引用本文:
刘方园, 徐鲁艺, 修阳, 王生杰. 非金属元素掺杂纳米二氧化钛[J]. 化学通报,
2021, 84(2): 108-119, 148.

Citation: Fangyuan Liu, Luyi Xu, Yang Xiu, Shengjie Wang. Non-Metallic Element Doped Titanium Dioxide[J]. Chemistry, 2021, 84(2): 108-119, 148.
Citation: Fangyuan Liu, Luyi Xu, Yang Xiu, Shengjie Wang. Non-Metallic Element Doped Titanium Dioxide[J]. Chemistry, 2021, 84(2): 108-119, 148.
非金属元素掺杂纳米二氧化钛
摘要:
二氧化钛在光电转化、光催化等众多领域具有重要的应用价值,但较宽的禁带宽度和较低的电子传递效率导致其光利用率较低。离子掺杂和纳米化是改变其能带结构、提高电子传输能力的有效策略,根据掺杂离子的性质,可分为金属离子掺杂和非金属元素掺杂。与传统二氧化钛相比,纳米二氧化钛具有特殊的表面效应和粒度效应,其化学活性、耐热性等都强于传统二氧化钛。本文主要对非金属元素掺杂纳米二氧化钛的研究进行评述,包括ⅢA、ⅣA、ⅤA、ⅥA、ⅦA族元素的单一元素掺杂以及和金属或非金属元素共掺杂,重点介绍了纳米二氧化钛的非金属掺杂与其能带结构、可见光响应和光催化性能之间的关系及其应用情况,并对其未来的发展趋势进行了展望。
English
Non-Metallic Element Doped Titanium Dioxide
Abstract:
Titanium dioxide attracts great attentions for its important applications in many fields, especially in photoelectronic conversion and photocatalysis due to its excellent light stability, non-toxicity and easy preparation. However, its relatively poor charge transport property and wide bandgap are two main limitations for its extensive application in light-responsive materials. To meet such challenges, two strategies including ion doping and going nanoscale are used and demonstrated to be effective in regulating its band gap structure and charge transport behavior. Ion doping can be divided into two categories, metal doping and nonmetal doping, according to the properties of the impurity elements. Besides, compared with traditional titanium dioxide, nano-sized titanium dioxide possesses larger surface areas and special nanosized effect, resulting in higher chemical activity and heat resistance. In this review, we focused on the research progress of non-metallic element doped titanium dioxide. Emphases was put on the relationship within the doping element, the band gap structure, visible light responsibility and photocatalytic performance of the hybrid titanium dioxide. Additionally, the application of non-metallic element doped titanium dioxide was also involved.
-
Key words:
- Titanium dioxide
- / Non-metal doping
- / Band gap structure
- / Photoelectric conversion
- / Photocatalysis
-
二氧化钛(TiO2)由于无毒、耐腐蚀、价格低廉和优异的光/化学稳定性而备受青睐,在存储材料、光电器件、太阳能电池、光催化、光解水制氢、光化学转化、传感器、空气净化、水处理等能源和环境相关的众多领域展示了重要的应用前景[1~12],但由于较宽的禁带宽度(锐钛矿和金红石结构分别为3.2和3.0 eV),必须采用紫外光(波长≤387.5nm)激发才能够使其发生电子跃迁,产生光生电子(e-)和空穴(H+)及后续的光催化、光电转化和光化学转化反应。而太阳光中紫外线比例只占4%~6%,因此利用率较低[13~17]。研究者做了大量工作来提高TiO2在可见光区的响应性,包括采用离子掺杂[18, 19]、表面贵金属沉积[20, 21]、与其他半导体复合[22, 23]和染料敏化[24, 25]等技术。其中,离子掺杂通过杂原子的引入在价带和导带之间形成中间能带,能够对能量更低的光子(可见光)产生响应,可作为“垫脚石”来有效地参与价带的电子传递,使得材料的光响应范围向长波方向移动,从而提高太阳光利用效率[25~27]。
根据掺杂物性质不同,主要分为金属离子掺杂和非金属元素掺杂。金属离子掺杂包括铁[28~35]、铜[36~38]、锰[39~42]、锌[43~45]、锡[46~49]、过渡金属[50]、稀土离子[51]掺杂等,已有专门的文献综述进行介绍[52, 53]。金属离子的掺入会代替TiO2晶格中部分钛原子,可能在TiO2半导体晶格中引入缺陷位置或改变结晶度,影响了电子与空穴的复合或改变其激发波长,从而改变TiO2的催化性能。而非金属掺杂TiO2,则是由非金属元素替代TiO2晶格中的部分氧原子,由于氧的2p轨道和非金属中能级与其能量接近的p轨道杂化后,价带宽化上移,禁带宽度相对减小,因此可见光响应范围拓宽,产生光生载流子从而发生氧化还原反应,增强催化活性。因此,本文主要对非金属元素掺杂纳米TiO2的研究进行评述,包括IIIA、IVA、VA、VIA、VIIA族元素的单一元素掺杂和与金属或非金属元素共掺杂的研究进展,以及掺杂方法的发展,例如退火、水热和溶胶-凝胶法等[54~56]。重点介绍了纳米TiO2的掺杂结构与其能带结构、可见光响应和光电转化性质的内在关系,以及掺杂材料在光催化和光电转化方面的应用情况,以助于对非金属元素掺杂TiO2的理解和认识。
1. 单一元素掺杂
1.1 IIIA族元素-硼(B)掺杂
由于B3+的离子半径(0.023nm)小于Ti4+的离子半径(0.064nm),因此,存在于TiO2表面、晶界或TiO2基质中的硼氧化物很容易融入到TiO2的骨架中。Lu等[57]在阳极氧化的钛片上以氮气为载气、硼酸三甲酯为硼源进行化学气相沉积处理,形成掺硼的纳米TiO2。其X射线光电子能谱分析表明,掺入的硼以Ti-B-O形式存在,硼掺杂的TiO2为锐钛矿和金红石相的混合物。Deependra等[27]以异丙醇钛、硼酸和去离子水采用溶胶凝胶法制备了B掺杂纳米TiO2。X射线衍射(XRD)结果表明,合成的纳米粒子存在锐钛矿相和金红石相。
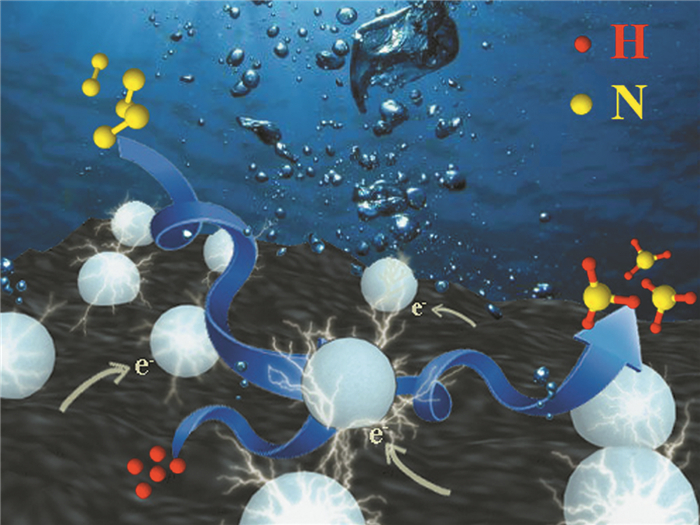
硼掺杂不会改变TiO2初始的形貌,而且比未掺杂的结构更均匀和紧凑。B掺杂会使TiO2禁带宽度减小,紫外光区和可见光区的吸收增强,这可能因为带隙能量降低和表面积增大所致[57, 58]。作为光电极材料使用时,B掺杂电极的饱和光电流密度约为未掺杂电极的1.6倍,即B掺杂电极上获得了升高的光电流密度,原因除了光吸收增强之外,B掺杂电极的混合晶相也可能有助于产生光电流。如图 1所示,Wang等[59]使用掺B硼TiO2在室温、常压的环境条件下将N2还原为NH3,而且NH3的产率优于未掺杂B的TiO2催化下的产率。掺B的TiO2用于光催化固氮,生成的氨是生物合成以及农业肥料中不可缺少的化学物质,而且光催化固氮可有效节约成本,清洁环保。
图 1
1.2 IVA族元素的掺杂
1.2.1 碳掺杂纳米TiO2
通常认为碳掺杂改善了催化剂表面上有机污染物分子的吸附。此外,碳掺杂可以提高TiO2的导电性,因为它可以促进电荷从TiO2结构内部转移到表面区域[60]。碳掺杂剂的存在形式不一,价态范围为-4(作为具有Ti-C键的碳化物)至+4(作为具有C-O键的碳酸盐)。
Lee等[61]通过简单的自组装混合物(P123-TTIP)一步碳化法制备了TiO2-xCx-AC纳米复合材料。通过XPS分析发现,掺入的碳以Ti-O-C形式存在,TiO2纳米粒子以锐钛矿形式存在。TiO2-xCx-AC纳米复合材料由较小尺寸的TiO2微晶组成,在氧化物形成过程中,Pluronic P123和TTIP的均匀混合抑制了晶粒生长和聚集。TiO2-xCx-AC纳米复合材料具有较高的比表面积,其中具有优异吸附能力的活性炭(AC)基团可以将有机分子浓缩在TiO2-xCx-AC附近,光照期间TiO2-xCx-AC纳米复合材料内的AC孔中亚甲基蓝(MB)和羟基自由基的逆流提高了有机物降解效率。如图 2所示,TiO2-xCx-AC纳米复合材料被设计用于光诱导的有机物降解,有效缓解环境污染问题。
图 2
 图 2. 由多孔AC包围的TiO2-xCx-AC纳米微晶的理想结构用于光诱导的有机分子降解:(A)含有TTIP的P123的自组装,(B)真空碳化下形成的TiO2-xCx-AC纳米复合材料,(C)光激发下在TiO2-xCx-AC纳米复合材料内的AC孔中的MB和羟基自由基的逆流流动[61]Figure 2. The desired schematic structure of TiO2-xCx-AC nanocrystallites surrounded by porous ACs for photo-induced degradation of organic molecules: (A) the self-assembly of P123 containing TTIP, (B) the formation of TiO2-xCx-AC nanocomposites under vacuum carbonization, (C) the countercurrent flows of MB and hydroxyl radicals in the pores of ACs within TiO2-xCx-AC nanocomposites under photo excitation[61]
图 2. 由多孔AC包围的TiO2-xCx-AC纳米微晶的理想结构用于光诱导的有机分子降解:(A)含有TTIP的P123的自组装,(B)真空碳化下形成的TiO2-xCx-AC纳米复合材料,(C)光激发下在TiO2-xCx-AC纳米复合材料内的AC孔中的MB和羟基自由基的逆流流动[61]Figure 2. The desired schematic structure of TiO2-xCx-AC nanocrystallites surrounded by porous ACs for photo-induced degradation of organic molecules: (A) the self-assembly of P123 containing TTIP, (B) the formation of TiO2-xCx-AC nanocomposites under vacuum carbonization, (C) the countercurrent flows of MB and hydroxyl radicals in the pores of ACs within TiO2-xCx-AC nanocomposites under photo excitation[61]Sakthivel等[62]通过四氯化钛与四丁基氢氧化铵水解合成了C-TiO2,并应用于降解4-氯苯酚。C-TiO2催化剂粉末具有较高的光催化活性,对通常方法难以降解的4-氯苯酚具有较好的光催化降解效果。Swapnil等[63]利用淀粉作为碳源合成的碳掺杂TiO2材料光降解效率显著提高。Zhou等[64]制备了CeO2和还原氧化石墨烯(rGO)共修饰的TiO2纳米管阵列(CeO2-TiO2-rGO),该复合材料可用于在可见光下降解四溴双酚A。通过在光电催化降解系统上施加偏压,电子-空穴分离效率显著提高,并且改善了降解效率。
Li等[65]将炭黑掺入水热合成的TiO2纳米线(NWs)中,从而合成了碳掺杂的TiO2纳米棒。结果表明,碳成功进入TiO2晶格,导致晶格畸变、带隙减小以及C-Ti-O键的形成,从而将TiO2光响应范围扩展至可见光区。与P25和锐钛矿型TiO2 NWs相比,掺杂炭黑后显示出更高的紫外光和太阳光催化活性。
Huang等[66]通过简单的一步溶剂热途径成功地制备了一种新型光催化剂锐钛矿TiO2纳米颗粒,其具有与g-C3N4和石墨烯同步掺入的暴露的(101)和(001)面。原位g-C3N4和rGO掺杂可以增强g-C3N4、TiO2(001对101)和rGO之间的相互作用,在g-C3N4/TiO2/rGO中产生更多的具有协同作用的异质结,可以促进界面处的快速电子转移,从而提高光催化活性。
1.2.2 硅掺杂纳米TiO2
Bao等[67]采用溶胶-凝胶法和离心纺丝相结合的方法,以四丁醇钛(TBOT)和四乙基正硅酸(TEOS)为前驱体,成功制备了由纳米颗粒和丰富的中孔组成的TiO2纤维。通过改变SiO2的含量和蒸汽热处理温度可以控制晶相、表面积和多孔结构。与纯TiO2相比,SiO2的添加是获得长纤维的关键因素,由于Ti-O-Si网络的形成,无定形SiO2以Si-O-Ti-O-Si网络的形式分散在TiO2中,无定形SiO2和Ti-O-Si键阻止了纤维前体的坍塌和破裂。适量的SiO2可能溶解在TiO2基质中,并抑制TiO2从锐钛矿相转变为金红石。除了在700℃下加热的样品之外,所有Si掺杂样品的XRD光谱均显示出完全的锐钛矿相。多孔结构允许在光照射期间产物快速扩散并提高光催化反应的速率。同时,带隙宽度减小,促进了TiO2在可见光区的吸收,增加了光催化效率。
Guo等[68]采用液相沉积法制备了一种新型Si掺杂分子印迹TiO2纳米光催化材料。分子印迹TiO2可以提高TiO2的光催化活性和分子识别能力,而Si掺杂分子印迹TiO2样品的光催化活性比分子印迹TiO2样品更高。Chen等[69]发现,通过Si掺杂与热处理工艺联用可以提高TiO2纳米棒阵列薄膜的结晶度并产生大量氧空位,从而增强亲水性以及改善电子传输性质。Du等[70]对Si掺杂TiO2纳米线的亲水性能作出解释,Si的掺杂产生更多的硅羟基,并通过Si-O-Ti键将光生电子加速传递到材料表面,这导致Si掺杂的TiO2纳米线的亲水性显著提高。
1.3 VA族元素的掺杂
1.3.1 氮掺杂纳米TiO2
N3-的离子半径为0.146nm,O2-半径为0.14nm,因此N与O具有相当的离子尺寸,而N的2p轨道为半饱和状态,较稳定,因此氮掺杂到TiO2基质中更有利。根据掺杂方式的不同,N掺杂可分为取代N掺杂和间隙N掺杂。研究表明,通常在TiO2晶格的氮掺杂过程中同时发生取代和间隙掺杂,而间隙氮掺杂和取代氮掺杂的概率则与掺杂方法有关。氮的存在可以改变TiO2的能带结构或抑制其光生电子-空穴的复合效率,从而导致TiO2在可见光区域的光催化能力增强[71]。
Danciu等[26]通过溶胶-凝胶法在乙酸或硝酸介质中获得N掺杂的TiO2光催化剂。N取代O原子,以Ti-N键形式存在,所制备的TiO2以锐钛矿和板钛矿形式存在,锐钛矿颗粒的直径随着热处理温度的升高而增加,并且在6.5~14 nm之间变化,而板钛矿纳米颗粒的尺寸在6.1~14.9 nm之间。随着粒径增加,促进了电子转移,因此可以获得增强的光催化活性。Wang等[72]证明,氮掺杂可以大大提高TiO2光阳极的光电化学池(PEC)性能,PEC性能的提高很大程度上归因于可见光吸收的增加和电荷分离效率的提高。如图 3所示,N掺杂提高了TiO2入射光子至电子转换效率(IPCE),表明N掺杂减小了电子和空穴的复合率,提高了电子的利用率。因此掺氮TiO2不仅增强了可见光区的吸收,而且还减少了电子和空穴的复合,大大提高了光催化性能[73, 74]。
图 3

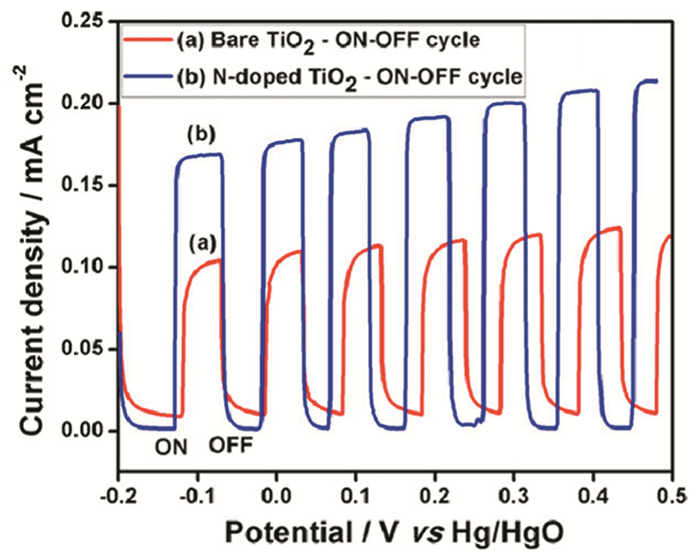
Kumar等[75]通过电化学阳极氧化工艺利用氮气作为氮源在TiO2纳米管上原位N掺杂,合成了多孔且富含缺陷的N掺杂TiO2纳米管。通过改变阳极氧化时间可以控制纳米管的尺寸和掺入TiO2晶格中的氮的量。如图 4所示,与未掺杂TiO2相比,N掺杂的TiO2纳米管的电流密度(0.2mA/cm2)增加了2倍。光电流密度的增加可以归因于氮元素在调节(减少)带隙、减少电子-空穴复合中的关键作用。
图 4

Jiang等[76]通过简便的三步法合成N掺杂TiO2/g-C3N4复合材料,其中N-TiO2纳米片在g-C3N4上均匀组装成层,导致异质结效应的最大化。所得杂化纳米材料的光催化性能显著提高,这归因于N掺杂TiO2与g-C3N4之间的异质结构增加了可见光的利用率和促进了光激发电子与空穴的分离。
1.3.2 磷掺杂纳米TiO2
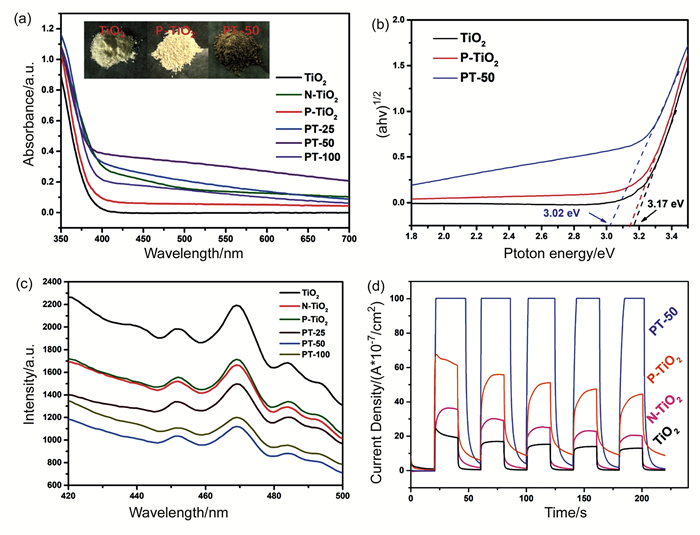
与其他非金属元素相比,磷可以显著提高TiO2的比表面积,防止锐钛矿相变为金红石,从而产生高光催化活性。Gopal等[77]通过溶胶-凝胶法制备了具有高可见光活性的磷掺杂TiO2纳米颗粒。P元素成功掺入TiO2晶格中,提高了TiO2的热稳定性。化学价态分析表明,掺杂的P取代晶格中Ti4+,其处于五价(P5+)的氧化态,形成Ti-O-P键。未掺杂样品在500℃时以锐钛矿相形态存在,并且在600℃开始出现金红石化,而掺P的样品保持其锐钛矿相结构的温度高达700℃。未掺杂样品的平均粒度随着煅烧温度的升高而显著增加,而掺P的样品粒度增加缓慢。P掺杂降低了粒径并且延迟了锐钛矿相向金红石的相变,也使其比表面积增加。P掺杂到TiO2中导致带隙变窄,因此吸收光谱带边红移。晶格中的一部分Ti4+被P5+取代,这表明掺P的样品中的电荷不平衡,这会降低光生电荷载流子的复合率。P掺杂的TiO2纳米粒子在可见光照射下表现出增强的光催化活性,优于未掺杂的TiO2和商业Degussa P25。增强的电荷分离与观察到的吸收边红移现象是可见光活性增强的主要原因。Feng等[78]研究表明,P掺杂会增加TiO2在可见光区的吸收,减小禁带宽度,而且光致发光光谱表明,P掺杂也会降低电子-空穴的复合效率,从而光电流密度增加(如图 5所示)。
图 5
 图 5. (a) TiO2、N-TiO2、P-TiO2和PT样品的紫外可见漫反射图谱(插图:样品的颜色变化);(b)TiO2、P-TiO2和PT-50的带隙;(c)TiO2、N-TiO2、P-TiO2和PT样品的PL光谱;(d)TiO2、N-TiO2、P-TiO2和PT样品的光电流响应[78]Figure 5. (a) The UV-vis DRS of TiO2, N-TiO2, P-TiO2 and PT samples. Inset image: the color change of samples; (b) band gap determination of TiO2, P-TiO2 and PT-50; (c) The PL spectra of TiO2, N-TiO2, P-TiO2 and PT samples; (d) Photocurrent responses of TiO2, N-TiO2, P-TiO2 and PT-50[78]
图 5. (a) TiO2、N-TiO2、P-TiO2和PT样品的紫外可见漫反射图谱(插图:样品的颜色变化);(b)TiO2、P-TiO2和PT-50的带隙;(c)TiO2、N-TiO2、P-TiO2和PT样品的PL光谱;(d)TiO2、N-TiO2、P-TiO2和PT样品的光电流响应[78]Figure 5. (a) The UV-vis DRS of TiO2, N-TiO2, P-TiO2 and PT samples. Inset image: the color change of samples; (b) band gap determination of TiO2, P-TiO2 and PT-50; (c) The PL spectra of TiO2, N-TiO2, P-TiO2 and PT samples; (d) Photocurrent responses of TiO2, N-TiO2, P-TiO2 and PT-50[78]Feng等[79]在超声和Pluronic P123表面活性剂辅助下进行磷掺杂,成功合成了具有稳定锐钛矿-板钛矿双相结构的磷掺杂TiO2催化剂。磷掺杂被证明是稳定锐钛矿-板钛矿双相结构并抑制晶粒生长的有效策略。而Pluronic P123有助于形成具有中孔结构和大表面积的催化剂颗粒,并防止颗粒聚集。在模型污染物MB的降解实验中,板钛矿相的低带隙使合成的P掺杂TiO2催化剂在太阳光和可见光照射下活性均优于商业P25。
1.4 VIA族元素的掺杂
1.4.1 硫掺杂纳米TiO2
与氮和氟相比,硫具有更大的原子半径,在氧位点掺杂硫可以显著改变TiO2的电子结构。Hosseinzadeh等[80]采用超声波辅助喷雾热解技术在玻璃基板上制备了具有高透明度的S掺杂TiO2薄膜。傅里叶变换红外光谱(FTIR)证实掺杂薄膜中存在S-Ti键和Ti-O-S键。锐钛矿是所制备薄膜的主要晶相,S掺杂的TiO2薄膜具有高单分散性和均匀性。通过荧光光谱研究了样品中电荷-载流子捕获的效率。S掺杂的TiO2薄膜具有比未掺杂的TiO2更低的PL强度,表明在S掺杂的TiO2中电荷转移和电子-空穴对的分离效率得以提升。S掺杂TiO2的结构氧空位和晶格缺陷可以作为载流子俘获中心,从而降低光诱导的电子-空穴复合率;另外,S掺杂使TiO2的带隙能量变窄,其吸收边红移,可见光催化活性增强[80, 81]。
硫的较低电负性(与氧相比)和Ti-S键的离子性较低(与Ti-O相比)可能会缩小带隙能量并提高TiO2的电导率。Zhang等[82]直接使用硫酸氧钛作为钛源、二乙烯三胺(DETA)作为络合剂、葡萄糖作为碳前驱体原位合成了固定在大面积碳片上的硫掺杂TiO2纳米粒子。硫掺杂和大面积碳片的结合可以大大提高TiO2的电子传导性,从而导致更快的电荷转移和增强的钠存储过程动力学。更重要的是,由TiO2与碳板之间的静电相互作用引起的强化学键杂化可能有利于钠离子在S-TiO2/CS界面的扩散,从而加速了钠存储赝电容过程。钠离子电池(SIB)被认为是锂离子电池(LIB)最具吸引力的替代电源,而氧化钛(TiO2)被认为是SIB最有潜力的阳极之一,但TiO2的倍率性能受到其缓慢的插层动力学和块体材料不良的电子迁移率的限制。对S-TiO2/CS复合材料的动力学研究表明,钠离子插层赝电容机制对优异的钠存储性能,特别是高循环寿命有很大帮助。
Wang等[83]使用水热法通过一步反应合成了TiO2-S/rGO杂化物。掺杂硫可有效缩小TiO2的带隙,从而显著提高可见光下的光催化反应速率。rGO的引入有助于加速电子转移并使电子-空穴对更有效地分离。因此,TiO2-S/rGO杂化催化剂在模拟太阳光下表现出高的光催化活性。这种TiO2-S/rGO杂化物有望在环境和能源领域得到更多应用。
1.4.2 硒掺杂纳米TiO2
Gurkan等[84]以SeCl4作为硒源,通过湿浸渍法制备了一系列Se掺杂的TiO2光催化材料。Se在晶格中以O-Se-O形式存在(如图 6所示),Se掺杂的TiO2具有锐钛矿和金红石两种结构。Se掺杂样品的高光催化活性归因于掺杂剂离子的能级。当掺杂剂离子的能级低于导带边缘(CB)时,它捕获受激电子;当能级高于价带边缘(VB)时,电子可以淬灭光生空穴。基于有利的能量水平(2.27eV),Se中心可以作为电子或空穴陷阱,以便光生电荷载体暂时分离。因此,除了表面捕获位点,吸附的O2和OH-、Se掺杂剂的存在为电子和空穴提供了更多的捕获位点。另一方面,光催化反应的速率与光催化剂吸收的光子数正相关。由于TiO2的吸光度随着Se掺杂而增加,因此产生更多的电子和空穴并参与表面氧化还原反应,导致引起污染物分子降解的羟基自由基数量增加。
图 6

Xie等[85]采用溶胶-凝胶法制备了一系列锐钛矿结构的Se掺杂TiO2纳米粒子,确认掺杂的Se离子主要处于+4价态,其在TiO2的带隙中提供额外的电子态。研究发现,较低浓度的Se掺杂可以改善TiO2的晶体结构,而较高浓度的Se掺杂使晶体结构劣化。另外,Se掺杂使带隙有效减小,理论计算表明[86],通过取代阴离子掺杂产生的杂质态恰好高于价带最大值(VBM),是造成其带隙减小的主要原因。在可见光照射下,TiO2光催化活性提高,这归因于Se掺杂带来的窄带隙以及由于最佳缺陷密度而抑制的电子-空穴复合的协同作用。
1.5 VIIA族元素的掺杂
与其他金属和非金属元素相比,卤素具有三重优势:改善UV活性;各种阳离子或阴离子替代TiO2基体中的Ti4+和/或O2-;缩小TiO2的带隙和调整价带或导带的位置。
1.5.1 氟掺杂纳米TiO2
Yu等[87]通过在NH4F·H2O溶液中水解四异丙醇钛制备了锐钛矿和板钛矿相的高光活性F掺杂TiO2纳米晶光催化剂。在F掺杂后,锐钛矿的结晶度得到改善。此外,随着F掺杂量的增加,F-不仅抑制了板钛矿相的形成,而且还防止了锐钛矿相变为金红石相。得到的TiO2粉末在400~700 ℃的煅烧温度范围内仅含有锐钛矿相。热能可以触发TiO2晶格中O被F取代。F掺杂TiO2的带隙减小,在可见光区有更强的吸收,其催化活性显著增强。Xu等[88]通过水解法制备的氟掺杂TiO2可以改善结晶度并抑制光催化剂的晶粒生长。氟掺杂可以促进TiO2表面上羟基的形成,从而提高苯酚的光催化降解速率。
Todorova等[17]采用溶胶-凝胶途径制备了F掺杂TiO2,发现锐钛矿/金红石结晶比取决于氟前体的量以及热处理的温度。纯金红石或锐钛矿/金红石混合物在低温下的结晶归因于氟前体的酸性特征。Shen等[89]以氢化TiO2纳米管为前驱体,通过水热途径制备了氟掺杂的锐钛矿TiO2纳米粒子,与未掺杂的TiO2相比,氟掺杂后能够大幅减少TiO2中的氧空位,在钙钛矿太阳能电池中应用时光电转化效率可提高40%以上。Wu等[90]在低温水热条件下在HF溶液中的Ti表面上直接生长F掺杂的花状TiO2纳米结构。合成的新型F掺杂TiO2与P25相比具有良好的结晶度,在水分解和光降解有机污染物方面具有较高的光电化学活性。
1.5.2 氯掺杂纳米TiO2
Wang等[91]通过简单的一步法在70℃下水/乙醇中超声处理钛酸四异丙酯和氯化钠溶液,制备了氯掺杂的锐钛矿TiO2纳米晶。通过Cl 2p XPS谱图证实氯化物的存在,Cl取代氧原子,以O-Ti-Cl形式存在。掺Cl样品的XRD图谱中所有峰对应于TiO2锐钛矿相。掺Cl的TiO2带隙变窄,光吸收明显红移,这有助于可见光催化活性的提高。掺杂的Cl原子可以通过电荷补偿将Ti4+转化为Ti3+,而Ti3+的存在可以降低电子-空穴复合率,进一步提高电子传输效率和光催化活性;但是较高的Cl掺杂剂导致过量的表面Ti3+离子,其充当e-/h+的表面重组中心,反而抑制了光催化活性。
Xu等[92]通过软界面法成功合成了高活性氯化金红石TiO2球形纳米棒光催化剂簇。软界面由两个不混溶的CHCl3和H2O相产生。这种有趣的软界面不仅产生了纳米棒的分层球形簇,而且还在TiO2中产生了Cl掺杂。当Cl被引入TiO2晶体时,其占据TiO2基体中的O位点,并且处于-1价态。Cl掺杂进一步扩展了TiO2的吸收边缘,导致TiO2表面酸性和晶体缺陷的增加,从而提高了光催化活性。
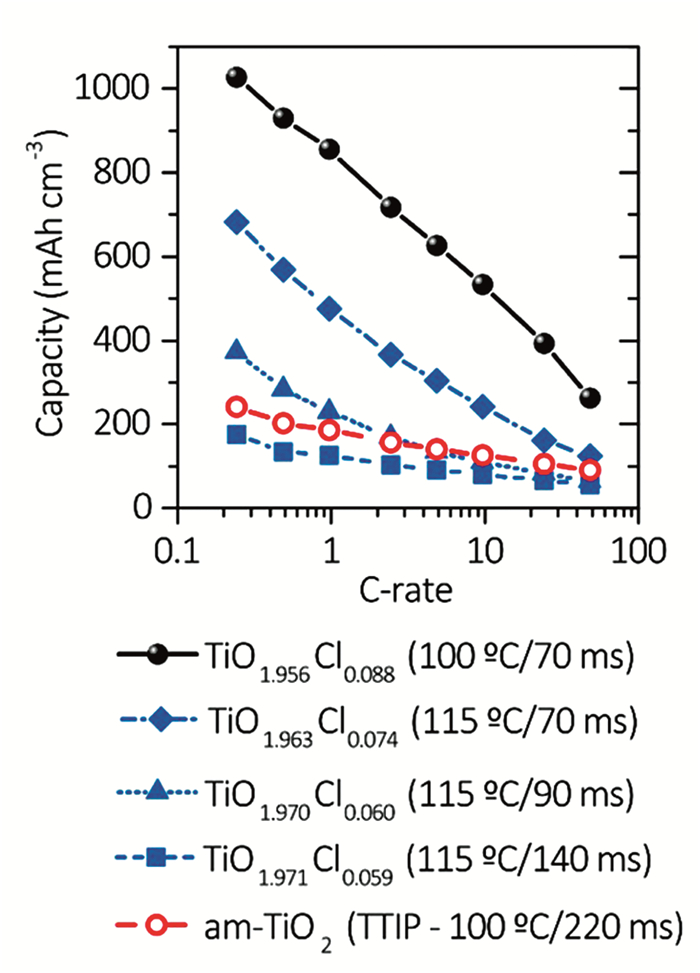
TiO2的理论容量为336mAh·g-1,每Ti单位插入1个Li。然而,块体TiO2电极离子导电性和电子导电性差,限制了其实际应用。尺寸小于20nm的纳米TiO2可以提高倍率性能和比容量,但其在1C下的容量不超过理论容量的75%。局部离子电荷分布的变化(由于较低负电性的氯的掺杂)可能会减少静电排斥,从而促进Li+扩散。Moitzheim等[93]采用空间原子层沉积(S-ALD)方法将TiCl4和H2O前驱体制备成非晶态TiO2-xCl2x薄膜。在一定范围内,TiO2-xCl2x中Cl的含量与最大容量呈线性关系(如图 7所示)。对于TiO1.956Cl0.088薄膜,在0.25C时其最大容量为1030mAh·cm-3和363mAh·g-1,是未掺杂TiO2膜的4倍多。Cl掺杂的TiO2可以作为新的锂电池材料,提供更高的存储容量和充电速率。
图 7
1.5.3 碘掺杂纳米TiO2
与其他卤素改性的TiO2体系不同,碘掺杂在化学结构上更复杂。碘会产生各种化学价态,例如-1、0、+5和+7。不同的化学状态占据碘掺杂TiO2的不同的位点,导致能带结构和TiO2光学特性的变化。
Zhang等[55]通过水热法合成了对可见光照射具有光催化响应性的碘掺杂TiO2(I-TiO2)纳米颗粒。XPS分析显示,碘的价态是I5+,以I-O-Ti键形式存在,I5+取代晶格中的Ti4+,结果产生Ti3+以平衡电荷。未掺杂和I掺杂的TiO2主要由锐钛矿和板钛矿两相组成,XRD分析显示,随着煅烧温度从375℃升高到450℃,锐钛矿相含量增加,板钛矿相减少;当煅烧温度升至550℃时,板钛矿含量随着少量金红石的出现而进一步降低。随着煅烧温度的升高,锐钛矿和板钛矿晶体的平均晶体尺寸增加。随着标称碘掺杂水平提高,晶体尺寸减小。另外,I掺杂使TiO2带隙减小,可见光区的吸收增强。I-TiO2在紫外-可见光照射下的活性增强可能是由于表面积略有增加、可见光吸收增加以及碘掺杂导致的电荷分离改善的组合效应。Liu等[94]使用四氟化钛作为TiO2前体、HIO4作为碘掺杂剂来合成纳米TiO2。大多数碘原子位于TiO2表面。I在TiO2晶体里的存在形式为I-O-I和I-O-Ti结构,I-TiO2光催化剂的可见光光催化活性远超未掺杂的TiO2。
对于不同的主族元素掺杂TiO2,掺杂剂的存在形式大部分是代替氧原子。大多数非金属元素掺杂TiO2能够增加比表面积,减小粒径,增加反应面积,提高催化效率,且都能不同程度地减小TiO2的带隙宽度,增强其在可见光区的吸收,或抑制电子与空穴的复合,从而提高光催化活性。
2. 共掺杂
与单元素掺杂剂体系相比,共掺杂的TiO2光催化剂由于在可见光区域的有利吸收而表现出更高的光催化活性。共掺杂的TiO2中的掺杂剂之间的电荷补偿通过增强氧化还原电势和增加光生载流子的迁移率来抑制体缺陷形成。
2.1 非金属与非金属元素共掺杂
2.1.1 碳和氮共掺杂
Chen等[95]通过溶胶-凝胶法制备具有不同氮和碳含量的TiO2纳米颗粒,四正丁基氧化钛(TTB)、尿素和四丁基氢氧化铵(TBAH)分别用作TiO2、氮和碳的前驱体。XRD分析显示所有样品都是锐钛矿相,TiO2中氮和碳的掺杂可以显著抑制晶体生长。碳掺杂的效果比氮掺杂的效果更明显,氮原子可以取代锐钛矿晶格中的氧原子位点,形成Ti-N键,这导致晶胞参数的增加。碳原子难以掺入到TiO2晶格中,而是存在于TiO2纳米颗粒的表面并形成由复合碳酸盐物质组成的膜层,导致TiO2晶体生长受抑制。C掺杂TiO2纳米颗粒比未掺杂的TiO2和N掺杂TiO2的聚集现象略有增加,说明掺杂的碳位于TiO2纳米颗粒的表面,在聚集的纳米颗粒之间起连接剂的作用。据XPS元素分析,C、N共掺杂的TiO2中的N原子数占总原子数的1.5%,是N掺杂TiO2样品中的N原子占比的2倍,表明TBAH可明显促进氮的吸收,推测TBAH可以抑制TiO2胶体溶液的水解,从而形成较小的TiO2纳米颗粒,由于较大的晶格应变和晶格畸变,使得氮吸收更容易。碳和氮掺杂都可以明显缩小TiO2的带隙,C、N共掺杂TiO2样品对可见光的吸收最强,其次是C掺杂TiO2和N掺杂TiO2。通过测量可见光照射下MB的分解速率来评价TiO2样品的光催化活性,C、N共掺杂、C掺杂TiO2和N掺杂TiO2均比未掺杂TiO2具有更高的光催化活性;C、N共掺杂的光催化活性高于C掺杂TiO2和N掺杂TiO2,表明存在碳和氮共掺杂的协同效应。此外,N含量越高,取代氧位点的氮原子数增加,从而引起氧空位和Ti3+增加,导致光催化活性增强;当N含量持续增加时,一些氧空位和Ti3+位点成为光生空穴和电子的复合中心,因此光催化活性开始下降。
Mani等[96]制备了纯锐钛矿C、N共掺杂纳米TiO2,在吸收光谱发现了明显的红移现象。除此之外,在系统中加入电子受体过氧单硫酸盐(PMS)以捕获电子从而阻止激子重组,并且在各种催化剂量下研究了其效果。在添加PMS后,发现甲基橙降解的速率常数几乎增加了4倍,这可能归因于电子空穴复合率的降低。
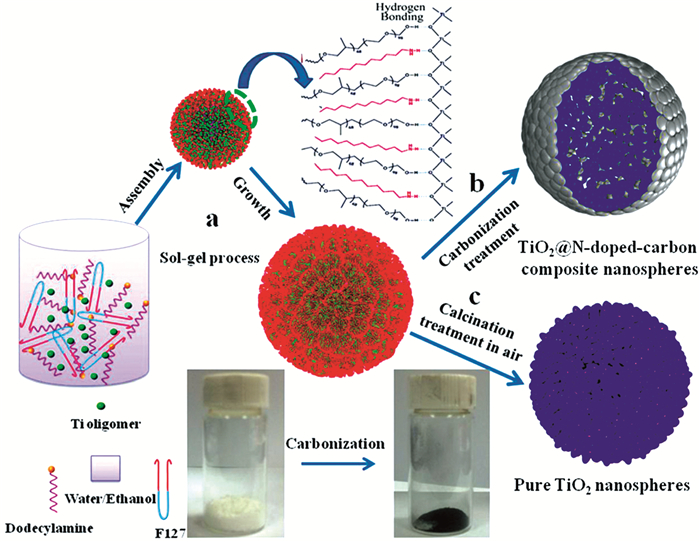
Zhu等[97]通过双表面活性剂辅助的组装溶胶-凝胶法(图 8)合成然后在高温下顺序碳化制备了单分散介孔TiO2@N掺杂的碳复合纳米球。该表面活性剂可以直接转化为连续的导电碳网络,有效地改善了TiO2纳米球的电化学性能。TiO2@N掺杂的碳复合纳米球具有大比表面积(~120m2·g-1)、大孔径(4~40nm);锐钛矿TiO2纳米晶体(约8nm)被一致地封装在N掺杂的碳层中。TiO2@N掺杂的碳复合纳米球电极在0.5C倍率下200个循环后显示出约167mAh·g-1的极高容量,被用作锂存储阳极材料。更重要的是,掺杂TiO2@N的碳复合纳米球表现出卓越的倍率性能,在10C下2000次循环后的比容量为117mAh·g-1。这些结果表明,介孔TiO2@N掺杂的碳复合纳米球有望用作下一代高性能锂离子电池材料。
图 8
 图 8. 均匀掺杂TiO2@N的碳复合纳米球的合成过程示意图[97]
图 8. 均匀掺杂TiO2@N的碳复合纳米球的合成过程示意图[97](a)双表面活性剂辅助的溶胶-凝胶法制备具有无机/有机混合球形结构的单分散无定形TiO2;(b)在N2气氛中在700℃下碳化处理具有无机/有机杂化结构的单分散无定形TiO2纳米球4h;(c)为了比较,将非晶态TiO2纳米球在空气中在相同温度下退火4h。插图是无定形TiO2胶体纳米球的数码照片和TiO2@N掺杂的碳复合纳米球在N2气氛中于700℃下碳化4h后的照片
Figure 8. Schematic of the synthesis process of uniform TiO2@N-doped carbon composite nanospheres[97]El-Sheikh等[98]通过水热法合成了甘氨酸助剂控制的可调节板钛矿/锐钛矿比的碳氮共掺杂介孔TiO2光催化剂,在不存在甘氨酸的情况下形成纯的板钛矿TiO2纺锤状颗粒,而当加入甘氨酸时,获得了由小的准球形锐钛矿和板钛矿纳米棒组成的介孔异质结TiO2纳米颗粒。XPS和FT-IR结果显示TiO2网格中存在C-C、C-H和N-H键。通过在可见光照射下光降解布洛芬(IBF)溶液研究了催化剂的光催化活性,结果表明,适当的掺杂使得其催化活性提高。原因归结于其高的比表面积、异质结构的形成以及可见光范围内的高吸收和电荷-载体的有效分离。
2.1.2 溴和氯共掺杂
Luo等[99]在混合氢溴酸-乙醇体系中使用氯化钛通过水热法合成溴和氯共掺杂的TiO2纳米晶体。通过加入HBr增加酸度,依次得到锐钛矿、混合锐钛矿/金红石、金红石、混合金红石/板钛矿、混合锐钛矿/金红石/板钛矿和金红石相的TiO2。Br、Cl共掺杂使TiO2的带隙减小,其吸收边移至可见光区。Br和Cl共掺杂的TiO2在光解水产H2和O2中具有较高催化活性,这种生产可再生氢的途径对于缓解未来的能源和环境问题意义重大。
2.1.3 氮和氟共掺杂纳米TiO2
Elbanna等[100]通过三乙醇胺的简单水热处理制备了N掺杂的TiO2颗粒,而在室温下通过简单搅拌TiO2颗粒与NaF可以容易地制备F掺杂的TiO2颗粒。TiO2颗粒的晶体结构和表面积不受N和F掺杂的影响。XRD分析显示样品为锐钛矿相,晶格中的O被N取代,N以Ti-N-O键或Ti-O-N键形式存在,F则吸附在颗粒表面。与N掺杂相比,N、F共掺杂时N原子掺入量略微减少,表明F在一定程度上限制了N的掺杂。N、F共掺杂TiO2的高活性归因于N和F掺杂的协同作用。掺杂的N原子增强了可见光吸收,导致带隙能量的减小,而F增加了羟基自由基的产生,并提高了电荷分离效率。Chen等[101]提出了一种通过钛络合物高温热解合成掺杂非金属元素的TiO2材料的简便方法。其因氮和氟掺杂的协同效应导致N-F-TiO2的高光催化活性。
纳米颗粒被广泛用于有毒化学物质的光催化降解和细胞的光动力治疗,涉及光诱导的活性氧(ROS)生成,然后由ROS诱导化学/生化转化。但是,设计纳米颗粒以捕获太阳光的可见光谱并生成可有效利用的高浓度ROS是一关键问题。氮和氟共-掺杂的TiO2纳米颗粒可捕获整个可见光谱并在光照射下产生ROS,在基于阳光的水净化和食品/环境消毒方面具有巨大的应用潜力。Mukherjee等[102]制备了N和F共掺杂的TiO2纳米颗粒作为抗真菌剂。胶体形式和适当的表面化学性质提供了与真菌细胞壁的有效相互作用,因纳米颗粒的光催化性质在真菌细胞壁的表面产生了ROS,导致真菌死亡。这项研究突出了环保型TiO2的潜在应用,新一代的光催化剂有望用于农业领域的植物病原体消毒。
2.2 非金属与金属元素共掺杂
2.2.1 钴和硫共掺杂
Siddiqa等[103]采用溶胶-凝胶法成功合成了钴、硫共掺杂TiO2纳米颗粒,其为锐钛矿相的单晶。随着钴含量的增加,颗粒逐渐减小,表明TiO2纳米颗粒的生长受到钴掺杂的抑制。掺杂S,可能存在一些S3p轨道与O2p轨道的混合,导致形成中间能级,从而使吸收扩展至可见光区域; 添加钴可以通过在导带附近引入其能级来促进可见光吸收。钴和硫离子掺杂在TiO2的晶体中,引起对称性的扭曲,导致框架结构中形成缺陷,产生氧空位并限制TiO2的晶体生长;而且共掺杂使TiO2的带隙宽度减小,使其吸收移至可见光区,光催化效率提高。
2.2.2 铁和硫共掺杂
Hamadanian等[104]以异丙醇钛、Fe(NO3)3·9H2O和硫脲为前驱体,采用改进的溶胶-凝胶法合成了一种新型的铁、硫共掺杂TiO2光催化剂。掺杂的Fe可能通过取代Ti4+而掺入TiO2基质中,掺杂的S取代TiO2晶格中的O原子或以Ti-O-S键存在。Fe、S共掺杂的TiO2在可见光区域的吸光度随着铁浓度的增加而增加。添加铁和硫后TiO2吸收初始值的红移表明掺杂剂降低了催化剂的带隙。Fe、S共掺杂TiO2光催化活性较高的原因是硫和铁在TiO2中的协同作用[104, 105]:与未掺杂的TiO2相比,Fe、S共掺杂TiO2的光吸收能力增强;铁掺杂也会阻碍TiO2颗粒的聚集和生长,从而导致晶粒尺寸减小和比表面积增加;Fe3+可以作为h+/e-陷阱来降低h+/e-的重组率并增强光催化活性。
2.2.3 钨和碳共掺杂
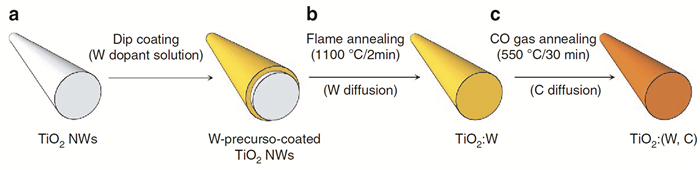
Lee等[56]报道了一种新的非原位掺杂方法制备钨和碳共掺杂TiO2。如图 9所示,在火焰和一氧化碳气体中顺序退火W前体涂层的TiO2纳米线。火焰退火的独特优势在于不损害纳米线形态和结晶度的情况下,高温(>1000℃)和快速加热火焰使W迅速扩散到TiO2中。掺杂剂浓度在样品表面附近较高,沿径向朝中心逐渐减小。研究表明,共掺杂的TiO2具有最高光电流密度和最高的IPCE值,原因可能是共掺杂可诱导高表面酸度和反应物吸附的增加,增强了电荷转移过程。通过共掺杂W(供体)和C(受体)的缺陷钝化效应,共掺杂比单掺杂能提供更高的电导率。
图 9

共掺杂两种阴离子,通常一种掺入TiO2晶格中,可以降低带隙,增加可见光区的吸收,而另一种则吸附在表面,增加光生电子迁移率等,二者的协同作用使光催化活性得以提高。同时掺杂阴离子和过渡金属离子(共掺杂)可以改变TiO2的导电性和光学性质。这种共掺杂可能引入与TiO2的导带或价带密切相关的新电子态。阴离子掺杂TiO2可以减小带隙宽度,增强可见光区的吸收,过渡金属可以作为TiO2晶格中的缺陷,抑制光催化剂表面上光生电子-空穴对的复合,从而提高光催化活性和光电转化效率。如表 1所示,不同的掺杂均可以提高TiO2的光电转化活性。
表 1
 表 1 不同掺杂材料改性前后的光电转化活性Table 1. Photoelectric conversion activity of different doped materials before and after modification
表 1 不同掺杂材料改性前后的光电转化活性Table 1. Photoelectric conversion activity of different doped materials before and after modification 下载:
导出CSV
下载:
导出CSV
TiO2在能源、环境、工业以及近年来蓬勃发展的医药领域均有广泛应用。非金属掺杂改性后的纳米TiO2由于显著提高了光电转化性能,可应用于太阳能电池或者锂离子电池;另外,这些材料在抗菌、防腐、防雾、生物医学等领域也展示出良好的应用价值。
3. 结语
本文评述了各种非金属离子掺杂纳米TiO2的方法、掺杂剂的存在形式和掺杂剂的作用。大多数非金属元素在TiO2的晶格中取代的是氧的位置。通过掺杂,一方面可以降低TiO2的带隙,提高可见光的响应性,另一方面通过掺杂(共掺杂)可以抑制电子-空穴的复合,提高光电分离和传输效率,从而提高其光催化或光电转化/光化学转化效率。
如金属离子掺杂的TiO2存在热稳定性差和光腐蚀一样,非离子掺杂的TiO2目前仍存在较多的问题。例如,非金属离子掺杂容易在TiO2的晶格中引入氧空位,成为电荷复合的中心,降低光催化剂的可见光催化效率[106];但也有研究者认为氧空位在催化反应中是有利的[107],因此如何改善氧空位,使其有利于电子-空穴的分离,提高催化性能,应该是接下来研究的重点。另外,非离子掺杂TiO2的制备过程通常需要较长时间的高温(400~500℃)处理,消耗较多的能量,而且在热处理过程中一些不稳定或有毒的非金属元素会以气化物的形式挥发出来,造成环境污染和掺杂效率降低,限制了阴离子掺杂TiO2的规模化生产[108]。因此,可精确控制掺杂位点和掺杂结构以及环境友好的非金属离子掺杂方法将是未来研发的重要方向。非金属掺杂的TiO2和相关的非金属掺杂的半导体用于催化光解水产氢以及用于人工光合作用以再循环CO2引起人们的关注,有助于解决未来可持续发展社会至关重要的问题。共掺杂由于不同元素之间的协同效应,金属与非金属的共掺杂、非金属之间的共掺杂可从光生电子的产生到转移和传输等方面提高电子或空穴的催化能力,因此共掺杂也将是后期重点研究的方向之一。
-
-
[1]
Hamad D, Dhib R, Mehrvar M. J. Polym. Environ., 2016, 24(1): 72~83.
-
[2]
Bustillo-Lecompte C F, Ghafoori S, Mehrvar M. J. Environ. Chem. Eng., 2016, 4(1): 719~732.
-
[3]
Nasirian M, Lin Y P, Bustillo-Lecompte C F, et al. Int. J. Environ. Sci. Technol., 2018, 15(9): 2009~2032.
-
[4]
Malakootian M, Mesdaghinia A, Rezaei S. Journal of Kerman University of Medical Sciences, 2017, 24(2): 147~158.
-
[5]
Wang S Q, Liu W B, Fu P, et al. Korean J. Chem. Eng., 2017, 34(5): 1584~1590.
-
[6]
Chen D, Caruso R A. Adv. Funct. Mater., 2013, 23(11): 1356~1394.
-
[7]
Wang H, Miyauchi M, Ishikawa Y, et al. J. Am. Chem. Soc., 2011, 133(47): 19102~19109.
-
[8]
Su J J, Li Z D, Zhang Y Q, et al. RSC Adv., 2016, 6(20): 16177~16182.
-
[9]
Ma Y, Wang X, Jia Y, et al. Chem. Rev., 2014, 114(19): 9987~10043.
-
[10]
Abdelhaleem A, Chu W. Chem. Eng. J., 2018, 338(15): 411~421.
-
[11]
Shehzad N, Tahir M, Johari K, et al. J. CO2 Util., 2018, 26: 98~122.
-
[12]
Sinhamahapatra A, Jeon J P, Yu J S. Energ. Environ. Sci., 2015, 8(12): 3539~3544.
-
[13]
Zheng X Z, Li D Z, Li X F, et al. Appl. Catal. B, 2015, 168~169: 408~415.
-
[14]
Khaki M R D, Shafeeyan M S, Raman A A A, et al. J. Environ. Manag., 2017, 198(2): 78~94.
-
[15]
Huang L W, Fu W Y, Zhang Z Y. Mater. Lett., 2017, 209(15): 585~588.
-
[16]
Behnajadym A, Eskandarloo H. Chem. Eng. J., 2013, 228(15): 1207~1213.
-
[17]
Todorova N, Giannakopoulou T, Romanos G, et al. Int. J. Photoenergy, 2008, 534038.
-
[18]
Mao C Y, Zuo F, Hou Y, et al. Angew. Chem. Int. Ed., 2014, 53(39): 10485~10489.
-
[19]
Shayegan Z, Lee C S, Haghighat F. Chem. Eng. J., 2018, 334(15): 2408~2439.
-
[20]
Yun J Y, Hwang S H, Jang J. ACS Appl. Mater. Inter., 2015, 7(3): 2055~2063.
-
[21]
Wei Z, Janczarek M, Endo M, et al. Appl. Catal. B, 2018, 237(5): 574~587.
-
[22]
Xie F Y, Li Y F, Dou J, et al. J. Power Sources, 2016, 336(30): 143~149.
-
[23]
Sengupta D, Das P, Mondal B, et al. Renew. Sustain. Energ. Rev., 2016, 60: 356~376.
-
[24]
Altin I, Sokmen M, Biykloglu Z. Desalin. Water Treat., 2016, 57(34): 16196~16207.
-
[25]
Shao G S. J. Phys. Chem. C, 2009, 113(16): 6800~6808.
-
[26]
Pap Z, Baia L, Mogyorósi K, et al. Catal. Commun., 2012, 17(5): 1~7.
-
[27]
Mulmi D D, Thapa D, Dahal B, et al. Int. J. Mater. Sci. Eng., 2016, 4(3): 172~178.
-
[28]
Zou M M, Xiong F Q, Ganeshraja A S, et al. Mater. Chem. Phys., 2017, 195(1): 259~267.
-
[29]
Primc D, Bartsch M, Barreca D, et al. Sustain. Energy Fuels, 2017, 1(1): 199~206.
-
[30]
Foura G, Chouchou N, Soualah A, et al. Catalysts, 2017, 7(11): 344.
-
[31]
Freyria F S, Compagnoni M, Ditaranto N, et al. Catalysts, 2017, 7(7): 213.
-
[32]
Shiba K, Kataoka T, Okuda M, et al. Royal Soc. Chem. Adv., 2016, 6(61): 55750~55754.
-
[33]
Husain S, Alkhtaby L A, Giorgetti E, et al. J. Luminescence, 2016, 172: 258~263.
-
[34]
Crisan M, Rǎileanu M, Drǎgan N, et al. Appl. Catal. A, 2015, 504(5): 130~142.
-
[35]
Santos R d S, Faria G A, Giles C, et al. ACS Appl. Mater. Inter., 2012, 4(10): 5555~5561.
-
[36]
Hinojosa-Reyes M, Camposeco-Olis R, Zanella R, et al. Chemosphere, 2017, 184: 992~1002.
-
[37]
Obregon S, Lee S W, Rodriguez-gonzalez Ⅴ. Mater. Lett., 2016, 173(15): 174~177.
-
[38]
Pham T D, Lee B K. Appl. Surf. Sci., 2014, 296(30): 15~23.
-
[39]
Zhi J T, Yu X Q, Bao J J, et al. Korean J. Chem. Eng., 2016, 33(6): 1823~1830.
-
[40]
Praveen P, Viruthagiri G, Mugundan S, et al. Spectrochim. Acta A, 2014, 120(24): 548~557.
-
[41]
Ning X W, Wang X X, Yu X F, et al. J. Alloys Compd., 2016, 658(15): 177~182.
-
[42]
Tshabalala Z P, Shingange K, Cummings F R, et al. J. Colloid Interf. Sci., 2017, 504(15): 371~386.
-
[43]
Salazar-villanueva M, Cruz-López A, Zaldívar-Cadena A A, et al. Mater. Sci. Semicon. Proc., 2017, 58: 8~14.
-
[44]
Wu M C, Chan S H, Jao M H, et al. Solar Energy Mater. Solar Cells, 2016, 157: 447~453.
-
[45]
Nair R G, Mazumdar S, Modak B, et al. J. Photochem. Photobiol. A, 2017, 345(1): 36~53.
-
[46]
Sui R H, Yong J L, Berlinguette C P. J. Mater. Chem., 2010, 20(3): 498~503.
-
[47]
Lübke M, Johnson L, Makwana N M, et al. J. Power Sources, 2015, 294(30): 94~102.
-
[48]
Li J L, Xu X T, Liu X J, et al. J. Alloys Compd., 2016, 679(15): 454~462.
-
[49]
Cai Q B, Zhang Y Q, Liang C, et al. Electrochim. Acta, 2018, 261, (20): 227~235.
-
[50]
Inturi S N R, Boningari T, Suidan M, et al. Appl. Catal. B, 2014, 144: 333~342.
-
[51]
Bhethanabotal V C, Russell D R, Kuhn J N. Appl. Catal. B, 2017, 201: 156~164.
-
[52]
Mazierski P, Mikolajczyk A, Bajorowicz B, et al. Appl. Catal. B, 2018, 233(5): 301~317.
-
[53]
Shwetharani R, Sakar M, Fernando C A N, et al. Catal. Sci. Technol., 2019, 9(1): 12~46.
-
[54]
Gao H T, Liu Y Y, Ding C H, et al. Int. J. Min. Metal. Mater., 2011, 18(5): 606~614.
-
[55]
Zhang Q Y, Li Y, Ackerman E A, et al. Appl. Catal. A, 2011, 400: 195~202.
-
[56]
Cho I S, Lee C H, Feng Y Z, Logar M, Rao P M, Cai L L, Kim D R, Sinclair R, Zheng X L. Nat. Commun., 2014, 5: 3204.
-
[57]
Lu N, Quan X, Li J Y, et al. J. Phys. Chem. C, 2007, 111(32): 11836~11842.
-
[58]
Simsek E B. Appl. Catal. B, 2017, 200: 309~322.
-
[59]
Wang Y, Jia K, Pan Q, et al. ACS Sustain. Chem. Eng., 2019, 7(1): 117~122.
-
[60]
Xiao Q, Ouyang L L. Chem. Eng. J., 2009, 148(2/3): 248~253.
-
[61]
Lee Y F, Chang K H, Hu C C, et al. J. Mater. Chem., 2010, 20: 5682~5688.
-
[62]
Sakthivel S, Kisch H. Angew. Chem. Int. Ed., 2003, 42(40): 4908~4911.
-
[63]
Warkhade S W, Warkhade G S, Zodape S P, et al. Mater. Sci. Semicon. Proc., 2017, 63(1): 18~24.
-
[64]
Zhou Q X, Xing A, Zhao D C, et al. Chemosphere, 2016, 165: 268~276.
-
[65]
Li W J, Liang R, Zhou N Y, et al. ACS Omega, 2020, 5(17): 10042~10051.
-
[66]
Huang M, Yu J H, Hu Q, et al. Appl. Surf. Sci., 2016, 389(15): 1084~1093.
-
[67]
Bao N, Wei Z T, Ma Z H, et al. J. Hazard. Mater., 2010, 174(1/2/3): 129~136.
-
[68]
Guo J F, Li S M, Duan L, et al. Integr. Ferroelectr., 2016, 168(1): 170~182.
-
[69]
Chen C L, Wei Y L, Yuan G Z, et al. Adv. Funct. Mater., 2017, 27(31): 1701575.
-
[70]
Du J, Li X Y, Li K, et al. J. Alloys Compd., 2016, 687(5): 893~897.
-
[71]
Ansari S A, Khan M M, Ansari M O, et al. New J. Chem., 2016, 40: 3000~3009.
-
[72]
Wang G M, Xiao X H, Li W Q, et al. Nano Lett., 2015, 15(7): 4692~4698.
-
[73]
Asahi R, Morikawa T, Ohwaki T, et al. Science, 2001, 293(5528): 269~271.
-
[74]
Kong X L, Peng Z B, Jia P P, et al. ACS Appl. Nano Mater. 2020, 3(2): 1373~1381.
-
[75]
Kumar M P, Jagannathan R, Ravichandran S. Energy Fuels, 2020, 34(7): 9030~9036.
-
[76]
Jiang G M, Cao J W, Chen M, et al. Appl. Surf. Sci., 2018, 458: 77~85.
-
[77]
Gopal N O, Lo H H, Ke T F, et al. J. Phys. Chem. C, 2012, 116(30): 16191~16197.
-
[78]
Feng X Y, Wang P F, Hou J, et al. J. Hazard. Mater., 2018, 351: 196~205.
-
[79]
Feng H J, Zhang M H, Yu L E. J. Nanosci. Nanotechnol., 2013, 13(7): 4981~4989.
-
[80]
Hosseinzadeh G, Rasoulnezhad H, Ghasemian N, et al, J. Aust. Ceram. Soc., 2019, 55(2): 387~394.
-
[81]
Ni J F, Fu S D, Wu C, et al. Adv. Mater., 2016, 28(11): 2259~2265.
-
[82]
Zhang Y, He X R, Tang J H, et al. ACS Appl. Mater. Inter., 2019, 11(47): 44170~44178.
-
[83]
Wang W L, Wang Z F, Liu J J, et al. Sci. Rep., 2017, 7: 46610.
-
[84]
Gurkan Y Y, Cinar Z. Chem. Eng. J., 2013, 214(1): 34~44.
-
[85]
Xie W, Li R, Xu Q U. Sci. Rep., 2018, 8: 8752.
-
[86]
Zheng J W, Bhattcahrayya A, Wu P, et al. J. Phys. Chem., 2010, 114(15): 7063~7069.
-
[87]
Yu J C, Yu J G, Ho W K, et al. Chem. Mater., 2002, 14(9): 3808~3816.
-
[88]
Xu J J, Ao Y H, Fu D G, et al. J. Phys. Chem. Solids, 2008, 69(10): 2366~2370.
-
[89]
Zhang X Q, Wu Y P, Huang Y, et al. J. Alloys Compd., 2016, 681: 191.
-
[90]
Wu G S, Wang J P, Thomas D F, et al. Langmuir, 2008, 24(7): 3503~3509.
-
[91]
Wang X K, Wang C, Jiang W Q, et al. Chem. Eng. J., 2013, 189/190: 288~294.
-
[92]
Xu H, Zhang Z, Zhang L Z, Zet al. J. Solid State Chem., 2008, 181(9): 2516~2522.
-
[93]
Moitzheim S, Balder J E, Poodt P, et al. Chem. Mater., 2017, 29(23): 10007~10018.
-
[94]
Liu G, Sun C H, Yan X X, et al. J. Mater. Chem., 2009, 19: 2822~2829.
-
[95]
Chen D M, Jiang Z Y, Geng J Q, et al. Ind. Eng. Chem. Res., 2007, 46(9): 2271~2746.
-
[96]
Mani A D, Muthusamy S, Anadan S, et al. J. Exp. Nanosci., 2015, 10: 115~125.
-
[97]
Zhu H, Jing Y, Pal M, et al. Nanoscale, 2017, 9: 1539~1546.
-
[98]
El-Sheikh S M, Khedr T M, Hakki A, et al. Sep. Purif. Technol., 2017, 173: 258~268.
-
[99]
Luo H M, Takata T, Lee Y, et al. Chem. Mater., 2004, 16(5): 846~849.
-
[100]
Elbanna O, Zhang P, Fujitsuka M, et al. Appl. Catal. B, 2016, 192: 80~87.
-
[101]
Chen D M, Jiang Z Y, Geng J Q, et al. J. Nanopart. Res., 2009, 11(2): 303~313.
-
[102]
Mukherjee K, Acharya K, Biswas A, et al. ACS Appl. Nano Mater., 2020, 3(2): 2016~2025.
-
[103]
Siddiqa A, Masih D, Anjum D, et al. J. Environ. Sci., 2015, 37: 100~109.
-
[104]
Hamadanian M, Reisi-Vanani A, Behpour M, et al. Desalination, 2011, 381(17): 319~324.
-
[105]
Bessergenev V G, Mateus M C, Vasconcelos D A, et al. International J. Photoenergy, 2012, 767054.
-
[106]
Dong F, Guo S, Wang H Q, Let al. J. Phys. Chem. C, 2011, 115(27): 13285~13292.
-
[107]
Zhao Y X, Zhao X F, Run S, et al. Adv. Mater., 2019, 31(16): 1806482.
-
[108]
Dong F, Wang H, Wu Z. J. Phys. Chem. C, 2009, 113(38): 16717~16723.
-
[109]
Rami R D, Joyashish D, Vijayamohanan K, et al. Sci. Rep., 2015, (4): 4897.
-
[1]
-

图 2 由多孔AC包围的TiO2-xCx-AC纳米微晶的理想结构用于光诱导的有机分子降解:(A)含有TTIP的P123的自组装,(B)真空碳化下形成的TiO2-xCx-AC纳米复合材料,(C)光激发下在TiO2-xCx-AC纳米复合材料内的AC孔中的MB和羟基自由基的逆流流动[61]
Figure 2 The desired schematic structure of TiO2-xCx-AC nanocrystallites surrounded by porous ACs for photo-induced degradation of organic molecules: (A) the self-assembly of P123 containing TTIP, (B) the formation of TiO2-xCx-AC nanocomposites under vacuum carbonization, (C) the countercurrent flows of MB and hydroxyl radicals in the pores of ACs within TiO2-xCx-AC nanocomposites under photo excitation[61]
图 5 (a) TiO2、N-TiO2、P-TiO2和PT样品的紫外可见漫反射图谱(插图:样品的颜色变化);(b)TiO2、P-TiO2和PT-50的带隙;(c)TiO2、N-TiO2、P-TiO2和PT样品的PL光谱;(d)TiO2、N-TiO2、P-TiO2和PT样品的光电流响应[78]
Figure 5 (a) The UV-vis DRS of TiO2, N-TiO2, P-TiO2 and PT samples. Inset image: the color change of samples; (b) band gap determination of TiO2, P-TiO2 and PT-50; (c) The PL spectra of TiO2, N-TiO2, P-TiO2 and PT samples; (d) Photocurrent responses of TiO2, N-TiO2, P-TiO2 and PT-50[78]
图 8 均匀掺杂TiO2@N的碳复合纳米球的合成过程示意图[97]
Figure 8 Schematic of the synthesis process of uniform TiO2@N-doped carbon composite nanospheres[97]
(a)双表面活性剂辅助的溶胶-凝胶法制备具有无机/有机混合球形结构的单分散无定形TiO2;(b)在N2气氛中在700℃下碳化处理具有无机/有机杂化结构的单分散无定形TiO2纳米球4h;(c)为了比较,将非晶态TiO2纳米球在空气中在相同温度下退火4h。插图是无定形TiO2胶体纳米球的数码照片和TiO2@N掺杂的碳复合纳米球在N2气氛中于700℃下碳化4h后的照片
表 1 不同掺杂材料改性前后的光电转化活性
Table 1. Photoelectric conversion activity of different doped materials before and after modification
 下载: 导出CSV
下载: 导出CSV
-












 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 34
- 文章访问数: 2669
- HTML全文浏览量: 663
