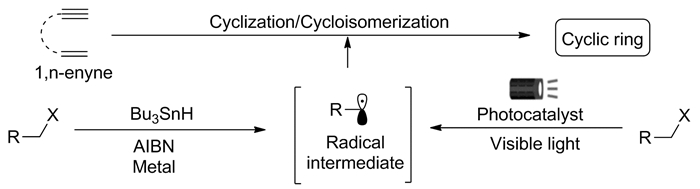
图式 1.
可见光诱导的1, n-烯炔的自由基串联反应
Scheme 1.
Visible-light-induced radical cascade cyclization of 1, n-enynes

1, n-烯炔的串联环化反应是构建重要的碳、氮杂环骨架的最有力和最直接的方法。由于其操作简单和具有高效、高原子经济性等特点,已经在天然产物、药物分子以及复杂分子的合成中展示了它独特的优越性。近年来,自由基促进的1, n-烯炔环化反应成为化学工作者的研究热点,其中大多是使用经典的氧化剂和过渡金属催化来实现的[1, 2]。尽管在这一领域取得了令人瞩目的进展,但由于使用过量的氧化剂产生大量废弃物、贵金属成本高、原子经济性不理想、化学选择性差等缺点,亟需开发绿色可持续的新方法来实现1, n-烯炔的新型自由基环化反应,合成多官能团碳氮杂环化合物。
在过去的十年中,可见光催化作为选择性小分子活化和化学键形成不可或缺的工具,已经成为有机合成化学的前沿领域之一[3, 4]。在光源的照射下,光敏剂可将光能转化为化学能,从而促进活性自由基中间体的产生,进而形成新的化学键。有机光催化的可持续性、可扩展性、温和的条件以及良好的化学选择性,为人们提供了一个可靠的替代传统氧化还原的新方法,充分展示了其在当今有机合成领域中的重要地位以及突出贡献,彰显了有机光化学这一领域的巨大创造力,为有机新反应发展、新物质的合成注入了新的活力。因此,通过绿色可持续的光氧化还原生成外部活性自由基,进而促进1, n-烯炔的自由基串联反应是一种可行的选择,也受到越来越多化学工作者的关注(图式 1)。本文综述了2013年以来一些研究组在可见光诱导的1, n-烯炔的自由基串联反应研究中取得的进展。
碳中心自由基促进的1, n-烯炔串联反应类型比较丰富,碳中心自由基的来源也多种多样。其来源可以是磺酰氯[5]、卤代酸酯[6, 10, 17]、酰氯[7]、三氟甲基亚磺酸钠[8]、二氯甲烷[9]、二甲基亚砜[11]等。由碳中心自由基促进的1, n-烯炔的串联反应,其一般反应机理为:在可见光的照射下,光催化剂由基态转变为激发态,并产生相应的自由基;接着,产生的自由基与1, n-烯炔发生加成环化,得到相应的产物。下面按时间顺序对碳中心自由基促进的1, n-烯炔串联反应进行概述。
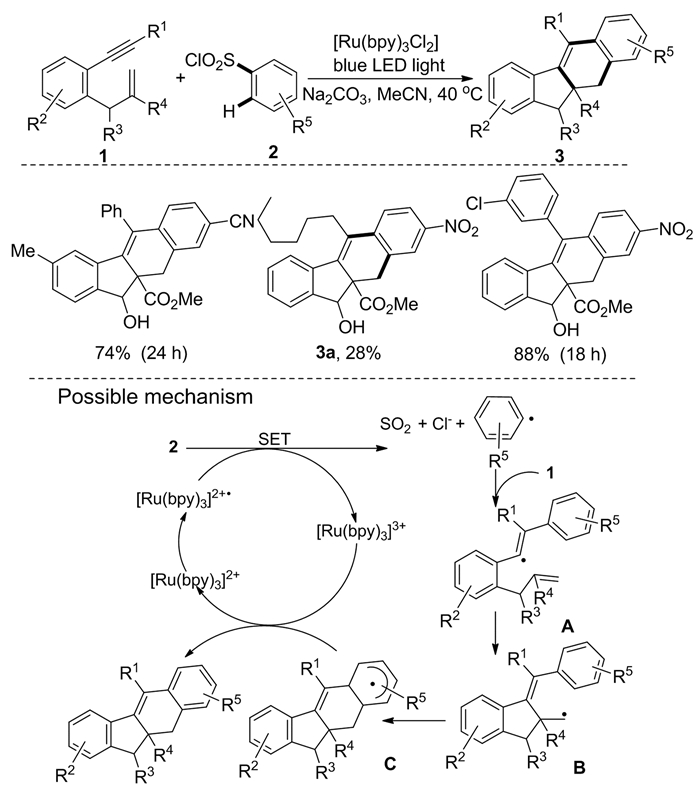
2013年,Deng等[5]利用1, 6-烯炔类化合物1作为底物与芳基磺酰氯2反应,在可见光照下得到了化合物3。优化反应条件为,Ru(bpy)3Cl2为催化剂、乙腈为溶剂,40℃下反应。值得注意的是,脂肪族炔能成功进行5-外环化(产物3a)和6-外环化。该反应的机理为:首先,芳基磺酰氯由激发态[Ru(bpy)3]2+·的单电子转移形成芳基自由基,随后芳基自由基与化合物1的碳碳叁键作用得到中间体A;A进而与化合物1的碳碳双键进行环加成得中间体B;B通过分子内环化得中间体C;最后,中间体C通过[Ru(bpy)3]3+的氧化及去质子化得化合物3(图式 2)。
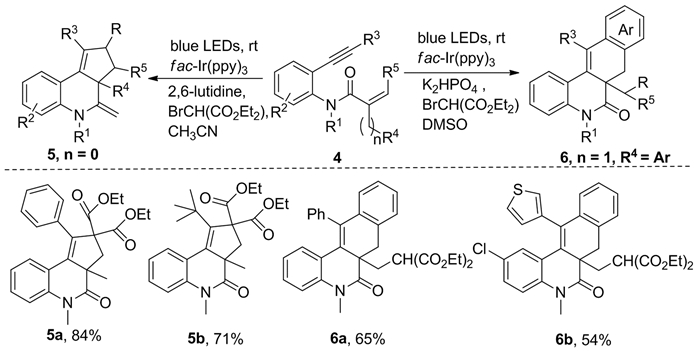
随后,Gao等[6]发现溴代丙二酸二乙酯作为碳中心自由基前体在添加剂2, 6-二甲基吡啶和光敏剂fac-Ir(ppy)3存在下,经蓝光照射可发生1, 7-烯炔的自由基串联反应,合成多环化合物5。此外,如果以芳基取代的1, 7-烯炔类为底物,可通过适当调整反应条件来合成化合物6(图式 3)。这种合成方法因操作简单、官能团耐受性良好、在温和的条件下具有高收率而备受关注。
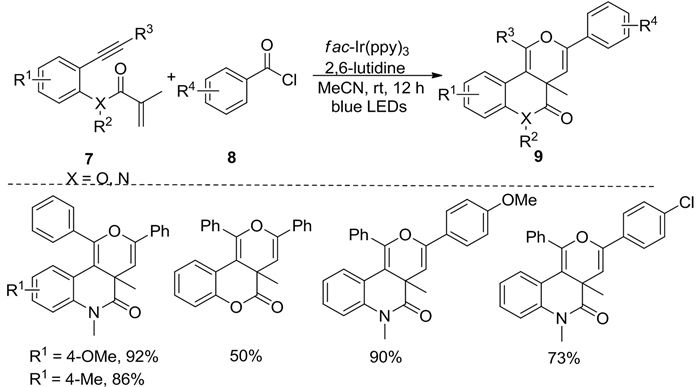
2017年,Li等[7]利用苯甲酰氯为酰基自由基源,在光催化下实现了1, 7-烯炔化合物7的串联环化反应,高效合成吡喃或吡啶类化合物9(图式 4)。当炔烃末端的取代基为含有甲氧基和甲基等给电子取代基的芳香环时具有良好产率。此外,当苯甲酰氯芳环上的取代基为富电子基团时产率较高,为缺电子基团时则得到中等产率。
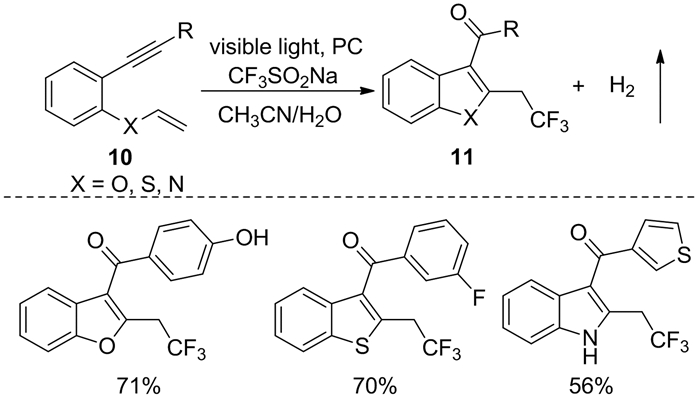
同年,Jana等[8]通过光氧化还原实现了1, 6-烯炔化合物10的氧-三氟甲基化反应[8],该反应以CF3SO2Na为三氟甲基自由基源、菲-9, 10-二酮(PQ)为光敏剂、水作为氧源,在温和条件下分别得到了三氟甲基苯并呋喃类、噻吩类和吲哚类化合物11(图式 5)。
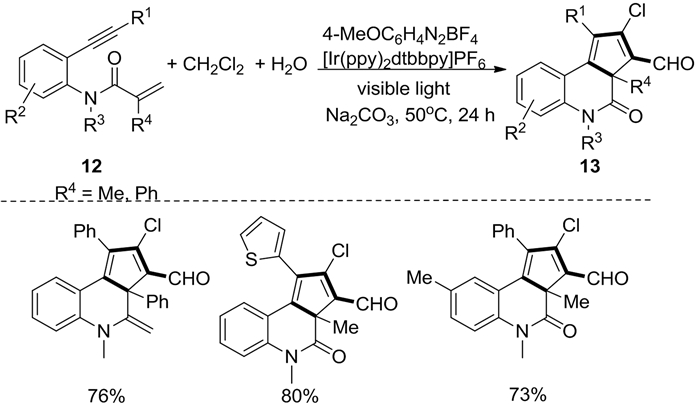
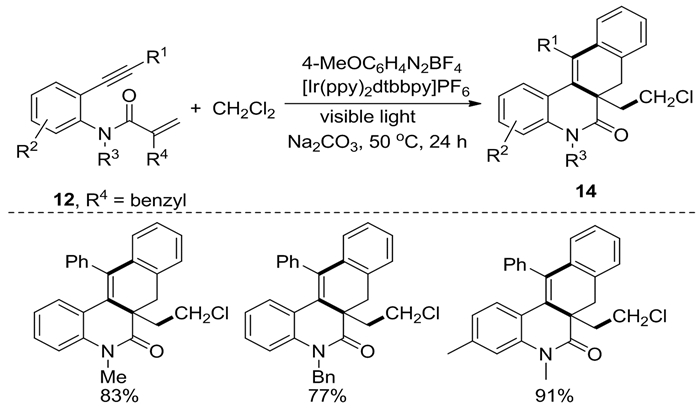
2018年,Liu等[9]以N-(邻乙炔基芳基)丙烯酰胺12为底物、CH2Cl2作为一碳单元,在芳香重氮盐、[Ir(ppy)2dtbbpy]PF6和可见光的作用下,通过多个C-Cl/C-H功能化和[2+2+1]串联环化反应合成了环戊[c]喹啉-4(5H)酮类化合物13(图式 6)。该反应对末端炔芳基上的Me、MeO、F、Cl、Br和CN等取代基表现出良好的官能团耐受性。此外,该体系还适用于由2-苄基取代的N-(邻乙炔基芳基)丙烯酰胺来合成苯并[j]菲啶-6(5H)-酮类化合物14(图式 7)。该转化以二氯甲烷为自由基源,具有很高的挑战性,在文献报道中极为少见。
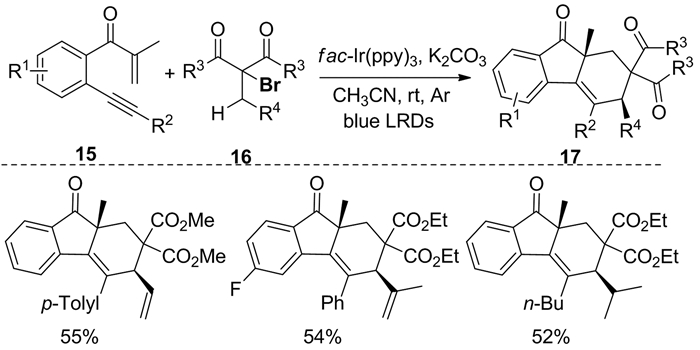
同年,Shen等[10]建立了一种新的可见光催化双环化串联反应,由易得的β-炔基丙酮15与α-溴代丙二酸二乙酯16反应,得到顺式芴-9-酮17(图式 8)。不过,该转化的效率不高,多数底物只能得到中等产率。
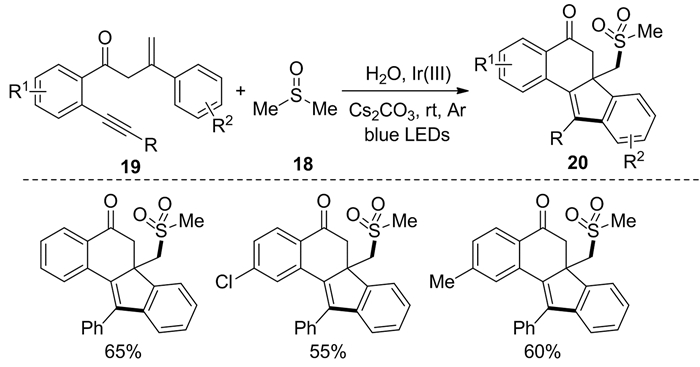
2018年,Huang等[11]报道了二甲基亚砜和1, 7-烯炔类化合物19在光催化的作用下合成甲磺酰化苯并[a]芴-5-酮20的反应(图式 9)。在此反应中,罕见地以二甲基亚砜作为碳自由基源,在光催化下实现了烯炔的环化过程,可一步构建多种S-O/C-S/C-C键。总的来说,该光催化反应底物适用范围广,环化效率较高,反应条件温和。
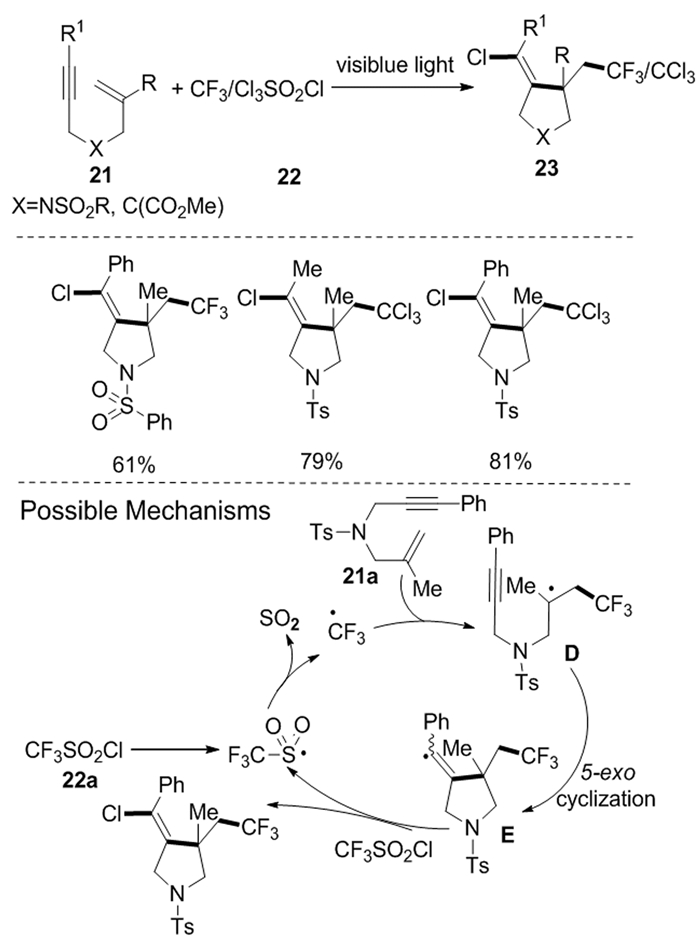
随后,Hou等[12]报道了可见光驱动的氯三氟甲基和氯三氯甲基化的烯炔环化反应。在光照下,首先产生三氟甲基和三氯甲基自由基,随后与1, 6-烯炔类化合物21的碳碳双键结合,得到中间体D,随后发生分子内自由基环化形成乙烯基自由基中间体E,最后,乙烯基自由基E和CF3SO2Cl(化合物22)发生氯原子转移反应得到了氯三氟甲基化和氯三氯甲基化吡咯烷类化合物23(图式 10)。
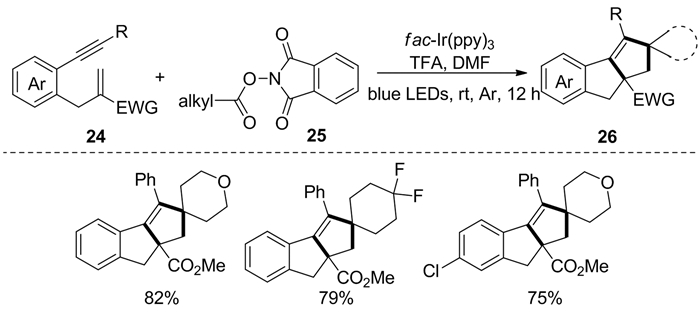
2019年,Jiao等[13]发现了一种底物范围广、反应条件温和、对官能团的耐受性极佳的光催化串联反应。该反应由N-羟基邻苯二甲酰亚胺酯25在光催化下脱羧形成烷基自由基,随后与1, 6-烯炔类化合物24经[2+2+1]串联环化反应生成环戊[a]茚和芴类似物26(图式 11)。体系中酸性添加剂能有效提高该反应的效率。此外,基于膦/碘化物的可见光介导的系统也可以用于该反应。
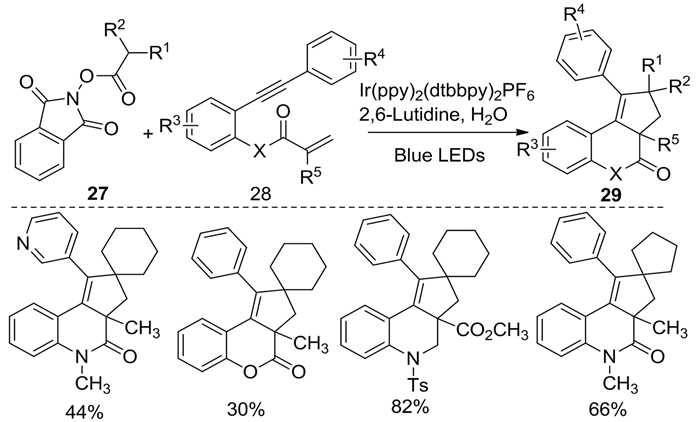
同年,上述类似的过程也被Silva等[14]公开报道。该转化也是由N-(酰氧基)邻苯二甲酰亚胺的在光催化下脱羧生成烷基自由基,随后诱导1, 7-烯炔类化合物发生串联环化反应,合成骨架多样的螺环戊酮[c]喹诺酮类化合物29(图式 12)。在此反应条件下,无论炔烃末端为吸电子基团或给电子基团均可获得良好的收率,表现出底物适用范围广和较高的官能团耐受性等优点。
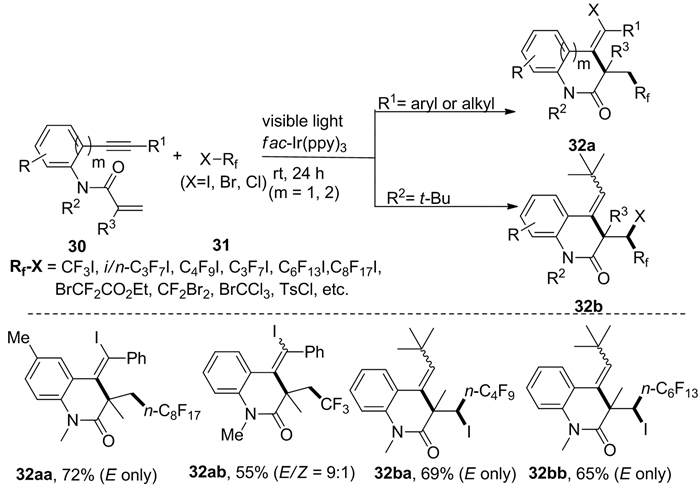
紧随其后,Wang等[15]报道了可见光诱导1, n-烯炔30(n=6, 7)的自由基加成与环化反应,可以合成卤代全氟氮杂环化合物32(图式 13)。值得一提的是,该反应不仅可以用全氟烷基卤化物31作为碳中心自由基源,其他多种全氟烷基卤化物(例如CF3I、CF2Br2、i/n-C3F7I、n-C4F9I等)均能参与反应,以高的顺反选择性实现全氟烷基和卤素基团的同时引入。
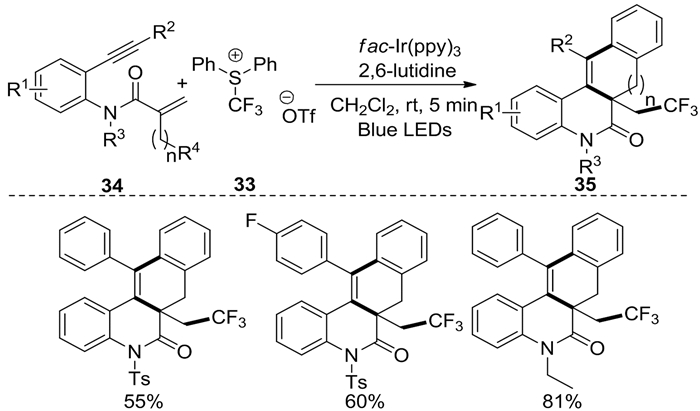
2020年,Zhuang等[16]使用一种容易制备的三氟甲基化试剂33在可见光诱导下完成了1, 7-烯炔34的三氟甲基化/环化反应,合成了大量CF3取代的苯并[j]菲啶和茚并[1, 2-c]-喹啉衍生物35(图式 14)。该反应表现出产率高、操作简单、官能团耐受性好等特点。
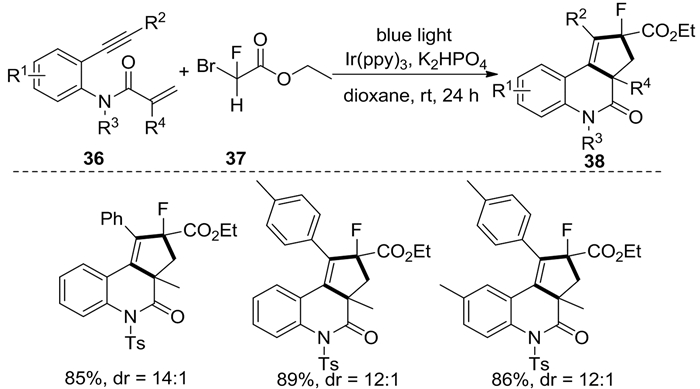
随后,Qu等[17]开发了可见光诱导的1, 7-烯炔36与氟溴乙酸乙酯37合成稠合的单氟化环戊[c]喹啉-4-酮衍生物38的[2+2+1]碳环化反应(图式 15)。当以Ir(ppy)3为催化剂、K2HPO4为碱、二氧六环为溶剂时,能得到高的产率和非对映选择性。该反应转化效率高,对于多种取代基的底物都能获得高的产率,且非对映选择性得到很好的保持。
上述碳中心自由基促进的1, n-烯炔串联反应中,碳自由基的来源很多样,实现的反应类型也很丰富。而硫中心自由基促进的1, n-烯炔串联反应类型则相对较少,硫自由基的来源主要集中在磺酸[18]、磺酰氯[19, 20]以及硫酚[21]等含硫化合物。对于硫中心自由基促进的1, n-烯炔串联反应,其反应机制一般为:首先,磺酸等含硫化合物在可见光照射下生成磺酰自由基;接着,磺酰自由基与1, n-烯炔发生一次或两次加成环合即可得对应产物。
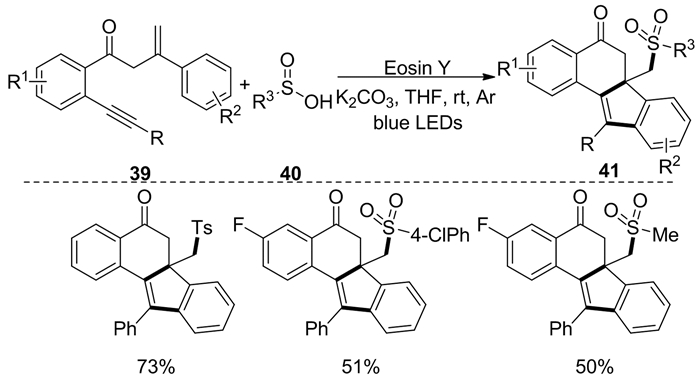
2017年,Huang等[18]报道了可见光催化1, 7-烯炔39与亚磺酸40的自由基环化反应,用于合成含砜的苯并[a]芴-5-酮41(图式 16)。在该反应中无论是使用亚磺酸或者磺酰氯,都可获得对应的环化产物。另外,该反应使用的光催化剂是曙红,它是一类有机染料,相对于金属光催化剂来说更廉价。总的来说,这种不含金属的方案底物适用范围广、官能团耐受性高、环化效率高且反应条件温和。
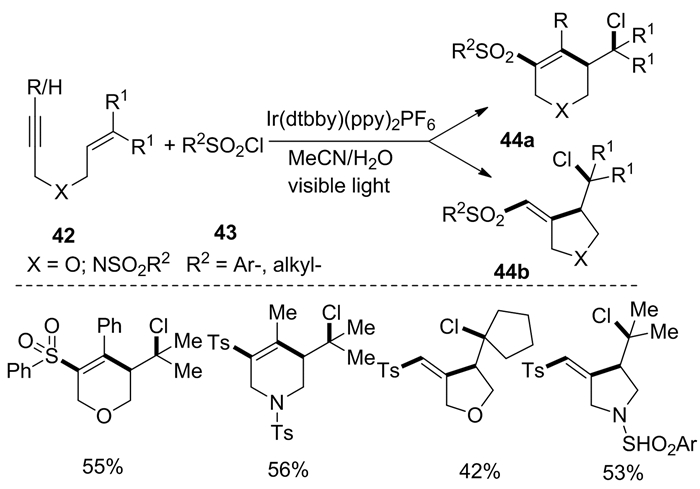
2018年,Hou等[19]发现了可见光介导的1, 6-烯炔42的氯-磺酰环化反应。以磺酰氯化物43作为自由基来源、[Ir(dtbbpy)(ppy)2]PF6(1.0(mol)%)为催化剂、MeCN和水为共溶剂,可以合成环状化合物44(图式 17)。该反应条件下,无论是氧联接的烯炔底物还是氮联接的烯炔底物,都能获得环化产物。不过,该反应的整体转化率为中等水平。
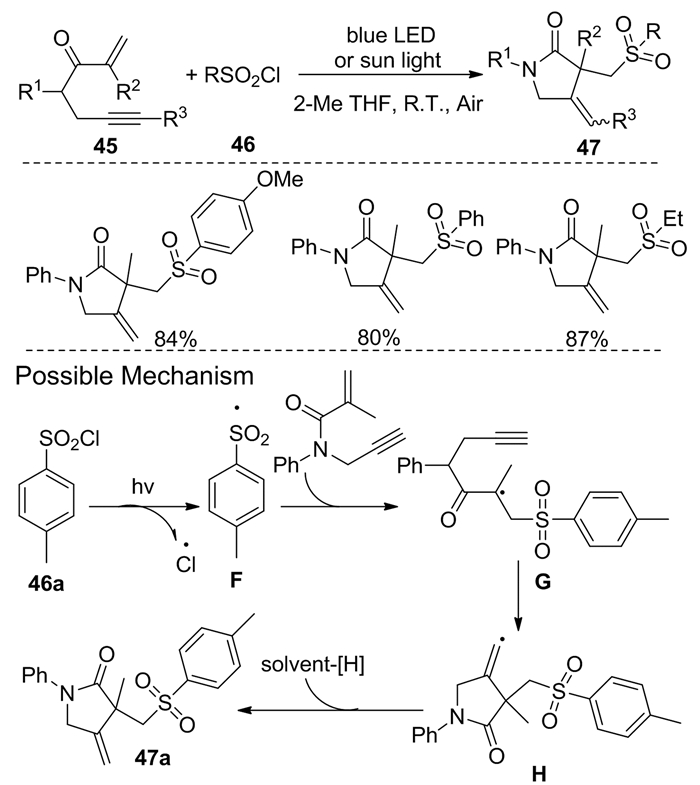
2020年,Meng等[20]在不使用催化剂和添加剂的情况下,开发了一种经济、环保和实用的策略,在磺酰氯化合物46和可见光的作用下,将1, 6-烯炔45进行区域选择性磺酰化,合成化合物47。其反应机理为:首先,在可见光照射下,磺酰氯化合物46a的S—Cl键进行选择性裂解形成磺酰自由基F。然后生成的磺酰自由基F向45a的碳碳双键进行区域选择性加成生成自由基G,进而与碳碳叁键进行环化生成乙烯基自由基H。最后,乙烯基自由基H从溶剂中提取氢以获得所需产物47a(图式 18)。该反应的整体产率较高,底物适用范围也较广。
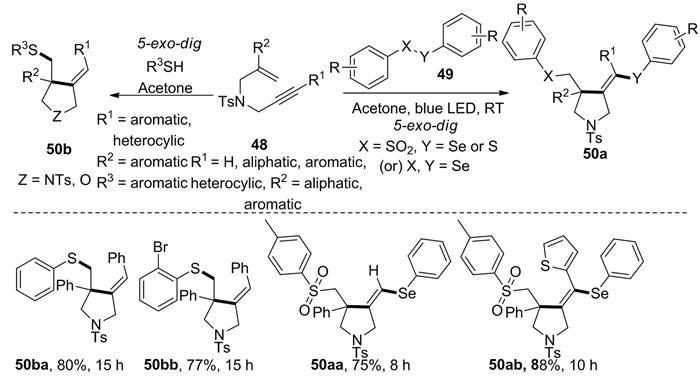
同年,Mutra等[21]在无金属和氧化剂存在情况下实现了非活化1, 6-烯炔48与硫族化合物的区域选择性自由基环化,合成了多取代吡咯烷衍生物50(图式 19)。有趣的是,当使用硫酚作为自由基源时,在非光照条件就能发生环化反应得到化合物50b;当使用化合物49作为自由基源时,则在光照下发生自由基环化反应生成化合物50a。该反应对各种类型的官能团均有良好的耐受性,其反应条件温和,对环境友好,不会产生有毒物质。
综上所述,光催化的1, n-烯炔的串联环化反应为复杂化合物的合成带来新的机遇,而且反应体系清洁,化学选择性好。然而,可见光诱导的1, n-烯炔的串联环化反应仍然存在一些局限,仍有较多问题需要解决,例如,烯炔底物的适应范围和自由基的多样性需要进一步提升以及需要开发不对称版本的可见光诱导的1, n-烯炔串联反应等。期待随着研究的深入,可见光诱导的1, n-烯炔串联反应为复杂分子的合成带来更多的惊喜。
Xuan J, Studer A. Chem. Soc. Rev., 2017, 46: 4329~4346. doi: 10.1039/C6CS00912C
Li Y, Pan G A, Luo M J, et al. Chem. Commun., 2020, 56: 6907~6924. doi: 10.1039/D0CC02335C
Xuan J, Xiao W J. Angew. Chem. Int. Ed., 2012, 51: 2~13. doi: 10.1002/anie.201107584
Prier C K, Rankic D A, Macmillan D W, et al. Chem. Rev., 2013, 113: 5322~5363. doi: 10.1021/cr300503r
Deng G B, Wang Z Q, Xia J D, et al. Angew. Chem. Int. Ed., 2013, 52: 1535~1538. doi: 10.1002/anie.201208380
Gao F, Yang C, Ma N, Gao G L, et al. Org. Lett., 2016, 18: 600~603. doi: 10.1021/acs.orglett.5b03662
Li C G, Xu G Q, Xu P F. Org. Lett., 2017, 19: 512~515. doi: 10.1021/acs.orglett.6b03684
Jana S, Verma A, Kadu R, et al. Chem. Sci., 2017, 8: 6633~6644. doi: 10.1039/C7SC02556D
Liu Y, Song R J, Luo S L, et al. Org. Lett., 2018, 20: 212~215. doi: 10.1021/acs.orglett.7b03561
Shen Z J, Shi H N, Hao W J, et al. Chem. Commun., 2018, 54: 11542~11545. doi: 10.1039/C8CC06086J
Huang M H, Zhu C F, He C L, et al. Org. Chem. Front., 2018, 5: 1653~1650.
Hou H, Tang D L, Li H X, et al. J. Org. Chem., 2019, 84: 7509~7517. doi: 10.1021/acs.joc.9b00842
Jiao M J, Liu D, Hu X Q, et al. Org. Chem. Front., 2019, 6: 3834~3838. doi: 10.1039/C9QO01166H
Correia J T M, Silva G P D, Andre E S, et al. Adv. Synth. Catal., 2019, 361: 1~8. doi: 10.1002/adsc.201801478
Wang S W, Yu J, Zhou Q Y, et al. ACS Sustain. Chem. Eng., 2019, 7, 10154~10162. doi: 10.1021/acssuschemeng.9b02178
Zhuang K Q, Cui Y S, Yuan X, et al. ACS Sustain. Chem. Eng., 2020, 8: 11729~11736. doi: 10.1021/acssuschemeng.0c03745
Qu Y, Xu W T, Zhang J J, et al. J. Org. Chem., 2020, 85: 5379~5389. doi: 10.1021/acs.joc.0c00087
Huang M H, Zhu Y L, Hao W J, et al. Adv. Synth. Catal., 2017, 13: 2229~2234.
Hou H, Li H X, Xu Y, et al. Adv. Synth. Catal., 2018, 22: 4325~4329.
Meng X X, Kang Q Q, Zhang J Y, et al. Green Chem., 2020, 22: 1388~1392. doi: 10.1039/C9GC03769A
Mutra M R, Kudale V S, Tsai W, et al. Green Chem., 2020, 22: 2288~2300. doi: 10.1039/D0GC00321B

图式 1 可见光诱导的1, n-烯炔的自由基串联反应
Scheme 1 Visible-light-induced radical cascade cyclization of 1, n-enynes

图式 3 α-溴代丙二酸二乙酯与1, 7-烯炔的环化反应
Scheme 3 Cyclization reaction of α-bromomalonic acid diethyl ester with 1, 7-enynes

图式 4 1, 7-烯炔与酰基氯化物的光催化串环化反应
Scheme 4 Photocatalytic cascade cyclization of 1, 7-enynes and acyl chloride

图式 6 N-(邻乙炔基芳基)丙烯酰胺的串联环化反应
Scheme 6 Tandem carbocyclization of the N-(o-ethynylaryl)acrylamides

图式 7 2-苄基取代的N-(邻乙炔基芳基)丙烯酰胺的串联环化反应
Scheme 7 Tandem carbocyclization of the 2-benzyl-N- (o-(arylethynyl)aryl)acrylamides

图式 10 可见光驱动三氟甲基和富氯甲基环化反应
Scheme 10 Visible-light-driven chlorotrifluoromethylative and chlorotrichloromethylative cyclizations of enynes

图式 11 1, 6-烯炔与N-羟基邻苯二甲酰亚胺酯光催化脱羧[2+2+1]环化合成反应
Scheme 11 Photocatalytic decarboxylation [2+2+1] annulation of 1, 6-enyne and N-hydroxyphthalimide

图式 12 1, 7-烯炔的光氧化还原脱羧烷基化反应
Scheme 12 Photoredox decarboxylative alkylation of 1, 7-Enynes

图式 13 全氟烷基卤化物与1, n-烯炔的原子转移自由基加成和环化反应
Scheme 13 Atom transfer radical addition and cyclization of perfluoroalkyl halides with 1, nenynes

图式 15 1, 7-烯炔与氟溴乙酸乙酯的[2+2+1]环化反应
Scheme 15 [2+2+1] Carbocyclization reactions of 1, 7-enynes with ethyl bromofluoroacetate

图式 16 1, 7-烯炔合成苯并[a]芴-5-酮的光催化双环化反应
Scheme 16 Photocatalytic bicyclization of 1, 7-enynes for the synthesis of benzo[a]fluoren-5-ones

图式 18 可见光引发1, 6-烯炔的区域选择性磺酰化/环化反应
Scheme 18 Visible-light-initiated regioselective sulfonylation/ cyclization of 1, 6-enynes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:






