引用本文:
石建兵, 蔡政旭. 面向本科生教学的开环聚合内容拓展[J]. 化学通报,
2020, 83(12): 1159-1163.

Citation: Shi Jianbing, Cai Zhengxu. Extending Content of Ring-Opening Polymerization for Undergraduate Course[J]. Chemistry, 2020, 83(12): 1159-1163.
Citation: Shi Jianbing, Cai Zhengxu. Extending Content of Ring-Opening Polymerization for Undergraduate Course[J]. Chemistry, 2020, 83(12): 1159-1163.
面向本科生教学的开环聚合内容拓展
English
Extending Content of Ring-Opening Polymerization for Undergraduate Course
Abstract:
The ring-opening polymerization is an important method along the same as the step polymerization and the chain polymerization. There are many superior characteristics for the ring-opening polymerization such as no by-products during polymerizing process, 100% atom utilization, high chain propagation rate, and controllable degradation/depolymerization. Here, the research progress of the ring-opening polymerization, including the successful ring-opening of γ-butyrolactone, living ring-opening polymerization, metathesis ring-opening polymerization, etc, was summarized as a useful supplement to the chapter of "ring-opening polymerization" in the Polymer Chemistry Course for undergraduates. This paper can expand undergraduates' understanding of cutting-edge knowledge and widen the international academic horizon of outstanding undergraduates. Moreover, some additional references were listed as suggestions for further reading.
-
开环聚合反应(Ring-opening polymerization,ROP),作为本科生《高分子化学》课程的重要一章,其单体特殊、聚合机理各异。其中,对环状单体可否发生开环聚合的判断是教学中的重点内容。环状化合物能否发生开环聚合反应以及其聚合能力的大小取决于热力学和动力学两方面的因素。在热力学方面,需要考虑环状单体与其所得线性聚合物的相对稳定性问题,也就是聚合过程中的自由能的变化(ΔG),它与焓变(ΔH)和熵变(ΔS)有关;从动力学角度上考虑,不含杂原子的环烷烃中不存在易受引发剂进攻的弱键,很难发生开环聚合反应。因此,发生开环聚合反应的单体大多含有杂原子,为环状单体打开环提供了动力学上的可能性。除一些六元环化合物外,其他环状单体都是热力学上允许发生开环聚合反应的单体。随着开环聚合研究的发展,新型开环聚合催化剂以及开环聚合机理不断涌现,为开环聚合注入了新的生机。本文将主要总结开环聚合中的一些研究进展,作为高分子材料及相关专业的本科生《高分子化学》教材外的有益补充,丰富他们在开环聚合上的认知以及对前沿科学问题的了解。
1. 五元环内酯的全新认识—可以发生开环聚合
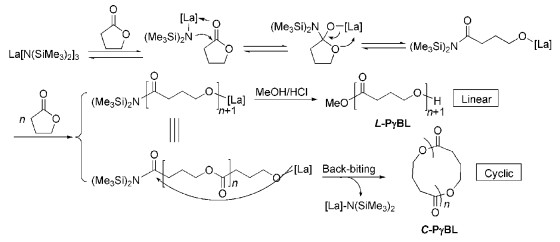
脂肪族聚酯材料具有生物相容性好、在一定条件下可降解等特性,在生物医药、可降解包装材料、生物组织工程等领域具有广泛的用途。环内酯单体,如四元环β-丁内酯(β-BL)、六元环δ-戊内酯(δ-VL)和七元环ε-己内酯(ε-CL)等存在环张力,可以发生开环聚合生成聚酯高分子。而五元环γ-丁内酯(γ-BL)存在的环张力很小,其具有很好的热力学稳定性,在《高分子化学》课本中通常被认为是不可发生聚合的环内酯单体[1]。2016年,Hong等[2]通过控制热力学和动力学条件,首次成功实现了γ-BL在常压下的开环聚合反应,高效合成了高分子量的聚(γ-丁内酯)(PγBL),如图式 1所示。该ROP能够成功实现的最主要原因是研究者找到用于该聚合反应的高效催化剂,在聚合温度为-40℃、单体起始浓度为10mol/L时,该类催化剂体系可以通过配位-插入机理高效地催化单体聚合,转化率高达90%,聚合物数均分子量(Mn)可以达到30.2kg/mol。该ROP反应既可以形成线性聚合物L-PγBL,也可以形成大环聚合物C-PγBL。更为有意思的是,无论是线性的还是环状的PγBL都可以经过加热实现定量的解聚反应,解聚时无任何副反应,单体回收率达到100%,方便实现聚合物材料的回收再利用,是真正的绿色高分子材料。考虑到金属催化剂在聚合物中的残留会影响PγBL在生物医药等领域的应用,他们又开发了非金属催化剂,如使用磷腈超强碱作为有机催化剂、苯甲醇等醇为引发剂同样实现了γ-BL的高效ROP[3]。在最优的聚合反应条件下,4h内的单体转化率高达90%,Mn可以达到25.0kg/mol,可与金属催化剂的效率相媲美。这些开拓性工作使得γ-BL及其衍生物的ROP研究如雨后春笋般涌现,具体研究工作可以进一步阅读综述性文献[4]。
图式 1
2. 活性开环聚合反应
在烯烃单体的聚合反应及功能材料制备领域,活性聚合研究是最为活跃的领域。然而,在环状单体的ROP研究领域,活性聚合的研究较少,而且仅限于少数的几个环状单体类型,并且该类单体中多数都含有烯键。
2.1 活性自由基开环聚合反应
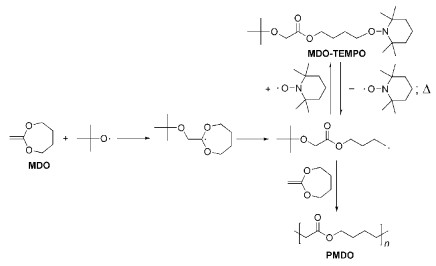
聚己内酯(PCL)可以通过ω-羟基己酸直接缩聚来合成,该方法合成路线简单、成本低;但是,缩聚过程往往需要较高的反应温度与较长的反应时间,而且所得聚合物的分子量不高。另外,通过ε-己内酯的开环聚合可以得到窄分布、高分子量聚合物,但是聚合条件苛刻,需要低温、高纯单体以及特殊催化剂等。1982年,Bailey等[5]使用2-亚甲基-1, 3-二氧环庚烷(MDO)单体实现了PCL的合成。该方法具有很多优势,如单体合成简单、聚合条件温和、可与乙烯基类单体共聚等。1996年,Wei等[6]使用2, 2, 6, 6, -四甲基-1-氧基哌啶(TEMPO)自由基捕捉剂实现了MDO的活性自由基开环聚合,如图式 2所示。TEMPO可以捕捉增长链自由基而生成MDO-TEMPO分子,其在加热条件下可以重新释放出链增长自由基和TEMPO,链增长自由基与单体MDO反应实现聚合物链增长。随后的研究表明,活性自由基聚合的方法都可以引入到MDO及其衍生物的活性自由基开环聚合中,如原子转移自由基开环聚合(ATRP-ROP)[7]、可逆加成断裂链转移自由基开环聚合(RAFT-ROP)[8]等,这些合成方法大大提升了PCL类聚合物的各种性能,扩大了其应用范围。进一步了解该领域的研究进展,可参阅综述文献[9]。
图式 2
2.2 活性开环易位聚合反应
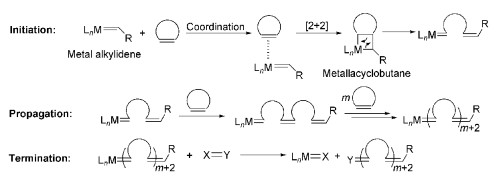
在环烯类单体中,降冰片烯(NBE)及其衍生物反应活性高,原料来源丰富,是研究环烯类开环聚合中常用的单体。早在20世纪50年代,Anderson等[10]就对NBE的聚合反应进行了研究。随后,开环易位聚合(ROMP)催化剂发展迅速,其中典型的是Schrock课题组开发的Mo系催化剂和Grubbs课题组开发的Ru系催化剂。这两个系列催化剂已经商业化,促进了这个领域的长足进步。如图式 3所示,ROMP反应包含链引发、链增长、链终止三个基元反应,是连锁配位增长机理[11]。首先,金属卡宾LnM=CHR(L为配体)是活性中心,它与环烯烃进行配位加成反应生成金属环丁烷中间体,随后该环裂解为具有引发活性的金属卡宾络合物,完成链引发过程;金属卡宾活性中心继续与单体发生类似反应,完成单体在活性中心的迅速加成,这就是链增长阶段;链增长会持续进行,直到单体消耗完毕或被终止剂强行终止。和其他聚合反应过程一样,ROMP存在的一些副反应,如分子内链转移和分子间链转移反应等,会增大最终聚合物的分子量分布指数。1989年,Grubbs课题组[12]采用Ti系催化剂实现了活性ROMP,数均分子量可以达到15900而多分散度为1.1。活性ROMP具有独特的优势,如聚合反应发生后主链上仍保留的不饱和双键有利于后功能化修饰并扩展聚合物性能[13, 14];Grubbs催化剂可在常温、常压下催化ROMP,催化活性高、反应时间短、官能团耐受性好等。因此,活性ROMP在合成功能性均聚物和共聚物领域得到了广泛的应用[15]。进一步了解该领域的发展脉络,可参阅综述文献[11]。
图式 3
2.3 活性阴离子开环聚合反应
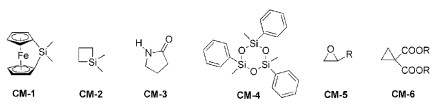
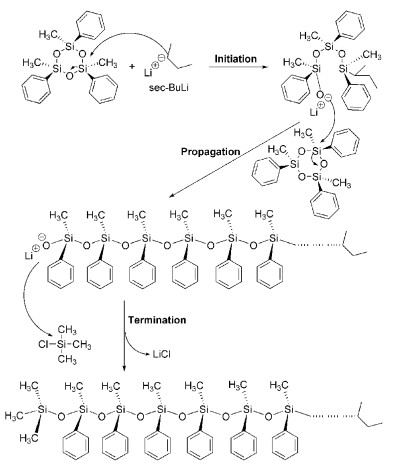
在阴离子聚合中,单体一旦经过引发剂引发形成阴离子活性种,就以相同的模式进行链增长。一般情况下,在链增长过程中不存在链转移和链终止反应,活性种的活性可以保持几天甚至几周,具有典型的活性聚合特征。因此,能发生阴离子开环聚合的环状单体众多(如图式 4所示),包括二茂铁二甲基环硅烷衍生物(CM-1)[16]、1, 1-二甲基硅杂环丁烷(CM-2)[17]、环酰内胺衍生物(CM-3)[18]、环三硅氧烷衍生物(CM-4)[19]、环氧乙烷衍生物(CM-5)[20]、环丙烷衍生物(CM-6)[21]等。活性阴离子ROP机理以CM-4为例(如图式 5所示),仲丁基锂引发剂引发单体开环形成单体活性种,之后开始链增长;阴离子聚合物增长链的终止或转移反应,需要外加终止剂/链转移剂,如三甲基氯化硅作为终止剂可以终止聚合反应。该活性阴离子聚合反应采用全顺式单体CM-4合成的聚合物以内消旋-内消旋为主,全同立构的产物比例可以达到80%以上。
图式 4
 图式 4. 能发生活性阴离子开环聚合的单体结构示例Scheme 4. The chemical structures of monomer for living anionic ROP
图式 4. 能发生活性阴离子开环聚合的单体结构示例Scheme 4. The chemical structures of monomer for living anionic ROP图式 5
2.4 活性阳离子开环聚合反应
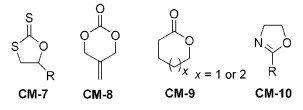
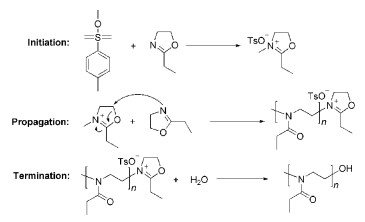
对于烯烃类单体的阳离子聚合研究很多,但是可用于聚合的单体很少。对于环状单体来说,可以发生阳离子聚合的也不多,尤其是可以发生活性阳离子聚合的单体非常有限。1998年,Endo课题组[22]报道了五元环黄原酸内酯衍生物(CM-7)(图式 6)的活性阳离子ROP。随后,他们[23]又开发了5-亚甲基-1, 3-二噁烷-2-酮(CM-8)的活性阳离子开环聚合反应,数均分子量达到21200,分子量多分散指数为1.28。2000年,Nomura等[24]使用三氟甲磺酸钪[Sc(OTf)3]作为催化剂,实现了内酯类化合物(CM-9)(己内酯ε-CL和戊内酯δ-VL)的活性阳离子ROP。2005年,Wiesbrock等[25]使用2-噁唑啉类单体(CM-10)在微波反应器中实现了活性阳离子ROP,其聚合反应机理如图7所示。在此基础上,2-噁唑啉类单体的活性阳离子开环聚合反应取得了系列进展[26~30]。
图式 6
 图式 6. 能发生活性阳离子开环聚合的单体结构示例Scheme 6. The chemical structures of monomer for living cationic ROP
图式 6. 能发生活性阳离子开环聚合的单体结构示例Scheme 6. The chemical structures of monomer for living cationic ROP图式 7

3. 结语
综上所述,环状单体的特殊性决定了开环聚合机理的多样性。活性开环聚合机理的深入研究对高性能与多功能高分子材料的制备提供理论与实践依据。由环状单体制备新型聚合物材料最有可能实现高分子材料的全链条绿色化,即绿色化生产、加工,材料使用完毕后可回收循环再利用,或无害化分解为单体再次聚合、加工,等等。因此,我们相信,随着开环聚合研究的深入与拓展,开环聚合制备的新型聚合物会占有越来越重要的地位。希望本文的总结有助于《高分子化学》课程中“开环聚合”一章内容深入学习与领会。
-
-
[1]
潘祖仁.《高分子化学》第5版.北京: 化学工业出版社, 2011: 215.
-
[2]
Hong M, Chen E Y X. Nat. Chem., 2016, 8: 42~49. doi: 10.1038/nchem.2391
-
[3]
Hong M, Chen E Y X. Angew. Chem. Int. Ed., 2016, 55: 4188~4193. doi: 10.1002/anie.201601092
-
[4]
袁鹏俊, 洪缪.高分子学报, 2019, 50(4): 327~337.
-
[5]
Bailey W J, Ni Z, Wu S R. J. Polym. Sci.: Polym. Chem. Ed., 1982, 20: 3021~3030.
-
[6]
Wei Y, Connors E J, Jia X R, et al. Chem. Mater., 1996, 8: 604~606. doi: 10.1021/cm950487o
-
[7]
袁金颖, 邹应芳, 潘才元.高等学校化学学报, 2000, 20(9): 1494~1496.
-
[8]
He T, Zou Y F, Pan C Y. Polym. J., 2002, 34: 138~143. doi: 10.1295/polymj.34.138
-
[9]
Tardy A, Nicolas J, Gigmes D, et al. Chem. Rev., 2017, 117: 1319~1406. doi: 10.1021/acs.chemrev.6b00319
-
[10]
张丹枫.高分子材料科学与工程, 2000, 16(1): 13~15.
-
[11]
Bielawski C W, Grubbs R H. Prog. Polym. Sci., 2007, 32: 1~29. doi: 10.1016/j.progpolymsci.2006.08.006
-
[12]
Risse W, Wheeler D R, Cannizzo L F, et al. Macromolecules, 1989, 22: 3205~3210. doi: 10.1021/ma00198a003
-
[13]
Hsu T W, Kim C, Michaudel Q. J. Am. Chem. Soc., 2020, 142: 11983~11987. doi: 10.1021/jacs.0c04068
-
[14]
Banwell M G, Liu X, Connal L A, et al. Macromolecules, 2020, 53: 5308~5314. doi: 10.1021/acs.macromol.0c01305
-
[15]
Varlas S, Lawrenson S B, Arkinstall L A, et al. Prog. Polym. Sci., 2020, 107: 101278. doi: 10.1016/j.progpolymsci.2020.101278
-
[16]
Rulkens R, Ni Y Z, Manners I. J. Am. Chem. Soc., 1994, 116: 12121~12122. doi: 10.1021/ja00105a090
-
[17]
Matsumoto K, Yamaoka H. Macromolecules, 1995, 28: 7029~7031. doi: 10.1021/ma00124a048
-
[18]
Hashimoto K. Prog. Polym. Sci., 2000, 25: 1411~1462. doi: 10.1016/S0079-6700(00)00018-6
-
[19]
Ahn H W, Clarson S J. Silicon, 2011, 3: 157~161. doi: 10.1007/s12633-011-9098-3
-
[20]
Wang G W, Fan X S, Huang J L. J. Polym. Sci. A, 2010, 48: 5313~5321. doi: 10.1002/pola.24331
-
[21]
Penelle J, Xie T. Macromolecules, 2000, 33: 4667~4672. doi: 10.1021/ma991173f
-
[22]
Choi W, Sanda F, Endo T. Macromolecules, 1998, 31: 9093~9095. doi: 10.1021/ma9805102
-
[23]
Sanda F, Fueki T, Endo T. Macromolecules, 1999, 32: 4220~4224. doi: 10.1021/ma981947c
-
[24]
Nomura N, Taira A, Tomioka T, et al. Macromolecules, 2000, 33: 1497~1499. doi: 10.1021/ma991580r
-
[25]
Wiesbrock F, Hoogenboom R, Leenen M A M, et al. Macromolecules, 2005, 38: 5025~5034. doi: 10.1021/ma0474170
-
[26]
Guerrero-Sanchez C, Hoogenboom R, Schubert U S. Chem. Commun., 2006, 3797~3799.
-
[27]
Hu F Y, Xie S L, Jiang L M, et al. RSC Adv., 2014, 4: 59917~59926. doi: 10.1039/C4RA11404C
-
[28]
Bouten P J M, Hertsen D, Vergaelen M, et al. Polym. Chem., 2015, 6: 514~518. doi: 10.1039/C4PY01373E
-
[29]
Petit C, Grassl B, Mignard E, et al. Polym. Chem., 2017, 8: 5910~5917. doi: 10.1039/C7PY01255A
-
[30]
Van Den Broeck E, Verbraeken B, Dedecker K, et al. Macromolecules, 2020, 53: 3832~3846. doi: 10.1021/acs.macromol.0c00865
-
[1]
-
图式 4 能发生活性阴离子开环聚合的单体结构示例
Scheme 4 The chemical structures of monomer for living anionic ROP
图式 6 能发生活性阳离子开环聚合的单体结构示例
Scheme 6 The chemical structures of monomer for living cationic ROP
-

 下载:
下载:

 下载:
下载:






 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 44
- 文章访问数: 3204
- HTML全文浏览量: 1135
