氰化物是电镀行业中常用的金属络合剂,对镀层性能的提升和稳定镀液起到显著作用,然而其毒性对环境和人体健康都是一个巨大的威胁[1 。此外,氰化物容易渗透到光刻胶中,导致光刻胶溶解或开裂[2 ,因此无论从环保还是进一步优化工艺的角度出发,开发新型的无氰电镀液都是十分重要的。乙内酰脲是一种低成本的有机杂环化合物,在宽温度范围的水溶液中具有良好的溶解度和稳定性[3 。其分子本身及衍生物近年来受到广泛关注,作为无氰络合剂表现出对许多金属良好的配位能力,在众多衍生物中有关5, 5-二甲基乙内酰脲(DMH)的报道较多[4 。
Ohtani等[5 对比了以DMH和5, 5-二甲基噁唑烷-2, 4-二酮(DMOD)为络合剂的两种镀金液的极化曲线,两种络合剂的添加均能提高阴极极化,DMH的沉积电位更负。Luo等[6 对比了pH为8、9和10的DMH体系电镀金液,发现无论使用Au+ 或是Au3+ 为金盐均能沉积得到均匀的镀层,其中pH=9时的镀层质量更好。杨潇薇等[7 对比了1, 3-二羟甲基-5, 5-二甲基乙内酰脲(DMDMH)和DMH体系镀金液的循环伏安曲线,发现金的沉积在DMDMH体系中更为困难,并且镀液可连续施镀两周。Huang等[8 配制了一种无氰Au-Sn镀液,室温下可密封保存至少三个月,所用Au3+ 的络合剂为DMH。
理论计算是一种可从分子水平理解络合剂行为的有效工具,在DMH的配位研究中得到广泛应用。Liu等[9 11 将DMH和烟酸作为复配络合剂应用于无氰镀银液,电沉积的镀层在颗粒大小和致密程度方面与氰化物体系的镀层接近,DMH体系镀银液在12个月的贮存过程中没有观察到沉淀或变色,说明其稳定性良好;此外,量子化学计算表明DMH与Ag+ 的配位能力比烟酸更强,并考察了DMH-Ag-DMH的配位结构。Ren等[12 报道了以DMH为络合剂的Au(III)体系无氰电镀金液,在pH=10条件下沉积了致密的金黄色镀金层,并通过量子化学计算和分子动力学模拟对乙内酰脲及其若干衍生物的最高占据分子轨道(HOMO)和最低非占据分子轨道(LUMO)以及吸附能进行分析,证明了DMH较强的给电子能力和较高的吸附能,然而其他衍生物也表现出较好的配位能力,却缺乏进一步的讨论。后来他们[13 通过阴极极化曲线发现,0.3mol/L的DMH可降低0.5V的还原电位,表明DMH与Au3+ 可较为稳定地配位,并通过键能计算确定了Au+ 和Au3+ 分别与DMH配位的最稳定配合物结构,但是在讨论的所有配位结构中却忽略了O原子的配位可能性。在这些研究中,对乙内酰脲及其衍生物的理论计算仅停留在分子整体性质的研究,然而当配位反应发生时需涉及到原子与中心原子(或离子)之间的成键,因此对分子中可能参与配位的原子进行分析有利于进一步理解实验结果。此外,以DMH为络合剂的镀液需在碱性条件操作才能保持配合物稳定,避免在电镀时发生置换反应[13 ,对优选这种酸碱性的原因却未曾有报道从原理上分析。
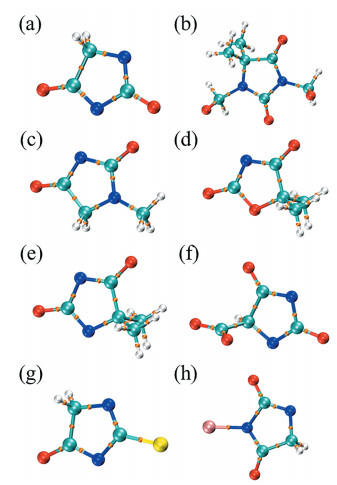
在本项研究中,以密度泛函理论(DFT)方法对乙内酰脲衍生物的反应活性进行了较为全面的探讨,并考虑了溶液酸碱性的影响,更为准确地模拟分子在镀液中的存在形式。所选择分子既包括前人实验中报道过的乙内酰脲、DMDMH、1-甲基乙内酰脲(MH)、DMOD和DMH,也包括未曾报道的羧基、硫原子和溴原子取代后的其他乙内酰脲衍生物,如乙内酰脲-5-乙酸(ACH)、2-硫代乙内酰脲(TH)和3-溴乙内酰脲(BH)。通过对溶解性、原子电荷、表面静电势(ESP)、原子对HOMO的贡献程度、平均局部离子化能(ALIE)、简缩局部软度、杂原子上H原子离去能力和分子稳定性的全面分析,重点考察各杂原子的配位活性。本工作既从多种计算参数结合较为可靠地解释了部分实验现象,也预测了一些衍生物的性质,为发展新的应用于无氰镀液的乙内酰脲衍生物提供了参考,同时全面的分析方法也为其他体系络合剂的理论计算提供借鉴。
1.
理论计算
所有乙内酰脲衍生物分子的结构优化均在Gaussian 09程序[14 进行。计算分子极性指数(MPI)和极性表面积所占总面积百分比(pApolar )在B3LYP/6-311G **[15 水平进行;原子电荷、ESP、原子对HOMO的贡献程度、ALIE和简缩局部软度的计算采用混合基组,通过杂原子上H原子的去质子化模拟碱性环境,由于H原子离去后的杂原子表现为带明显的负电荷,因此对这些原子加上弥散函数,在B3LYP/6-311+G **[15 水平上计算;其他原子在B3LYP/6-311G **水平上计算,以IEFPCM溶剂模型中的水为溶剂。运用Multiwfn程序[16 对Gaussian 09程序的计算结果进行处理,得到本文所有最终计算结果。本文所有分子结构图均由VMD程序[17 绘制。
MPI源于18碳环的极性预测的报道[18 ,其定义如下,式中V 是分子静电势,A 是分子表面积。
极性表面积和非极性表面积以静电势绝对值10kcal/mol为分界线,大于该值为极性部分,小于该值为非极性部分。
原子电荷没有唯一的定义,因此计算方法多种多样。原子偶极矩校正的Hirshfeld布居(ADCH)电荷在电荷的合理性、基组依赖性、偶极矩重现性和静电势的重现性上都表现较好,电荷计算速度也很快[19 21 ,因此本文选择ADCH电荷进行探讨。
静电势定义为r
由式可知,静电势是由原子核和电子共同作用的结果,Z A 是原子A的核电荷,R ρ 是电子密度。范德华表面上ESP分布图是以电子密度为0.001a.u.的等值面作为分子表面,格点间距为0.25Bohr。利用定量分子表面分析算法获得分子表面静电势的极值点位置和数值[22 。
有多种计算分子轨道中原子的成分的方法,本文采用的是基于自然原子轨道概念(NAOMO)的方法,其具有计算速度快、结果合理、基组依赖性很小等优点[23 。虽然该方法不适合分析LUMO轨道,但是本文计算的是HOMO轨道,因此不影响计算结果的准确性[24 。
ALIE是Sjoberg等[25 在1990年提出的,它可以被理解为空间中任何一点的电子的电离能。它的定义如下:
其中,ρ 代表总密度,ρ i i 个占据的分子轨道所对应的电子密度,ε i i 个分子轨道的能量。范德华表面上ALIE分布图中以电子密度为0.005a.u.的等值面作为分子表面。利用定量分子表面分析算法获得分子表面ALIE的极值点位置和数值[22 。
由Parr和Yang提出的Fukui函数是DFT中的一个重要概念,已被广泛用于预测反应位点[26 28 。简缩Fukui函数仅能对比分子内各原子的反应活性,涉及到分子间原子反应活性的对比,需对简缩Fukui函数进一步计算,如下式所示[29 30 。
s A - 称为原子A的简缩局部软度,S 是垂直电离能与垂直电子亲和能之差的倒数[31 ,f A - 为原子A的简缩Fukui函数。Hirshfeld电荷十分适合于简缩Fukui函数的计算[32 ,因此该部分使用的原子电荷为Hirshfeld电荷。
杂原子上H的离去难易程度是通过考察各杂原子与H原子的解离常数(pK a)来比较的。pK a的计算公式如下式所示[33 。H原子离去后的杂原子计算水平为B3LYP/6-311+G **,其他原子计算水平为B3LYP/6-311G **。
通过基于“分子中的原子”理论(AIM)的拓扑分析研究分子的环张力[34 35 。通过分子动力学模拟研究水溶液中分子的热力学稳定性,计算水平为B3LYP/6-311G **,时间步长0.5fs,总模拟时间1.27ps,模拟温度373K[36 。
2.
计算结果与分析讨论
2.1
溶解性
大多数镀液是以水为溶剂,为了保证有足够数量的络合剂分子与金属离子络合以及考虑商用浓缩镀液的配制,要求络合剂在水中应当有较好的溶解性。水是极性分子,根据“相似相溶”的原理,若络合剂分子极性越大,一般来讲在水中溶解度也越高。MPI和pApolar 均能反映分子的极性,这两个参数数值越大,则分子极性越大。MPI和pApolar 的结果如表 1
表 1
在所考察的分子中,乙内酰脲的MPI和pApolar 都较大,表现为明显的极性,因此具有较好的溶解性。当乙内酰脲部分氢原子被甲基或羟甲基取代后生成MH、DMH和DMDMH,MPI和pApolar 都有不同程度的降低,并随着非极性部分数目的依次增加,降低的程度依次增大,但是仍然表现为较高的极性,与Yu等[37 的实验结果一致。ACH的MPI和pApolar 与乙内酰脲相比仅略有降低,这是由羧基与水分子形成强的氢键所致。TH的极性与乙内酰脲相比略有下降,可能是由于S原子与O原子相比不易与水形成氢键。结合分子极性的预测可知,从总体上看,乙内酰脲及所考察的衍生物的极性较大,可认为在水中具有较好的溶解性,具有进一步研究的意义。由于乙内酰脲本体溶解性较好,因此尽管有一些非极性基团进行取代也仍满足镀液对络合剂浓度的要求,0.26mol/L DMDMH[7 、0.6mol/L DMH[13 、0.24mol/L MH和0.24mol/L DMOD[5 组成的镀液已有报道。这些使用浓度不代表溶解度,但可知溶解性都比较好,反映出极性较高,与计算结果基本相符。值得注意的是,大体积的非极性基团的引入可能造成乙内酰脲本体在分子中所占比例显著降低,造成溶解性明显下降,这类衍生物不适合作为水体系镀液中金属离子的配体。
2.2
静电相互作用
根据软硬酸碱理论[38 39 ,属于软酸的金属离子倾向于与软碱配体形成共价键,因此通过配体的给电子能力考察配位能力至关重要。静电作用通常是硬酸与硬碱的结合方式,但也影响着配位共价键的成键过程,对原子的配位活性的比较起到补充作用。静电相互作用将探讨金属离子向络合剂的哪个原子靠近,该原子未必是配位原子,但配位原子一定在该原子附近,只有当金属离子与配位能力较强的原子距离较近时才有可能进一步形成配位共价键。
2.2.1
原子电荷
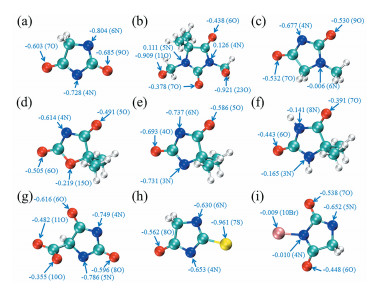
原子电荷是对化学体系中电荷分布最简单、最直观的描述方式之一[19 。电荷越负越可能吸引带正电的亲电试剂进攻而发生反应。图 1 3 杂化轨道的电子已与C原子形成共价键,因此原子电荷相比于其他杂原子较正,但不是很明显的正值,可能与N原子的一个sp3 杂化轨道仍存在一对孤对电子有关。乙内酰脲的两个N原子的原子电荷比O原子更负,表明N原子更容易吸引金属离子接近。DMDMH的羟甲基的H原子解离后,O原子具有极明显负值的原子电荷,对金属离子的吸引能力较强。MH、DMOD和DMH的N原子的原子电荷都比各自分子内的O原子更负,其中DMH的两个N原子的原子电荷为-0.731和-0.737,比MH和DMOD的更负。对比DMH的H原子解离后的电荷可知,解离后的N和O原子都比解离前的更正,N原子的变化更为显著,解离前只有-0.165和-0.141,极大地降低了吸引金属离子的能力,表明碱性条件对DMH吸引金属离子接近进而发生配位反应极其重要。在ACH分子中,N原子和羧基O原子的H原子均解离后相比,N原子表现为更负的带电性,原子电荷为-0.749和-0.786,而O原子只有-0.485。TH的S原子的原子电荷为-0.961,在所有分子的杂原子中是电荷最负的,因此表现为对金属离子最强的静电相互作用。在BH分子中,构成N-Br键的两个原子电荷均接近0,该分子中5N的原子电荷最负,为-0.6526,在所考察的分子中对金属离子的吸引能力一般。
图 1
2.2.2
静电势
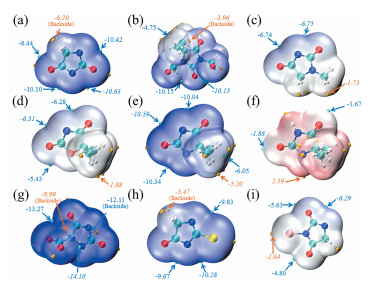
配体与金属离子的相互吸引是依靠静电相互作用进行的,静电势分析是研究这种弱相互作用的有效手段。空间中某一点的静电势为负,说明此处静电势由电子主导,通常认为距离分子表面静电势最低的点最近的原子是亲电反应位点。图 2 图 2
图 2
乙内酰脲9O原子附近两个极小值点均较负,分别为-10.63和-10.42 eV,因此金属离子较容易往这两个位点靠近;然而这两个极小值点附近也有N原子存在,当靠近至一定程度,是通过与N原子配位或是与O原子配位,还需要进一步分析讨论。DMDMH的两个羟甲基显现出较负的静电势,极小值均为-10.15eV,环状结构附近的表面颜色较淡,ESP较正。MH、DMOD和BH基本为白色,静电相互作用不如其他衍生物强,较小的极小值大约为-6.5eV左右,均出现在已解离的N原子附近。DMH存在较负的3个极小值点,最负的两个(-10.59和-10.34eV)分布在4O原子两边,值得注意的是,该位置也是与N原子的作用交界处,因此尚不能得出O原子优先配位的结论。可观察到处于酸性或中性条件的DMH与其他分子不同的颜色,淡红色的着色表示该分子的表面出现不同程度的正值ESP,但是并不是明显的阳离子特性,最大的极大值为2.29eV,该值较低。两个数值接近的极小值点出现在C=O键所在直线上,因此在非碱溶液中,金属离子将向O原子靠近,同时如果O原子的配位能力良好,可能优于N原子配位。ACH是所有衍生物中ESP最负的,这从其深蓝色的表面着色就可直观地观察出,较小的极小值点主要出现在两个N原子附近,数值为-14.10eV的极小值点十分接近5N,羧基处的极值点主要是极大值点。与乙内酰脲类似,TH的4N和6N与7S交界处也出现极小值点,分别为-10.28和-9.83 eV,可初步判断这片区域与金属离子的静电相互作用更显著,然而单从ESP难以判断配位原子。
2.3
原子给电子能力
2.3.1
HOMO原子成分
前线分子轨道理论[40 认为,在众多分子轨道中,HOMO和LUMO与分子的反应活性密切相关。HOMO与分子给电子能力有关,HOMO能量越高,分子给电子能力越强;LUMO与分子得电子能力有关,LUMO能量越低,分子得电子能量越强。对于配位反应,研究配体的给电子能力尤为重要,根据HOMO中原子的成分可以判断配位位点,虽然难以比较不同分子之间的原子的给电子能力,但是对于判断分子内的反应位点尤为重要。
表 2 [23 。对比每个分子内的N原子和O原子对HOMO的贡献可知,除DMDMH和DMOD外,N原子的贡献普遍更大,表明多数情况下N原子的给电子能力强于O原子。乙内酰脲的6N占据了58.57%,4N只有10.49%,表明当乙内酰脲发生配位时,6N将优先于4N反应。DMDMH的两个羟甲基O原子电荷相近,但是对HOMO的贡献差异明显,11O贡献了80.89%,而23O由于低于阈值0.5%未显示在表中。MH的所有原子没有超过50%的,O原子和两个N原子较为均等地对HOMO产生贡献,总体上6N的贡献更高。DMH的3N给电子能力最强,贡献了59.51%,未解离的DMH发生配位时也是该N原子优先配位,其他原子的贡献程度也变化不大,表明溶液的酸碱性对DMH的HOMO成分影响较小。当DMH的3N被O原子取代,即DMOD,可能的配位原子变为O原子,具体地讲是DMOD的5O,贡献了55.31%。ACH的5N贡献了55.38%,是该分子的可能配位原子。TH的7S和6N分别贡献42.46%和39.73%,这两个原子的给电子能力接近,都可能是配位位点,但是S原子可能性稍高一些。对于BH,已解离的N原子贡献59.87%,给电子能力显著强于Br原子和与其成键的N原子。
表 2
2.3.2
平均局部离子化能
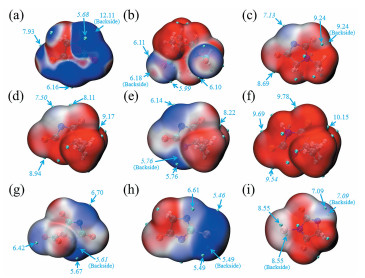
ALIE可以解释为在分子空间的任何特定点电离一个电子所需的平均能量。因此,具有最低值ALIE的位置就是电子被束缚得最弱,即活性最高的位置,最容易发生亲电反应[25 。配位反应是由配体提供孤对电子到金属离子的空轨道而形成的,因此要求配体的反应位点应当具有活泼的电子;周围具有活泼电子的原子,原理上也可反过来认为是配位反应最可能发生的位置。ALIE值越低代表电子活性越高,越容易发生配位,因此本文重点关注这些区域。
图 3
图 3
由图 3 图 3
2.3.3
简缩局部软度
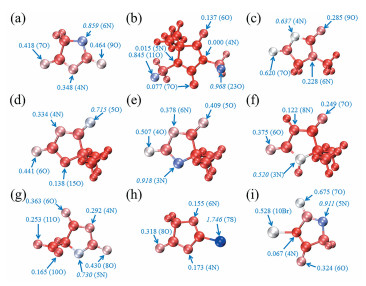
根据软硬酸碱理论[38 39 ,配体原子的软度越大,越易与软酸类型的金属离子成键。图 4
图 4
乙内酰脲的6N软度在该分子中为最高(0.859),其他N和O原子的软度明显比6N的小,大约只有其数值的1/2,表明当乙内酰脲与软酸配位时,6N是最可能的配位原子,与ALIE分布图得出的结果一致。DMDMH的简缩局部软度也表明羟甲基中的O原子比环上的杂原子更软,更可能是配位位点,两个O原子都有较高的软度,分别为0.845和0.968。MH的4N和7O软度接近,分别为0.637和0.620,从软度的角度考虑,这两个原子具有相似的配位能力,但是ALIE表明电子在4N的活性略高于7O,因此综合这两个参数可知MH的配位原子为4N。DMOD的N原子的软度与其他分子相比是最小的,5O原子虽然软度最大,是DMOD最可能的配位原子,但是其软度数值不高,仅为0.715,该处附近的ALIE也较大,总体上DMOD的配位能力不强。DMH与乙内酰脲相比,其原子的软度有一定提高,最软的原子位于3N,数值为0.918,因此对软酸也有较强的配位能力。当DMH处于酸性或中性时,原子软度有了明显下降,表明溶液的酸碱性对简缩局部软度也有影响,同时也表明DMH在碱性条件配位能力更强。ACH的羧基上的两个O原子与5N相比软度小得多,表明羧基不太可能参与配位,5N的软度为0.730,比乙内酰脲的6N略小。TH的S原子具有极高的软度,为1.746,不仅在本身的分子中显著高于其他杂原子,与其他所有分子的原子相比也具有最高的软度,这表明得益于S原子的存在,TH具有优异的配位能力。有报道指出含S原子的络合剂在置换镀金液稳定性和镀层性能表现良好[41 。当Br作为取代基时,虽然Br原子配位能力不如N原子,但对N原子的软度有一定提高效果,使得5N的软度能够达到0.911。
综合考虑2.2和2.3两部分研究内容,将乙内酰脲及其衍生物的配位能力以打分的形式汇总于表 3 [5 7 的实验结果。此外,该部分的计算结果也表明DMH的优先配位原子为3N,与Liu等[9 和Ren等[12 通过键能计算DMH-Ag-DMH和DMH-Au-DMH的配位结构的结果一致。
表 3
2.4
H原子离去能力
表 4 K a 值越小,表示H原子更易解离。ACH羧基的pK a为3.10,比其他pK a都大,因此该H原子最容易解离,但该处O原子配位能力不如N原子。乙内酰脲及其衍生物含有两个N原子,同两个碳基相邻的N原子的pK a比与一个碳基相邻的N原子的小,H原子更易离去,原因在于两个碳基的吸电子效应更强,增大N-H键的极性,使其更易断开。N原子的pK a还随着分子取代基团的变化而发生改变。DMOD、ACH、TH和BH由于分子中增加了具有吸电子效应的O、S或Br原子,因此相比于乙内酰脲,对应N原子的pK a均减小,表明H原子离去能力提高。而MH和DMH相比乙内酰脲增加了甲基,既无诱导效应也无共轭效应使得H原子离去后的结构更为稳定,因此pK a基本不变。以DMH为络合剂的镀液常在pH=8~10的条件下反应,很可能该pH范围内H原子已基本离去,DMH配位能力较好,因此可以预测,其他衍生物无需在强碱条件下即可达到去质子化状态,从而减小对光刻胶的破坏程度。
表 4
2.5
分子稳定性
2.5.1
结构稳定性
作为配体的乙内酰脲衍生物,其本身的结构应当是稳定的,否则将影响配合物的性质进而导致电镀液的组成发生改变,电沉积的镀层质量也随之改变。乙内酰脲衍生物的结构特征为环状结构,环张力将对结构稳定性有较大影响,当环张力较大,分子能量较高,分子便不稳定。以AIM拓扑分析判断环的张力的计算结果见图 5 图 5
图 5
2.5.2
热稳定性
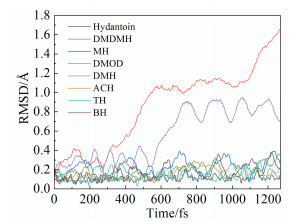
电镀液通常在加热条件下操作,因此配体在施镀温度下也应当是稳定的。电镀液以水为溶剂,施镀温度不超过373K,图 6
图 6
3.
结语
本文通过DFT对乙内酰脲及其衍生物的溶解性、配位能力、去质子化难易程度和分子稳定性进行计算,具体结论如下:
(1) 所考察的分子的MPI在13~20 kcal·mol-1 范围之间,pApolar 均大于65%,表明这些分子较易溶于水,与曾报道的实验结果基本一致。
(2) TH的S原子的原子电荷为-0.961,在所有分子的杂原子中是电荷最负的,ESP值整体上仅正于ACH,最小的极小值位于S原子附近,为-10.28eV,表现为对金属离子较强的静电相互作用。TH的S原子对HOMO贡献了42.46%,其附近的ALIE极小值为5.46和5.49 eV,比其他分子的极小值点更小,而S原子的软度为1.746,又是所有分子中最软的原子,因此TH的S原子具有很强的配位能力,其作为氰化物替代品的可能性有待进一步研究。
(3) 综合考虑静电相互作用和原子给电子能力,DMDMH、DMH、DMOD和MH的配位能力依次减弱,从微观层面解释了前人的实验结果。
(4) DMH去质子化后的两个N原子的原子电荷更负,ESP分子表面着色由红色变为蓝色,表明H原子解离后N原子对金属离子的静电吸引力显著提高。去质子化后ALIE数值变小,软度变大,因此配位能力提高,表明碱性条件的去质子化状态有利于DMH的配位。
(5) 所考察的分子各杂原子的pK a与DMH接近或减小,可能各衍生物与DMH类似,酸碱性为弱碱性即可达到去质子化。
(6) 各分子的环张力较小,环结构较稳定。DMDMH的热力学稳定性较差,不适合应用于操作温度过高的电镀液,而其他衍生物的热力学稳定性较好,在373K下结构未发生明显变化。

 下载:
下载:

 下载:
下载:







