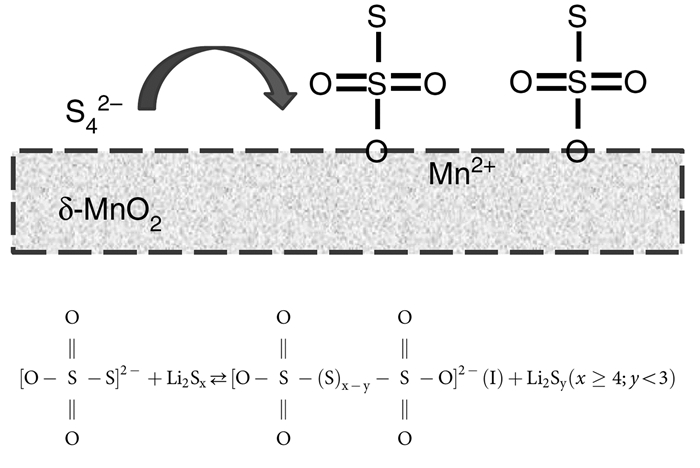
锂硫电池在放电过程中,正极会发生一系列的分步反应,由单质硫分步放电形成不同链长的Li2Sn(n>2),其中长链的Li2Sn易溶解于电解液中;另外,长链Li2Sn反应过程较多,从而为长链Li2Sn的穿梭提供条件。研究人员发现,Li2Sn会在一些正极材料表面形成化学官能团,能够使长链Li2Sn快速分解成溶解性很差的固态Li2S2或Li2S[101]。Nazar等[102]提出通过化学反应来减弱多硫分子溶解,以GO作模板和KMnO4合成层状δ-MnO2纳米片,再和纳米尺寸硫单质在155℃下渗硫得到S/MnO2复合物。以这种材料作锂硫电池正极,放电过程中Li2Sn会和材料表面形成一种与硫代硫酸根类似的中间体,这种中间体能加快长链Li2Sn反应形成短链的Li2S2或Li2S,沉积在正极材料表面,从而减弱穿梭效应,提高电池的循环稳定性。在中等倍率放电,电池容量可达1300mAh/g,2000次循环后电池容量每循环衰减率仅为0.036%。中间体形成机理及催化反应原理如图 13所示。
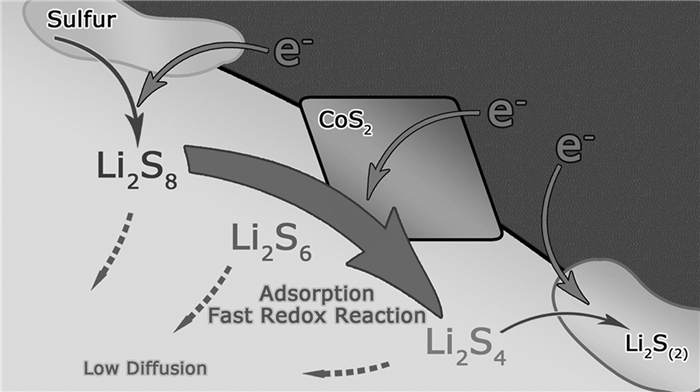
Faber等[103]已经证明黄铁矿型CoS2对Li2Sn还原有很高的催化活性,在300K下具有6.7×103S/cm的电导率。Zhang等[104]将一种亚磺酸半金属性黄铁矿CoS2电催化剂引入锂硫电池碳/硫正极中,通过推进多硫化物氧化还原来驱动Li-S电池性能。半金属性的CoS2和Li2Sn之间具有很强的化学吸附作用。而且,在静电亲和力驱动下提供有效的电子途径和对Li2Sn氧化还原反应的高电催化活性(其原理示意图如图 14所示)。电池的电化学性能大幅改进,在0.5C倍率下的初次放电容量为1368mAh/g,2C倍率下每个循环电池容量衰减率仅0.034%。
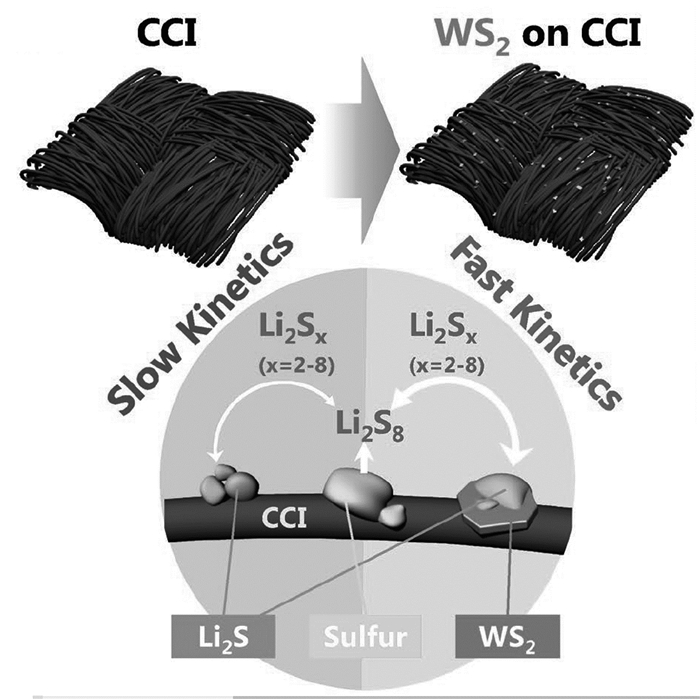
二硫化钨(WS2)是一种常见的用于加氢脱硫的催化剂,它对硫和硫化物具有很强的吸附能力[105, 106]。Goodenough等[107]提出将层状WS2负载在阴极集电器和碳布中间层上的双功能电极结构,WS2能够有效吸附Li2Sn,而碳布中间层既作为一种物理屏障阻碍Li2Sn的迁移,又为阴极集电器和吸附在WS2颗粒亲水边缘位点的Li2Sn提供快速的电子转移,加快Li2Sn还原形成不溶的短链Li2Sn,如图 15所示。这种电极结构渗硫作正极在0.5C倍率下500次循环后仍能获得近1000mAh/g放电容量,在5C高倍率下仍能达到750mAh/g放电容量。
在过去的几十年中,科研人员在锂硫电池正极材料的研制和探索中取得很大进展,在解决Li2Sn穿梭效应方面取得一定成效,使锂硫电池的实际放电比容量和循环性能有很大改善,性能优良的正极复合材料即使在1000次循环后,仍保持很低的电池容量衰减率,表现出良好的循环性能。但是对于锂硫电池的实用化仍然有很大的挑战:一是锂硫电池正极材料在充放电过程中Li2Sn固液转换引起的体积膨胀的问题;二是锂硫电池正极导电性能差使得硫有效成分利用效率较低,此外Li2Sn的溶解流失易引起穿梭效应;三是正极复合材料中硫有效物质含量较低等问题都亟待解决。
如果要在锂硫电池正极材料设计中进一步取得更大突破,可以在以下几个方面展开探索。其一,通过适量引入良好的导体改善正极材料的导电性能;开发尝试新型的渗硫方法(如电解法)进一步减小硫颗粒尺寸;增加硫和正极导电材料的接触率,提高硫的利用效率。再是,设计导电性、机械性能良好的正极硫包覆材料,并合理调控材料的尺寸,其中介孔和大孔材料可作为硫的载体,微孔材料可以作为束缚Li2Sn溶解的有效屏障。另外尝试将导电性优良的材料(如导电聚合物、碳纤维布材料等)引入正极材料的吸附体系中,减少添加剂在正极材料中所占比率,提高电池容量。还有,探索具有加快Li2Sn分解的催化活性物质尤其是未来研究的重要方向,其核心可集中在Li2Sn中硫链的捕捉及电子的输送方面。高效催化剂可加快长链Li2Sn分解成难溶的短链Li2S2/Li2S,缩短长链Li2Sn的存在时间,有效抑制多硫化物在电解液中的溶解,提高硫的利用率;而且在一定程度上缓解充放电过程中硫正极的体积膨胀,改善锂硫电池的循环寿命。
J Wang, J Yang, J Xie et al. Adv. Mater., 2002, 14: 963~965. doi: 10.1002/1521-4095(20020705)14:13/14%3C963::AID-ADMA963%3E3.0.CO;2-P/full
H Jiang, Y Fu, Y Hu et al. Small, 2014, 10: 1096~1100. http://europepmc.org/abstract/med/24532322
J Mun, T Yim, J H Park et al. Sci. Rep., 2014, 4: 5802. http://europepmc.org/articles/PMC4148675
J Jeong, H Lee, J Choi et al. Electrochim. Acta, 2015, 154: 149~156. http://www.sciencedirect.com/science/article/pii/S0013468614024864
B Jin, E M Jin, K H Park et al. Electrochem. Commun., 2008, 10: 1537~1540. http://www.sciencedirect.com/science/article/pii/S1388248108003469
X Xiong, Z Wang, G Yan et al. J. Power Sources, 2014, 245: 183~193. http://www.sciencedirect.com/science/article/pii/S0378775313011403
H Chen, J A Dawson, J H Harding. J. Mater. Chem. A, 2014, 2: 7988~7996. http://pubs.rsc.org/en/Content/ArticleLanding/TA/2014/C4TA00637B#!divAbstract
H Jiang, Y Fu, Y Hu et al. Small, 2014, 10: 1096~1100. doi: 10.1002/smll.201302177
S Lee, Y Cho, H K Song et al. Angew. Chem. Int. Ed., 2012, 51: 8748~8752. http://europepmc.org/abstract/MED/22865641
S Zhang. NPJ Comput. Mater., 2017, DOI: 10.1038/s41524-017-0009-z.
A Urban, D-H Seo, G Ceder. NPJ Comput. Mater., 2016, 2: 16002. http://www.nature.com/articles/npjcompumats20162
S H Chung, A Manthiram. Adv. Mater., 2014, 26: 7352~7357. http://europepmc.org/abstract/MED/25219844
B Jin, J-U Kim, H-B Gu. J. Power Sources, 2003, 117: 148~152. http://www.sciencedirect.com/science/article/pii/S0378775303001137
Y V Mikhaylik, J R Akridge. J. Electrochem. Soc., 2003, 150: A306~A311. http://jes.ecsdl.org/content/150/3/A306.short
X Ji, L F Nazar. J. Mater. Chem., 2010, 20: 9821~9826. http://pubs.rsc.org/en/content/articlelanding/2010/jm/b925751a#!divAbstract
G Zheng, Y Yang, J J Cha et al. Nano Lett., 2011, 11: 4462~4467. http://europepmc.org/abstract/MED/21916442
L Ji, M Rao, H Zheng et al. J. Am. Chem. Soc., 2011, 133: 18522~18525. http://europepmc.org/abstract/MED/22017295
Z Xiao, Z Yang, L Zhang et al. ACS Nano, 2017, 11: 8488~8498. http://europepmc.org/abstract/MED/28745863
Y V Mikhaylik, J R Akridge. J. Electrochem. Soc., 2004, 151: A1969~A1976. http://jes.ecsdl.org/content/151/11/A1969.abstract
Kolosnitsyn V, Karaseva E, Ivanov A. Russ. J. Electrochem., 2008, 44: 564~569. doi: 10.1134/S1023193508050091
L Wang, T Zhang, S Yang et al. J. Energy. Chem., 2013, 22: 72~77. http://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&filename=TRQZ201301011&dbname=CJFD2013
Y Diao, K Xie, S Xiong et al. J. Electrochem. Soc., 2012, 159: A1816~A1821. http://jes.ecsdl.org/content/159/11/A1816.abstract
S Evers, L F Nazar. Chem. Commun., 2012, 48: 1233~1235. http://pubs.rsc.org/en/content/articlelanding/2012/cc/c2cc16726c#!divAbstract
J R Akridge, Y V Mikhaylik, N White. Solid State Ionics, 2004, 175: 243~245. http://www.sciencedirect.com/science/article/pii/S0167273804005958
S Chen, X Huang, H Liu et al. Adv. Energy Mater., 2014, 4: 1079~1098. doi: 10.1002/aenm.201470040/full
G Zheng, Y Yang, J J Cha et al. Nano Lett., 2011, 11: 4462~4467. doi: 10.1021/nl2027684
Q Li, Z Zhang, K Zhang et al. J. Power Sources, 2014, 256: 137~144. http://www.sciencedirect.com/science/article/pii/S0378775314000871
S Dörfler, M Hagen, H Althues et al. Chem. Commun., 2012, 48: 4097~4099. http://pubs.rsc.org/en/content/articlehtml/2012/cc/c2cc17925c
J Schuster, G He, B Mandlmeier et al. Angew. Chem., 2012, 124: 3651~3655. doi: 10.1002/ange.201107817
X Yang, L Zhang, F Zhang et al. ACS Nano, 2014, 8: 5208~5215. doi: 10.1021/nn501284q
C Xu, Y Wu, X Zhao et al. J. Power Sources, 2015, 275: 22~25. http://www.sciencedirect.com/science/article/pii/S0378775314018333
F Liu, J Liang, C Zhang et al. Results Phys., 2017, 7: 250~255. http://www.sciencedirect.com/science/article/pii/S2211379716305770
X Ji, K T Lee, L F Nazar. Nat. Mater., 2009, 8: 500~506.
N Jayaprakash, J Shen, S S Moganty et al. Angew. Chem., 2011, 123: 6026~6030. doi: 10.1002/anie.201100637
J Guo, Y Xu, C Wang. Nano Lett., 2011, 11: 4288~4294. doi: 10.1021/nl202297p
S Xin, L Gu, N H Zhao et al. J. Am. Chem. Soc., 2012, 134: 18510~18513. doi: 10.1021/ja308170k?journalCode=jacsat
Zhang B, Qin X, Li G et al. Energ. Environ. Sci., 2010, 3: 1531~1537. http://pubs.rsc.org/en/content/articlelanding/2010/ee/c002639e#!divAbstract
D W Wang, G Zhou, F Li et al. Phys. Chem. Chem. Phys., 2012, 14: 8703~8710. http://europepmc.org/abstract/MED/22618882
Z Li, Y Jiang, L Yuan et al. ACS Nano, 2014, 8: 9295~9303. doi: 10.1021/nn503220h
Y Zhao, W Wu, J Li et al. Adv. Mater., 2014, 26: 5113~5118. doi: 10.1002/adma.201401191/pdf
D S Jung, T H Hwang, J H Lee et al. Nano Lett., 2014, 14: 4418~4425. doi: 10.1021/nl501383g
X Ye, J Ma, Y S Hu et al. J. Mater. Chem. A, 2016, 4: 775~780. http://pubs.rsc.org/-/content/articlelanding/2016/ta/c5ta08991c#!divAbstract
H J Peng, J Q Huang, M Q Zhao et al. Adv. Funct. Mater., 2014, 24: 2772~2781. doi: 10.1002/adfm.201470126/pdf
B He, W C Li, C Yang et al. ACS Nano, 2016, 10: 1633~1639. doi: 10.1021/acsnano.5b07340
Y X Yin, S Xin, Y G Guo et al. Angew. Chem. Int. Ed., 2013, 52: 13186~13200. doi: 10.1002/anie.201304762/pdf
Z Li, L Yuan, Z Yi et al. Adv. Energy Mater., 2014, 4: 1634~1642. doi: 10.1002/aenm.201301473
F Jin, S Xiao, L Lu et al. Nano Lett., 2015, 16: 440~447. doi: 10.1021/acs.nanolett.5b04105?journalCode=nalefd
K Mi, Y Jiang, J Feng et al. Adv. Funct. Mater., 2016, 26: 1571~1579. doi: 10.1002/adfm.201504835/pdf
C Zhang, H B Wu, C Yuan et al. Angew. Chem., 2012, 124: 9730~9733. doi: 10.1002/ange.201205292
Y Fu, A Manthiram. RSC Adv., 2012, 2: 5927~5929. http://pubs.rsc.org/en/content/articlepdf/2012/ra/c2ra20393f
Y Fu, Y-S Su, A Manthiram. J. Electrochem. Soc., 2012, 159: A1420~A1424. http://jes.ecsdl.org/content/159/9/A1420.abstract
C Y Chen, H J Peng, T Z Hou et al. Adv. Mater., 2017, 29(23): 1606802. doi: 10.1002/adma.201606802
L Xiao, Y Cao, J Xiao et al. Adv. Mater., 2012, 24: 1176~1181. doi: 10.1002/adma.201103392/full
F Wu, J Chen, R Chen et al. J. Phys. Chem. C, 2011, 115: 6057~6063. doi: 10.1021/jp1114724
X Liang, Z Wen, Y Liu et al. J. Power Sources, 2012, 206: 409~413. http://www.sciencedirect.com/science/article/pii/S0378775312002261
S Gope, D Dutta, A J Bhattacharyya. Chem. Select., 2016, 1: 728~735. doi: 10.1002/slct.201600185/pdf
Q Yu, Y Lu, T Peng et al. J. Phys. D: Appl. Phys., 2017, 50(11): 115002. doi: 10.1088/1361-6463/aa5ade
W Qian, Q Gao, H Zhang et al. Electrochim. Acta, 2017, 235: 32~41. http://www.sciencedirect.com/science/article/pii/S0013468617305297
S Moon, J-K Yoo, Y H Jung et al. J. Electrochem. Soc., 2017, 164: A6417~A6421. http://jes.ecsdl.org/content/164/1/A6417.full
J Liu, D G D Galpaya, L Yan et al. Energy. Environ. Sci., 2017, 10: 750~755. http://pubs.rsc.org/-/content/articlelanding/2017/ee/c6ee03033e#!divAbstract
C Wang, H Chen, W Dong et al. Chem. Commun., 2014, 50: 1202~1204. http://europepmc.org/abstract/med/24326574
J Li, B Ding, G Xu et al. Nanoscale, 2013, 5: 5743~5746. http://pubs.rsc.org/en/content/articlelanding/2013/nr/c3nr01393f#!divAbstract
W Xue, Q-B Yan, G Xu et al. Nano Energy, 2017, 38: 12~18.
X Liang, L F Nazar. ACS Nano, 2016, 10: 4192~4198. doi: 10.1021/acsnano.5b07458
Z Liang, G Zheng, W Li et al. ACS Nano, 2014, 8: 5249~5256. doi: 10.1021/nn501308m
Z Li, J Zhang, B Guan et al. Nat. Commun., 2016, 7: 13065. http://europepmc.org/articles/PMC5080434/
J Y Hwang, H M Kim, S K Lee et al. Adv. Energy Mater., 2016, 6(1): 1501480. doi: 10.1002/aenm.201501480
B Campbell, J Bell, H H Bay et al. Nanoscale, 2015, 7: 7051~7055. http://europepmc.org/abstract/med/25712745
Z W Seh, W Li, J J Cha et al. Nat. Commun., 2013, 4: 2327. http://www.nature.com/ncomms/2013/130902/ncomms3327/full/ncomms3327.html
H Cheng, S P Wang, D Tao et al. Funct. Mater. Lett., 2014, 07: 1450020. doi: 10.1142/S1793604714500209
L-C Yin, J Liang, G-M Zhou et al. Nano Energy, 2016, 25: 203~210. http://www.sciencedirect.com/science/article/pii/S2211285516301094
Q Zhang, Y Wang, Z W Seh et al. Nano Lett., 2015, 15: 3780~3786. doi: 10.1021/acs.nanolett.5b00367
T A Pascal, K H Wujcik, J Velasco-Velez et al. J. Phys. Chem. Lett., 2014, 5: 1547~1551. doi: 10.1021/j100884a020?journalCode=jpchax
G Zhou, L-C Yin, D-W Wang et al. ACS Nano, 2013, 7: 5367~5375. doi: 10.1007/978-981-10-3406-0_4.pdf
X Liu, J Q Huang, Q Zhang et al. Adv. Mater., 2017, 29(20): 1601759. doi: 10.1002/adma.201601759/pdf
Q Pang, X Liang, C Y Kwok et al. Nat. Energy, 2016, 1: 16132. http://www.nature.com/articles/nenergy2016132
F Sun, J Wang, D Long et al. J. Mater. Chem. A, 2013, 1: 13283~13289. http://pubs.rsc.org/en/content/articlelanding/2011/jm/c1jm11292a/unauth#!divAbstract
Q Li, Z Zhang, K Zhang et al. J. Solid State Electrochem., 2013, 17: 2959~2965. doi: 10.1007/s10008-013-2203-3
Y Zhang, L Wang, A Zhang et al. Solid State Ionics, 2010, 181: 835~838. http://www.sciencedirect.com/science/article/pii/S0167273810001785
Y Zhang, X Wu, H Feng et al. Int J. Hydrogen Energy, 2009, 34: 1556~1559. http://www.sciencedirect.com/science/article/pii/S0360319908016698
L Z Tan, F Zheng, S M Young et al. NPJ Comput. Mater., 2016, 2: 16026. https://www.nature.com/articles/npjcompumats201626
Y Shan, Z Zheng, J Liu et al. NPJ Comput. Mater., 2017, DOI: 10.1038/s41524-017-0008-0.
M-S Song, S-C Han, H-S Kim et al. J Electrochem. Soc., 2004, 151: A791~A795. http://jes.ecsdl.org/content/151/6/A791
L Q Lu, L J Lu, Y Wang. J. Mater. Chem. A, 2013, 1: 9173~9181. http://onlinelibrary.wiley.com/resolve/reference/XREF?id=10.1039/c3ta11255a
X Wang, Z Wang, L Chen. J. Power Sources, 2013, 242: 65~69. http://www.sciencedirect.com/science/article/pii/S0378775313008422
X Yao, N Huang, F Han et al. Adv. Energy Mater., 2017, 7(17): 1602923. doi: 10.1002/aenm.201602923
S Evers, T Yim, L F Nazar. J. Phys Chem. C, 2012, 116: 19653~19658. doi: 10.1021/jp304380j
X Tao, J Wang, Z Ying et al. Nano Lett., 2014, 14: 5288~5294. doi: 10.1021/nl502331f
M Yu, W Yuan, C Li et al. J. Mater. Chem. A, 2014, 2: 7360~7366. http://pubs.rsc.org/en/content/articlelanding/2014/ta/c4ta00234b#!divAbstract
Q Fan, W Liu, Z Weng et al. J. Am. Chem. Soc., 2015, 137: 12946~12953. doi: 10.1021/jacs.5b07071
K Han, J Shen, S Hao et al. ChemSusChem., 2014, 7: 2545~2553. doi: 10.1002/cssc.201402329
H J Peng, T Z Hou, Q Zhang et al. Adv. Mater. Interf., 2014, 1(7): 1400227. doi: 10.1002/admi.201470042
K A See, Y-S Jun, J A Gerbec et al. ACS Appl. Mater. Interf., 2014, 6: 10908~10916. doi: 10.1021/am405025n
J Lee, S-K Park, Y Piao. Electrochim. Acta, 2016, 222: 1345~1353. https://www.sciencedirect.com/science/article/pii/S001346861632446X
Y Tan, Z Zheng, S Huang et al. J. Mater. Chem. A, 2017, 5: 8360~8366. http://pubs.rsc.org/en/content/articlelanding/2017/ta/c7ta01346a#!divAbstract
K Mi, S Chen, B Xi et al. Adv. Funct. Mater., 2017, 27(1): 1604265. doi: 10.1002/adfm.201604265
G Zhou, E Paek, G S Hwang et al. Nat. Commun., 2015, 6: 7760. http://europepmc.org/articles/PMC4518288
Q Pang, J Tang, H Huang et al. Adv. Mater., 2015, 27: 6021~6028. http://med.wanfangdata.com.cn/Paper/Detail?id=PeriodicalPaper_PM26314378
S Yuan, J L Bao, L Wang et al. Adv. Energy Mater., 2016, 6(5): 1501733. doi: 10.1002/aenm.201501733/pdf
Z Wang, Y Dong, H Li et al. Nat. Commun., 2014, 5: 5002. http://europepmc.org/abstract/MED/25255431
G C Yang, S Q Shi, J H Yang et al. J. Mater. Chem. A, 2015, 3: 8865~8869. http://pubs.rsc.org/en/Content/ArticleLanding/2015/TA/C5TA00499C#!divAbstract
X Liang, C Hart, Q Pang et al. Nat. Commun., 2015, 6: 5682. http://www.nature.com/ncomms/2015/150106/ncomms6682/abs/ncomms6682.html
M S Faber, R Dziedzic, M A Lukowski et al. J. Am. Chem. Soc., 2014, 136: 10053~10061. http://europepmc.org/abstract/med/24901378
Z Yuan, H J Peng, T Z Hou et al. Nano Lett., 2016, 16: 519~527. doi: 10.1021/acs.nanolett.5b04166
E M Fernández, P G Moses, A Toftelund et al. Angew. Chem. Int. Ed., 2008, 47: 4683~4686. http://europepmc.org/abstract/MED/18484577
H G Füchtbauer, A K Tuxen, P G Moses et al. Phys. Chem. Chem. Phys., 2013, 15: 15971~15980. http://europepmc.org/abstract/med/23959329
J Park, B C Yu, J S Park et al. Adv. Energy Mater., 2017, 7(11): 1602567. doi: 10.1002/aenm.201770059

图 1 Li-S电池的工作原理示意图
Figure 1 Schematic operation proposed for the rechargeable Li-S battery










表 1 不同尺寸和类型碳结构负载硫复合阴极
Table 1. Different sizes and types of carbon structural load sulfur composite cathodes
| 正极材料 | 合成方法 | 形态 | 硫:载体质量比/硫负载率/(wt)% | 电压窗口/V (vs.Li+) |
初次放电容/(mAh/g) (容量保持%,循环次数/倍率) |
渗硫方法 | 尺寸/nm | 文献 |
| 3D超支化中空碳纳米棒封装硫(CNR-S)纳米复合阴极 | 化学气相传输/冷凝法和模板法 | 超支化中空碳纳米棒 | -/71.5 | 1.7~2.6 | 1378(84.8%,C275/0.1C); 990(94.4%,C500/1C)) | 熔体扩散 | - | [25] |
| 高度有序的中-微孔核壳碳结构(MMCS)负载硫复合阴极 | 模板法和纳米喷涂表面涂覆法 | 高度有序的中孔/微孔碳核壳结构 | 3:1/72 | 3.0~1.0 | 1037/C2(80%,C200/C2,/0.5C) | 熔体扩散 | 微孔、介孔 | [39] |
| 中空多孔CNT封装MWNT-硫(TTCN-S)复合阴极 | 模板法 | CNT包覆CNT结构 | 4:1/71 | 1.95~2.7 | 1274(72%,C50/500mAg-1); 750(72.2%,C200/2Ag-1) | 熔体扩散 | 微孔、介孔 | [40] |
| 分层多孔碳-硫(HPC-S)复合阴极 | 超声波喷雾热解 | 微孔包覆中孔和大孔分层多孔碳结构 | 1:1/46 | 1.7~2.6 | 1412(77%,C500/2.4C) | 熔体扩散 | 微孔、介孔、大孔 | [41] |
| 3D高效导电MWCNT多孔微球-硫(MS-C/S)复合阴极 | 喷雾干燥 | MWCNT缠结构建三维多孔微球 | 7:3/70 | 1.8~2.6 | 983(87.2%,C100/0.2C); 921(87.5%,C100/0.5C) | 熔体扩散 | - | [42] |
| 纳米结构石墨烯/SWNT@活化多孔碳-硫(GSH@APC-S)复合阴极 | 化学气相渗透化学气相沉积 | 多孔碳原位生长在导电石墨烯/CNT混合支架上的全碳纳米结构 | 1.5:1、5:1/50、77 | 1.5~3.0 | 50(wt)%,1010 (86.8%,C150/1C);77wt%,914(72%,C150/1C) | 熔体扩散 | 0.5~5 | [43] |
| 微孔碳纳米片组装的自支撑整体碳-硫复合阴极 | - | 微孔碳纳米片组装的自支撑整体碳 | -/70 | 1.7~2.8 | 1068(70.8%,C100/0.5C) | 电解法 | 微孔 | [44] |
| 中空碳纳米纤维-硫(HCNF-S)复合阴极 | 混合溶剂法制备 | 中空碳纳米纤维 | 1.5:1/60.8 | 1.5~3.0 | 1090(55.1%,C100/1C) | 熔体扩散 | - | [27] |
| 中空碳纳米纤维-硫复合阴极 | 模板法 | 中空碳纳米纤维 | -/75 | 1.7~2.6 | 900,(81.1% C30/C150/0.2C) | 熔体扩散 | - | [26] |
| 高度有序介孔碳-硫(CMK-3-S)复合阴极 | 模板法和纳米浇注法 | 高度有序介孔碳 | 7:3/70 | - | 1320,(83.3%,C20) | 熔体扩散 | 3~4 | [33] |
| 高孔隙率球形有序介孔碳纳米粒子-硫(S-BMC/S)复合阴极 | 模板法 | 球形有序介孔碳 | -/49.7、61.4、70.0 | 1.5~3.0 | 49.7(wt)%:1200(60.8%,C100/1C); 61.4&70.0(wt)%:1070(65.4%,C100/1C) | 熔体扩散 | 3.1~6 | [29] |
| 石墨烯层状多孔碳-硫(L-GPCS)复合阴极 | 水热法原位聚合 | 层状石墨烯多孔碳 | -/68 | 1.7~2.6 | 885.5(70%,C100/0.5) | 熔体扩散 | 1~10 | [30] |
| 空心碳胶囊/硫(C@S)复合阴极 | 模板法 | 空心碳球 | 7:3/70 | 1.7~3.1 | 1071(91%,C100/0.5C) | 熔体扩散 | 3 | [34] |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们