Login In
Login In
DFT Study on the Structure and Properties of Hydantoin and Its Derivatives in the Alkaline Aqueous Solution
- Corresponding author: Zhifeng Hao, haozf@gdut.edu.cn Chengqiang Cui, cqcui@gdut.edu.cn
Figures(6)
Citation:
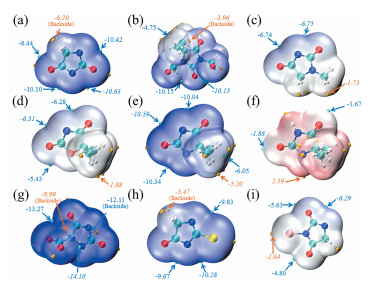
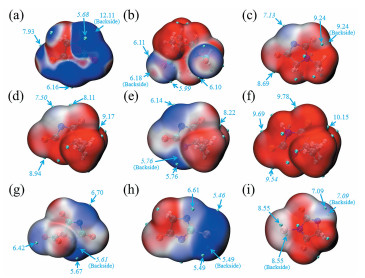
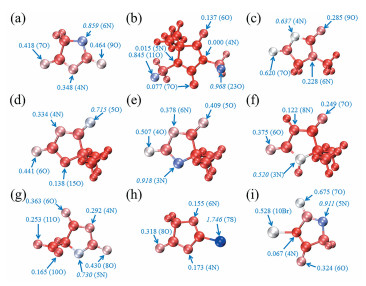
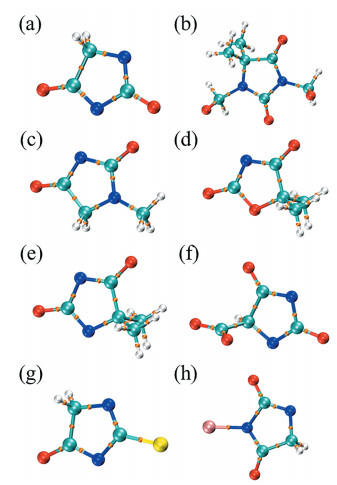
Bo Wu, Jingmeng Huang, Guizhen Tan, Zhifeng Hao, Guanghui Hu, Jiye Luo, Chengqiang Cui. DFT Study on the Structure and Properties of Hydantoin and Its Derivatives in the Alkaline Aqueous Solution[J]. Chemistry,
;2021, 84(6): 610-619.

Figures(6)





Honma H, Hagiwara K. J. Electrochem. Soc., 1995, 142(1): 81.
Meusel M, Gütschow M. Org. Prep. Proced. Int., 2004, 36(5): 391~443.
Ohtani Y, Sugawara K, Nemoto K, et al. Hyomen gijutsu., 2004, 55(12): 933~936.
Huang F F, Huang M L. J. Electrochem. Soc., 2018, 165(3): D152.
Liu A M, Ren X F, An M Z, et al. Sci. Rep., 2014, 4(1): 1~10.
Liu A M, Ren X F, Wang B, et al. RSC Adv., 2014, 4(77): 40930~40940.
Liu A M, Ren X F, Zhang J, et al. RSC Adv., 2016, 6(9): 7348~7355.
Ren X F, Song Y, Liu A M, et al. RSC Adv., 2015, 5(80): 64997~65004.
Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 09, Revision A. 02, Gaussian, Inc. Wallingford CT, 2009.
Humphrey W, Dalke A, Schulten K. J. Mol. Graphics., 1996, 14(1): 33~38.
Lu T, Chen F W. J. Theor. Comput. Chem., 2012, 11(1): 163~183.
Sjoberg P, Murray J S, Brinck T, et al. Can. J. Chem., 1990, 68(8): 1440~1443.
Parr R G, Yang W. J. Am. Chem. Soc., 1984, 106(14): 4049~4050.
Yang W, Mortier W J. J. Am. Chem. Soc., 1986, 108(19): 5708~5711.
Parr R G, Pearson R G. J. Am. Chem. Soc., 1983, 105(26): 7512~7516.
Wang B, Rong C, Chattaraj P K, et al. Theor. Chem. Acc., 2019, 138(12): 124.
Alongi K S, Shields G C. Annual reports in computational chemistry, Amsterdam: Elsevier, 2010: 113~138.
Kassaee M Z, Musavi S M, Akhavan A, et al. Struct. Chem., 2009, 20(5): 839~846.
Yu C Y, Chen M Y, Lin B, et al. J. Chem. Eng. Data, 2020, 65(2): 814~820.
Fukui K. Orientation and Stereoselection. Springer, 1970: 1~85.
Wu B, Huang J M, Lv Z M, et al. RSC Adv., 2020, 10(17): 9768~9776.
Hao XU , Ruopeng LI , Peixia YANG , Anmin LIU , Jie BAI . Regulation mechanism of halogen axial coordination atoms on the oxygen reduction activity of Fe-N4 site: A density functional theory study. Chinese Journal of Inorganic Chemistry, 2025, 41(4): 695-701. doi: 10.11862/CJIC.20240302
Chenxu Gong , Weizhen Wang , Ruiying Zhang , Wenfeng Wang , Yuanming Li , Yaofeng Yuan , Keyin Ye . Computational Chemistry-Assisted Organic Structure Analysis (CCAOSA): A Case Study of Propeller-Shaped Hexabenzotriphenylene. University Chemistry, 2026, 41(4): 438-446. doi: 10.12461/PKU.DXHX202503076
Kaifu Zhang , Shan Gao , Bin Yang . Application of Theoretical Calculation with Fun Practice in Raman Spectroscopy Experimental Teaching. University Chemistry, 2025, 40(3): 62-67. doi: 10.12461/PKU.DXHX202404045
Jie ZHAO , Sen LIU , Qikang YIN , Xiaoqing LU , Zhaojie WANG . Theoretical calculation of selective adsorption and separation of CO2 by alkali metal modified naphthalene/naphthalenediyne. Chinese Journal of Inorganic Chemistry, 2024, 40(3): 515-522. doi: 10.11862/CJIC.20230385
Jie ZHAO , Huili ZHANG , Xiaoqing LU , Zhaojie WANG . Theoretical calculations of CO2 capture and separation by functional groups modified 2D covalent organic framework. Chinese Journal of Inorganic Chemistry, 2025, 41(2): 275-283. doi: 10.11862/CJIC.20240213
Weina Wang , Lixia Feng , Fengyi Liu , Wenliang Wang . Computational Chemistry Experiments in Facilitating the Study of Organic Reaction Mechanism: A Case Study of Electrophilic Addition of HCl to Asymmetric Alkenes. University Chemistry, 2025, 40(3): 206-214. doi: 10.12461/PKU.DXHX202407022
Tongqi Ye , Yanqing Wang , Qi Wang , Huaiping Cong , Xianghua Kong , Yuewen Ye . Reform of Classical Thermodynamics Curriculum from the Perspective of Computational Chemistry. University Chemistry, 2025, 40(7): 387-392. doi: 10.12461/PKU.DXHX202409128
Wei Sun , Yongjing Wang , Kun Xiang , Saishuai Bai , Haitao Wang , Jing Zou , Arramel , Jizhou Jiang . CoP Decorated on Ti3C2Tx MXene Nanocomposites as Robust Electrocatalyst for Hydrogen Evolution Reaction. Acta Physico-Chimica Sinica, 2024, 40(8): 2308015-0. doi: 10.3866/PKU.WHXB202308015
Xiaochen Zhang , Fei Yu , Jie Ma . Cutting-Edge Applications of Multi-Angle Numerical Simulations for Capacitive Deionization. Acta Physico-Chimica Sinica, 2024, 40(11): 2311026-0. doi: 10.3866/PKU.WHXB202311026
Xinwan Zhao , Yue Cao , Minjun Lei , Zhiliang Jin , Tsubaki Noritatsu . Constructing S-scheme heterojunctions by integrating covalent organic frameworks with transition metal sulfides for efficient noble-metal-free photocatalytic hydrogen evolution. Acta Physico-Chimica Sinica, 2025, 41(12): 100152-0. doi: 10.1016/j.actphy.2025.100152
Yuai Duan , Xuanyu Gan , Yao Fu , Yingjie Cao , Hongliang Han , Zhanfang Ma . Application and Innovative Design of Digital Technology in the Preparation Experiment of Cis(Trans)-Diglycine Copper Complexes. University Chemistry, 2026, 41(1): 373-381. doi: 10.12461/PKU.DXHX202504048
Shuangshuang Mao , Juhua Luo , Bingjie Han , Jiahuan Shi , Yujia Gu . Covalent organic framework-derived Fe3C/NC/TiO2 heterostructures for high-performance electromagnetic wave absorption. Acta Physico-Chimica Sinica, 2026, 42(7): 100290-. doi: 10.1016/j.actphy.2026.100290
Hao Wu , Fengqi Li , Xinwei Shi , Haifeng Bian , Qing Zhou , Shunshun Jia , Yujie Ma , Jian Gu , Jingzi Zhang , Shuijian He , Xiangkang Meng . Machine-learning guides discovery of multi-principal element alloys as electrocatalyst for hydrogen evolution reaction. Acta Physico-Chimica Sinica, 2026, 42(8): 100227-0. doi: 10.1016/j.actphy.2025.100227
Weiheng Liu , Juhua Luo , Jiahuan Shi , Di Lan , Shuangshuang Mao , Yu Xie . Honeycomb-like BiCo@NC composites derived from bimetallic organic frameworks for high-efficiency electromagnetic wave absorption. Acta Physico-Chimica Sinica, 2026, 42(8): 100313-0. doi: 10.1016/j.actphy.2026.100313
Yang Wang , Shuangliang Liu , Jianbo Zhao . Exploring the mechanism of Diels-Alder reaction: a computational chemistry experiment for undergraduate students. University Chemistry, 2026, 41(7): 257-265. doi: 10.12461/PKU.DXHX202504061
Hongfei Yin , Mengling Hong , Jinyang Zhang , Wentao Wang , Wei Chen , Guozhi Wu . 氧空位介导的2D/2D Bi2MoO6/Bi2O2S S型异质结用于高效CO2光还原. Acta Physico-Chimica Sinica, 2026, 42(9): 100332-. doi: 10.1016/j.actphy.2026.100332
Meifeng Zhu , Jin Cheng , Kai Huang , Cheng Lian , Shouhong Xu , Honglai Liu . Classical Density Functional Theory for Understanding Electrochemical Interface. University Chemistry, 2025, 40(3): 148-152. doi: 10.12461/PKU.DXHX202405166
Yupeng TANG , Haiying YANG , Fan JIN , Nan LI . Hydrogen storage properties of C6S6Li6: A density functional theory study. Chinese Journal of Inorganic Chemistry, 2025, 41(9): 1827-1839. doi: 10.11862/CJIC.20240460
Yuhui Yang , Jintian Luo , Biao Zuo . A Teaching Approach to Polymer Surface and Interface in Undergraduate Polymer Physics Courses. University Chemistry, 2025, 40(4): 126-130. doi: 10.12461/PKU.DXHX202408056
Maitri Bhattacharjee , Rekha Boruah Smriti , R. N. Dutta Purkayastha , Waldemar Maniukiewicz , Shubhamoy Chowdhury , Debasish Maiti , Tamanna Akhtar . Synthesis, structural characterization, bio-activity, and density functional theory calculation on Cu(Ⅱ) complexes with hydrazone-based Schiff base ligands. Chinese Journal of Inorganic Chemistry, 2024, 40(7): 1409-1422. doi: 10.11862/CJIC.20240007
 DownLoad:
DownLoad: