引用本文:
裴晓燕, 李志勇, 史云磊, 曹新华. 环境响应型纳米粒子在不互溶两相间的相转移[J]. 化学通报,
2021, 84(3): 204-214.

Citation: Xiaoyan Pei, Zhiyong Li, Yunlei Shi, Xinhua Cao. Phase Transfer of Environmental Responsive Nanoparticles between Two Immiscible Liquid Phases[J]. Chemistry, 2021, 84(3): 204-214.
Citation: Xiaoyan Pei, Zhiyong Li, Yunlei Shi, Xinhua Cao. Phase Transfer of Environmental Responsive Nanoparticles between Two Immiscible Liquid Phases[J]. Chemistry, 2021, 84(3): 204-214.
环境响应型纳米粒子在不互溶两相间的相转移
摘要:
纳米粒子在不互溶两相间的相转移在催化剂的循环利用、药物输送等领域发挥着重要的作用。环境响应性纳米粒子因兼具纳米粒子的优点和刺激响应的特性而受到了广泛的关注。环境响应型纳米粒子相转移的出现使相转移过程更为高效、可逆且智能化,已展现出了广阔的应用前景。本文综述了近年来环境响应型纳米粒子在不互溶两相间转移的研究进展,主要内容包括pH、CO2、温度、光照、离子强度、配体交换和离子交换等刺激诱导的纳米粒子的相转移及其在催化、反应分离耦合等方面的应用,并分析了在环境刺激过程中纳米粒子的界面效应、自组装行为以及溶剂化效应等对相转移过程起到的关键性作用;同时,对目前该领域所面临的主要问题和进一步的研究工作提出了建议。
English
Phase Transfer of Environmental Responsive Nanoparticles between Two Immiscible Liquid Phases
Abstract:
Phase-transfer processes of nanoparticles between two immiscible liquid phases play an important role in many aspects such as recycling of catalysts, drug delivery and preparation of nanoparticles. Environmental responsive nanoparticles have been widely developed because of their advantages of nanoparticles and stimuli-responsive properties. The phase transfer of environmental responsive nanoparticles makes the phase transfer process more efficient, reversible and intelligent, which has shown a broad application prospect. In this paper, the recent progress in the phase transfer of environmental responsive nanoparticles between two immiscible phases is reviewed. The main contents include the phase transfer of nanoparticles triggered by stimuli such as pH, CO2, temperature, light, ionic strength, ligand exchange and ionic exchange, and their applications in sustainable catalysis and reaction separation coupling. The key influence of interface effect and self-assembly behavior of nanoparticles and solvation effect on the phase transfer process is analyzed. At the same time, the main problems and further research work in this field are proposed.
-
Key words:
- Stimuli-responsive
- / Phase transfer
- / Interface effect
- / Self-assembly behavior
- / Solvation effect
-
纳米粒子由于具有独特的物理化学性质,在传感[1, 2]、催化[3, 4]、生物成像[5, 6]、能量转换[7, 8]等领域展现出广阔的应用前景。随着纳米科学的不断发展,人们能够根据所需尺寸、形状及组成等进行纳米粒子的设计与制备[9, 10]。然而,其中的许多合成路线仅在特定介质中可用,利用这些方法制备的纳米粒子在不同极性介质中分散性的差异限制了其更广泛的应用。例如,表面包覆有疏水性稳定剂的纳米粒子可以分散在有机溶剂中使用,但不能在水中分散和使用,反之亦然。因此,纳米粒子在不互溶两相间的相转移显得十分重要。尤其是在纳米粒子的催化应用中,由于避免了催化剂不可逆聚集,纳米粒子的相转移过程对催化剂的循环利用极为有利[11, 12]。因此,人们致力于理解和控制纳米粒子的相转移行为。
迄今为止已报道的纳米粒子相转移策略可分为两类:改变稳定剂的润湿性和改变相介质组成。这两种方法分别通过改变稳定剂或介质的性质来调控纳米粒子在不互溶液相之间的转移。然而,为了使相转移过程更为高效、可逆且智能化,环境响应型纳米粒子的相转移得以迅速发展,其转移过程是通过改变周围环境来调控纳米粒子亲水-疏水性而实现的,成为纳米粒子合成、功能化和应用的一个重要领域[13, 14]。本文主要总结了环境响应型纳米粒子相转移发展过程中的研究成果。在这一发展过程中,研究者通过pH、CO2、温度、光照、离子强度、配体交换和离子交换等刺激手段调控纳米粒子在不互溶两相间的转移,不仅实现了纳米粒子的智能化,而且对于充分发挥纳米粒子的潜在应用价值具有重要的意义。
1. 环境响应型纳米粒子相转移
环境响应型纳米粒子是指对周围环境变化敏感的粒子,在受到环境刺激后,纳米粒子表面的结构发生改变,导致其相关性质也发生变化。因兼具纳米粒子的优点和环境响应的特性,环境响应型纳米粒子的相转移一直是研究的热点。根据刺激信号的不同可以将环境响应型纳米粒子的相转移分为pH响应型、CO2响应型、温度响应型、光响应型、离子强度响应型、配体交换响应型、离子交换响应型等不同的类型。pH、CO2、温度等刺激手段与纳米粒子表面作用后会使其发生物理或化学变化,进而改变粒子的润湿性,使人们能够调控纳米粒子在不互溶两相之间的转移。
1.1 pH响应型
pH是一种常见且容易实现的外部刺激[15, 16],根据响应基团的不同,可以将pH响应纳米粒子分为酸类和碱类。这些纳米粒子表面含有大量的质子酸或碱性基团,如羧基或氨基等,通过调节体系的pH,pH响应官能团发生质子化/去质子化,从而改变纳米粒子的润湿性,实现纳米粒子亲水性-疏水性的转变,促使纳米粒子从一相转移到另一相。
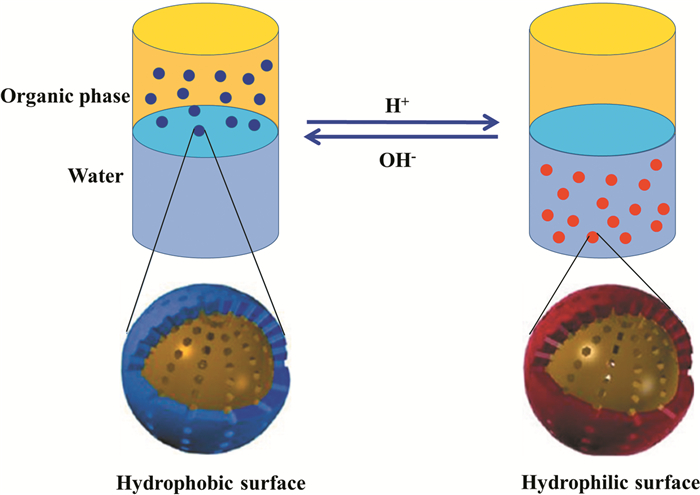
为了探索相转移过程在纳米粒子分离与回收利用方面的创新应用,Wang等[17]设计并制备了pH响应的辛基-三胺双功能化的介孔SiO2纳米材料。当调节水相pH为3~4时,SiO2穿过油/水界面,从有机相转移到水相(图 1);调节水相pH为9~10时,SiO2又从水相转移到有机相。针对上述现象,进一步的机理分析可知,SiO2表面的胺在酸性条件下被质子化,使SiO2变得亲水,这种亲水性降低了粒子与水之间的界面能,促使SiO2向水中迁移。在碱性条件下,SiO2去质子化并恢复疏水性,对有机物的亲和力以及溶剂化效应驱使粒子进入有机相。基于上述相转移策略,将SiO2作为载体负载Pd催化剂,用于催化醚/水体系中的苯乙烯加氢反应,并通过改变体系的pH,可以有效地实现产物与催化剂的分离以及对该类化学反应的高效调控。该方法不仅提供了一种性能可控的新型纳米复合材料,而且为纳米催化剂的原位分离与回收开辟了一条新的途径,可以有效避免离心、过滤等传统方法所遇到的障碍。
图 1
Kitchens等[18, 19]以十八胺为相转移剂,通过改变体系的pH,研究了巯基十一酸(MUA)和硫代聚丙烯酸(PAA-SH)功能化的金纳米粒子(AuNP-MUA、AuNP-SPAA)在水和氯仿之间的相转移。实验发现,分别向AuNP-MUA和AuNP-SPAA体系中加入HCl调节pH≈8,由于相转移剂十八胺发生了质子化,并与AuNPs表面的羧酸根形成离子对使AuNPs更具疏水性,导致粒子从水相转移到氯仿相。当调节体系pH ≈ 11时,离子对解离使得AuNPs恢复亲水性,又从氯仿相回到水相。将上述功能化AuNPs用于4-硝基苯酚的还原反应,并通过调节体系的pH,实现了AuNPs的循环利用。值得注意的是,MUA在AuNP表面覆盖率为100%时无催化作用,而部分覆盖保留了AuNPs的催化活性,但却降低了AuNP-MUA的回收率和稳定性,研究表明MUA覆盖率90%为较佳覆盖水平。对于AuNP-SPAA,增加聚合物主链上硫醇部分的数量可以极大地提高催化剂在苛刻还原环境下的稳定性,然而还原剂诱导配体从AuNPs表面的解吸会导致部分催化剂聚集并失去活性。
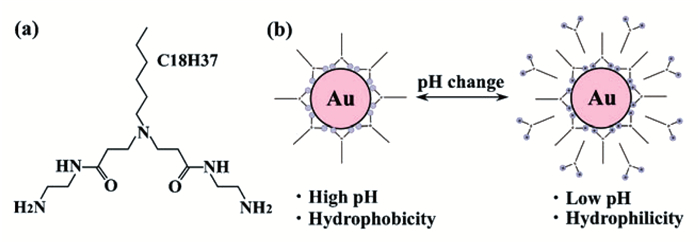
Imura等[20]报道了pH诱导长链胺衍生物(C18AA)负载的AuNPs在水和氯仿之间的相转移。当调节水相pH为13时,AuNPs从水相转移到氯仿中;调节水相pH为8时,粒子又从氯仿相转移到水中。通过调节体系的pH,AuNPs能够在水和氯仿之间可逆相转移至少6次。机理研究表明,该相转移过程可能是由C18AA末端胺基的质子化/去质子化促进的(图 2)。进一步分析可知,从水到氯仿相转移的第一阶段,AuNPs被C18AA分子包覆,且部分C18AA分子可能吸附在AuNPs表面的阴离子位点上,使得AuNPs在氯仿中形成稳定的分散体成为可能。当调整水相pH至8时,末端胺基被质子化,C18AA分子获得更高的亲水性,并从氯仿相转移到水相以达到新的平衡。C18AA分子的这种运动导致AuNPs从氯仿向水的转移。美中不足的是,该相转移过程需要较长时间才能达到平衡,而且纳米粒子在可逆转移时并不完全,这在一定程度上限制了其在更广泛领域内的应用和进一步的发展。
图 2

Zhao等[21]利用化学共沉淀法合成了亲水性的油酸钠包覆的CoFe2O4纳米粒子,研究了pH调控该纳米粒子在水和氯仿之间的相转移。当向体系中加入HCl调节水相pH=5时,CoFe2O4表面的油酸钠发生质子化转变为疏水性的油酸,导致CoFe2O4从水相转移到氯仿相,并可得到CoFe2O4的有机溶胶。加入三乙胺后,疏水性的CoFe2O4去质子化变为亲水性,又从有机相回到水相。该过程中,纳米粒子从水相转移到有机相得到的有机溶胶可用于形成自组装的纳米粒子层,而有机相转移到水相形成的水溶胶则适合于生物医学应用。
Gui等[22]以十二胺接枝的聚丙烯酸(PAA-g-DDA)两亲聚合物为模板,利用光还原策略合成了pH响应的PAA-g-DDA包覆的发光银纳米晶(L-AgNCs)。研究发现,当体系pH=2.5时,PAA-g-DDA中的羧酸根被中和,使得L-AgNCs表面变得疏水并分散在氯仿中。调节体系pH=8.0,PAA-g-DDA中的羧基去质子化,导致L-AgNCs表面存在大量负电荷并由疏水性转变为亲水性,进而从氯仿相转移到水中。从这一现象可以发现,pH对L-AgNCs在不同极性溶剂中溶解度的调控促使其从水相转移到氯仿相。基于这一设计可进一步开发成像试剂或治疗性纳米粒子,并有望用于可通过微小pH变化控制溶解度的功能纳米晶的合成和应用。
Peng等[23]报道了pH诱导4-氨基苯硫酚包覆的AuNPs在水和甲苯之间的相转移。实验过程中,通过将水相的pH由3.0增大到7.0,并剧烈搅拌,4-氨基苯硫酚包覆的AuNPs能够从水相转移到甲苯相。相转移前后粒子之间的唯一区别在于AuNPs表面电荷的不同。在pH≈3.0的水中,AuNPs表面的氨基发生质子化,形成了高电荷密度的粒子表面,使AuNPs具有亲水性,降低了其与水之间的界面能,因而很好地分散在水中;当水相pH≈7.0时,AuNPs表面的氨基去质子化,AuNPs表现为疏水性,对甲苯的亲和力驱使AuNPs转移到甲苯相。
需要指出的是,pH作为一种有用的刺激手段,可以在较宽的范围内调控纳米粒子的相转移。然而,该过程中酸碱中和反应产生的无机盐会导致盐的累积,对体系造成污染,这不利于pH响应体系更广泛的应用。
1.2 CO2响应型
与pH相比,常压CO2作为环境刺激的“开关”具有特殊的优越性和重要性[11, 24],其优点如下:(1)无毒、无腐蚀性、含量丰富、价格便宜、易于除去且在体系中不积累;(2)不需要特殊的高压装置;(3)反应生成的铵盐能够循环利用,不会因为高浓度盐的产生而引起水环境污染的问题。因此CO2是一种有优势的刺激手段。
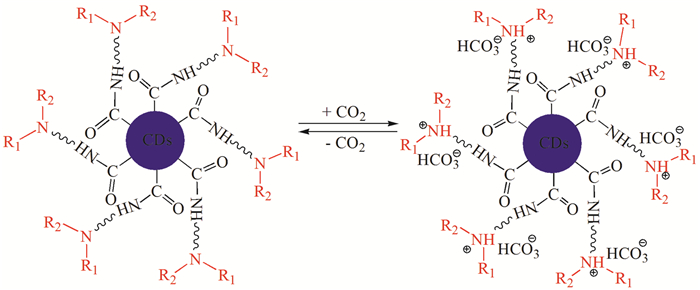
笔者课题组[25]利用水热法合成了CO2响应的疏水性胺基功能化碳量子点(CDs),研究了CO2驱动CDs在水和有机溶剂之间的相转移。当向体系中鼓入CO2,CDs从有机相转移到水相。之后,鼓入N2,CDs又从水相回到有机相。通过交替地鼓入CO2/N2,CDs能够在水和有机溶剂之间可逆地相转移。进一步的机理分析表明,CDs表面的胺基与CO2发生可逆反应,引发了CDs疏水性-亲水性的转变,导致CDs的相转移(图 3)。该相转移策略可用于Knoevenagel等碱催化的反应,并通过交替鼓入CO2/N2,实现均相反应、异相分离、CDs循环利用的有效耦合以及对该类化学反应的高效调控。
图 3

Xiong等[26]通过对富含胺的CDs进行改性,制备了脒基功能化的CDs,并对CO2驱动该CDs相转移以及相应的发光变化进行了研究。CDs最初只能分散在有机相(甲苯、正辛醇等),鼓入CO2后,由于脒基与原位生成的H+反应在CDs表面形成了离子部分,使CDs变得亲水并从有机相转移到水相。同时,原位生成的H+还可以改变CDs表面的发射态,使其在相转移过程中出现相应的由蓝色到青绿色的发光变化。进一步鼓入N2以完全除去原位生成的H+,CDs的表面化学和发射状态得以恢复,又从水相转移到有机相,并出现相应的从青绿色到蓝色的发光变化。这种智能纳米粒子的出现可以极大地改善发光纳米粒子在传感、油墨和细胞示踪等领域的应用,为开发通过表面态控制发光行为的其他智能发光材料提供参考。
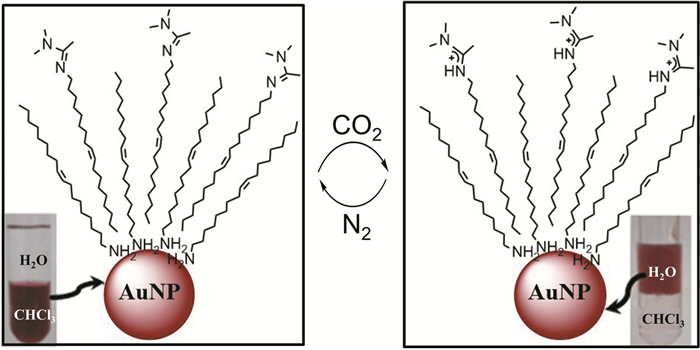
Pocoví-Martínez等[27]报道了在N′-(9-十八烯基)-N, N-二甲基乙脒(OAA)存在下,油胺包覆的AuNPs在水和氯仿之间的相转移。OAA以壳层形式自组装在油胺包覆的AuNPs表面,使得AuNPs很好地分散在氯仿中。当向体系中鼓入CO2,OAA的脒基与CO2发生反应生成了亲水性的碳酸氢盐,导致AuNPs变得亲水,因此从氯仿相转移到水相。鼓入N2后,OAA恢复疏水性,AuNPs又从水中转移到氯仿相。通过向体系中交替地鼓入CO2或N2,实现了油胺包覆的AuNPs在水和氯仿中的可逆相转移(图 4)。然而,在没有OAA的情况下,即使向体系中鼓入CO2,AuNPs也不能从有机相转移到水相;而且,纳米粒子在两相之间转移所需的时间随着循环次数增加而增加,成为它实际应用的不利因素。
图 4

Kim等[28]设计并制备了叔胺功能化的共轭聚合物纳米粒子,研究了其对CO2响应的特性。当向含有共轭聚合物纳米粒子的水-正辛醇体系中鼓入CO2,聚合物侧链上的胺与体系中生成的碳酸发生反应,导致该粒子变得亲水并均匀地分散在水中。随后,鼓入N2移除CO2,聚合物纳米粒子去质子化又恢复原来的状态,并从水相转移到正辛醇中。在该过程中,由于共轭聚合物纳米粒子中的胺基与CO2发生可逆反应,引发了该纳米粒子亲水性-疏水性的转变,导致相转移的发生。
值得一提的是,虽然这些有用的策略不能减少CO2的排放,但是通过循环利用废气中的CO2气体,使其变得有用,这对实现社会的可持续发展有重要的意义。
1.3 温度响应型
温度是一种研究较广且容易控制的环境刺激手段。温度刺激不会直接影响体系的化学组成,因此受到了研究者的广泛关注。温敏型纳米粒子可以通过在纳米粒子表面或内部引入具有上临界溶解温度(UCST)或下临界溶解温度(LCST)的物质或官能团制备而成,其显著特点是微小的温度变化会引起粒子结构可逆性的变化。目前,已报道的温敏型纳米粒子中研究较多的是LCST型粒子。当体系温度低于其LCST时,纳米粒子中的亲水基团与水形成的氢键起关键作用,纳米粒子表现为亲水性;当温度高于其LCST时,氢键作用被破坏,纳米粒子的疏水作用增强,导致其在水和有机相之间转移[16]。
Zhao等[29]首先报道了温度驱动LCST型聚(甲基丙烯酸甲氧基三(乙二醇)酯)(PTEGMMA)功能化的SiO2纳米粒子在水和乙酸乙酯中的相转移(图 5),且该过程是定量的、可逆的并可以重复多次。随后,他们[30]又研究了温度诱导温敏性聚合物刷[聚甲基丙烯酸(甲氧基聚(乙二醇)酯)(PPEGMMA)、PTEGMMA、聚甲基丙烯酸甲氧基二(乙二醇)酯(PDEGMMA)以及聚甲基丙烯酸乙二醇酯与甲基丙烯酸甲氧基三(乙二醇)酯共聚物(P(PEGMMA-co-TEGMMA)-82和P(PEGMMA-co-TEGMMA)-74)]接枝的SiO2在水和疏水性离子液体1-乙基-3-甲基咪唑双(三氟甲磺酰亚胺)([EMIM][TFSI])之间的相转移。研究表明,PPEGMMA接枝的SiO2的相转移过程是一个熵驱动的过程,受水中温敏性聚合物LCST控制。对于P(PEGMMA-co-TEGMMA)-82、P(PEGMMA-co-TEGMMA)-74和PTEGMMA接枝的纳米粒子,在体系加热到70℃以及冷却至0℃时也能够在水和[EMIM][TFSI]之间可逆地转移。然而,PDEGMMA接枝的纳米粒子在加热时虽然也能够自发地从水相转移到离子液体中,但是降低温度时,粒子却不能回到水中。这些含有低聚乙二醇侧链重复单元的热敏性聚合物在水中LCST的跃迁主要取决于其与水形成氢键产生的负ΔH值以及聚合物链周围形成的更有序的结构水引起的负ΔS值。因此,选用合适的热敏性聚合物或调节两种或多种不同单体的共聚,将有助于我们设计具有所需转移温度的能够在水和离子液体之间转移的热驱动粒子。
图 5

Qin等[31]研究了温度驱动2-(N, N-二乙基氨基)乙硫醇(DEAET)稳定的CdTe纳米粒子在水和甲苯中的相转移。结果发现,当体系温度为0℃时,CdTe很好地分散在甲苯中;升高温度至70℃,CdTe几乎全部转移到水中(图 6)。之后,将体系温度降为0℃,CdTe又从水相回到甲苯相。因此,该转移过程是可逆的,且可以重复多次而不会显著改变纳米粒子光致发光峰的强度和位置。理论研究表明,与DEAET分子不同,DEAET稳定的CdTe纳米粒子具有较大尺寸,在高温下这种尺寸足以破坏粒子周围水之间的氢键网络,从而增加其在水中的分散性。通过热力学模型的构建以及对体系混合自由能的计算,进一步表明,CdTe纳米粒子间及其与水之间的作用力(包括氢键、偶极引力、静电作用和净疏水力等)随温度的变化而变化,促使其在界面聚集和随后的相转移。这些发现有助于我们进一步了解纳米粒子独特的理化性质,并有利于其在不同极性溶剂中的应用。
图 6

Ma等[32]利用聚乙烯吡咯烷酮(PVP)在水中的溶解度随温度升高而降低的特性实现了温度诱导PVP稳定的金、银、铂纳米粒子在水和正丁醇之间的相转移。PVP稳定的金、银、铂纳米粒子最初分散在水相,升高体系温度到80℃时,由于PVP在水中溶解度的降低导致这些粒子从水相转移到正丁醇中。相转移过程中,因为PVP链的收缩和纳米粒子类流体的表面性质,金和银纳米粒子分别在容器壁上形成了大量的金纳米晶和致密的银粒子膜。由于铂纳米粒子没有类流体的性质,即使铂粒子之间存在聚集,也不会形成较大粒子。所以,铂纳米粒子的相转移过程与金和银粒子的相转移过程略有不同,没有在容器壁上形成铂纳米粒子膜,而是直接穿过正丁醇/水界面,从水相转移到正丁醇相,且保持原来的尺寸和形貌。需要指出的是,80℃时水与正丁醇部分互溶,这在一定程度上促进了纳米粒子的相转移。
上述体系均通过调节温度实现了纳米粒子在不互溶两相之间的转移,该过程不需要额外的添加剂,因此整个过程是清洁的,但是温度的变化需要能量的输入,能量的消耗会增加生产成本,使其在实际应用中受到一定限制。
1.4 光响应型
光作为一种“清洁”的环境刺激方法,其信号稳定且方便易得。由于光信号可以被准确定位,因此在纳米科学技术和药物控释等方面具有重要的意义[33~38]。当光照进行时,光敏性分子或基团在吸收光能量后会发生分子内或分子间结构的变化,进而表现出光响应性能。常见的感光基团主要包括偶氮苯、三苯基甲烷、螺吡喃等。光响应型纳米粒子中含有能够对外界光刺激(如紫外光、红外光等)做出快速响应的感光基团,这些基团在光照条件下会发生相应的物理或化学变化,使得纳米粒子的结构得以调控。
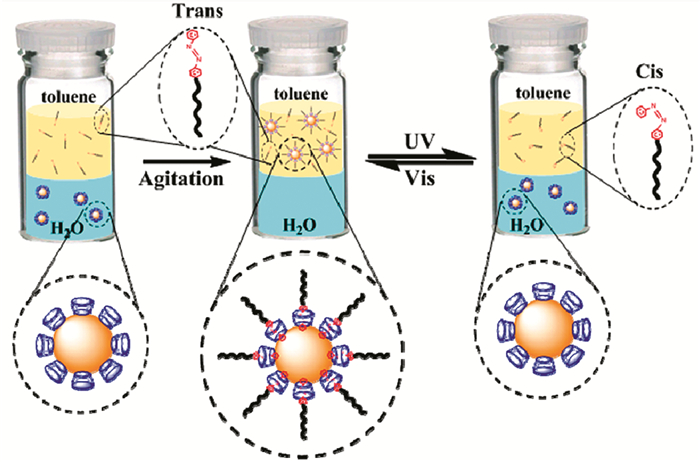
Peng等[39]利用偶氮苯类化合物与α-环糊精(α-CD)之间光可控的主客体化学反应,研究了α-CD包覆的AuNPs在水和甲苯之间的相转移。研究表明,AuNPs最初分散在水中,由于反式偶氮苯类化合物与AuNPs表面的α-CD结合,将疏水烷基链引入到粒子外围,形成反胶束结构,AuNPs从水相转移到甲苯中。在紫外光照射下,偶氮苯类化合物由反式异构体转变为顺式异构体,顺式偶氮苯类化合物不能与α-CD形成超分子结构,导致其从环糊精腔体内脱出,使得AuNPs恢复亲水性,从甲苯相转移到水相(图 7)。对上述体系进行可见光照射,顺式偶氮苯类化合物又可逆地转变为反式,AuNPs从水相又转移到甲苯相。通过对体系进行紫外/可见光的照射,偶氮苯类化合物可逆的光致异构化使得AuNPs能够在水和甲苯之间进行多次可逆的相转移。这种基于光控分子识别的可逆相转移方法可用于反应的调控及催化剂的回收利用,并为纳米粒子的表面修饰提供了新的思路,从而有利于提高和扩大纳米粒子在不同领域的应用。
图 7

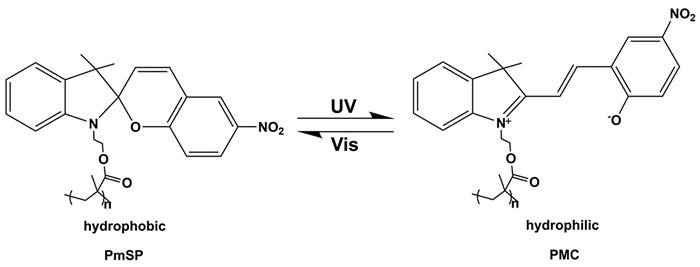
Wu等[40]通过将光敏性的聚甲基丙烯酸螺吡喃酯(PmSP)负载到SiO2表面制备了光响应的PmSP/SiO2复合胶体。研究发现,紫外光照射时,PmSP经历了C—O键的光化学裂解而形成部花菁(PMC)两性离子,并由疏水性转变为亲水性(图 8),导致PmSP/SiO2从甲苯相转移到水相。进一步使用可见光照射,PmSP重新形成且恢复疏水性,PmSP/SiO2从水相又回到甲苯中。在进行多次紫外/可见光的照射后,由于聚合物刷经历了疏水到亲水状态的可逆变化,PmSP/SiO2也实现了在甲苯相和水相之间可逆的转移。在照射的中间阶段,复合胶体的疏水性与亲水性组分共存,而具有两亲性。将该两亲性复合胶体作为乳化剂可用于形成Pickering乳液,在短时间的紫外光照射时,PmSP/SiO2是疏水性的,得到的Pickering乳液是W/O型的。长时间的紫外光照射后,该复合胶体变为亲水性的,得到O/W型Pickering乳液。
图 8

光源清洁、安全、易于使用和控制,如果将光响应型纳米材料作为催化剂或药物载体等,这些相转移策略则有望在相转移催化、药物输送等领域具有重要的应用。
1.5 离子强度响应型
离子强度是影响纳米粒子物化性质的关键环境因素,在一定的暴露条件下会引起纳米粒子的相转移。然而,高离子强度的溶液趋向于减弱纳米粒子之间的斥力,降低纳米粒子表面的双电层,最终导致纳米粒子的聚集。因此,选用合适的盐及一定的离子强度来驱动纳米粒子的相转移是十分必要的。
Wang等[41]通过将低聚(乙二醇)甲基丙烯酸甲酯(OEGMA)和2-(2-甲氧基乙氧基)甲基丙烯酸乙酯(MEO2MA)的均聚物与无规共聚物修饰到AuNPs表面,制备了离子强度响应型AuNPs。实验发现,向含有OEGMA均聚物修饰的AuNPs水分散液中加入NaCl,AuNPs自发地从水相转移到甲苯中。机理分析表明,这是由于AuNPs与盐水之间的界面能(2.1±0.8mN·m-1)大于其与甲苯之间的界面能(1.3±0.1mN·m-1)进而优先被甲苯相润湿所致。之后,使用柠檬酸水溶液代替NaCl水溶液以增强粒子与水溶液之间的氢键作用,实现了AuNPs从甲苯相到水相的转移。对于MEO2MA均聚物和OEGMA-co-MEO2MA共聚物修饰的AuNPs,在加入NaCl后也能够从水相转移到甲苯相。然而,当使用柠檬酸水溶液代替盐水后粒子却无法从甲苯相回到水中,这可能与这些粒子表面聚合物中的甲基丙烯酸酯主链和聚乙二醇侧链均被甲苯润湿有关。这种纳米粒子-聚合物复合材料具有作为药物跨越生物屏障的载体、生物系统的显影剂以及研究有趣界面现象的探针等潜在的用途。
随后,为了实现MEO2MA与OEGMA共聚物修饰的AuNPs在水和甲苯之间的相转移,该课题组[42]制备了摩尔比为90∶10的MEO2MA与OEGMA共聚物(MEO2MA90-co-OEGMA10)修饰的AuNPs(Au@MEO2MA90-co-OEGMA10)。研究表明,在室温下,向体系中加入盐,由于AuNPs与盐水之间的界面能增大,导致粒子从盐水相转移到甲苯中。当体系温度降低至5℃以下时,由于冷却可以改善Au@MEO2MA90-co-OEGMA10与水分子之间的水合作用,AuNPs从甲苯相回到盐水中。当体系温度升至室温时,AuNPs又从盐水相转移至甲苯相。然而在没有盐的情况下,即使升高温度至室温,AuNPs也无法从水相转移至甲苯中,这表明温度本身不能引起纳米粒子表面亲水-疏水转变,且与周围介质的温度相比,纳米粒子对离子强度的变化更敏感。
1.6 配体交换响应型
配体交换在调控纳米粒子相转移方面发挥着重要的作用。涉及配体交换的相转移同时也是合成具有理想性质和官能团纳米粒子的一个重要方法[9]。虽然这种方法涉及了两个步骤,并使用了额外的稳定剂,且循环性和可逆性较差,但它能够实现对纳米粒子粒径、形状和分散性的控制。人们可以构建具有易于配体交换官能团的纳米粒子库,并根据需要将这些纳米粒子转移到不同极性介质中。
Dorokhin等[43]通过配体交换作用实现了二茂铁硫醇包覆的CdSe/ZnS量子点在氯仿和水中的相转移。研究发现,CdSe/ZnS最初分散在氯仿相,向体系中加入β-环糊精(β-CD)后,CdSe/ZnS从氯仿相转移到水相。这主要是由于量子点表面的二茂铁配体与β-CD结合,嵌入其圆形腔体内部,形成包合物,而β-CD分子外部以羟基为主,具有亲水性,导致CdSe/ZnS发生相转移。之后,向体系中加入萘盐(NAS)或金刚烷衍生物(AdCOOH),由于β-CD与NAS或AdCOOH之间发生了更强的络合作用而从量子点表面解离,CdSe/ZnS恢复疏水性从水相回到氯仿相。然而,为了将CdSe/ZnS从水相转移到氯仿相,需要在水中加入10倍于β-CD的NAS或100倍于β-CD的AdCOOH,这很难实现相转移的多次循环。另外,与NAS不同的是,使用AdCOOH可以很大程度上避免量子点在氯仿相的聚集,是一种更有效的β-环糊精的络合剂。因此,要建立量子点从水相到有机相的可逆转移,选择合适的解离剂至关重要。
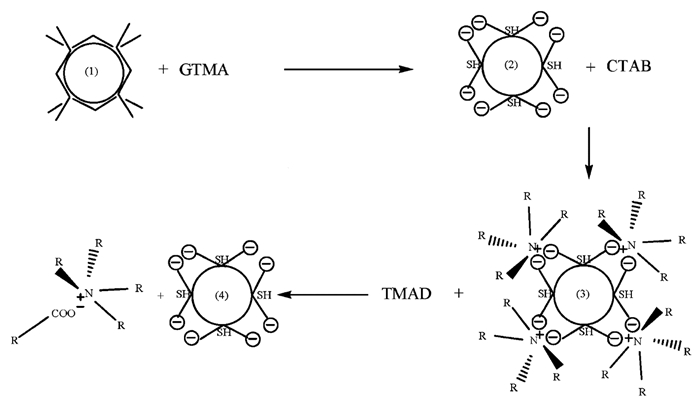
Wei等[44]以甲醇为介导剂,通过配体交换和静电作用实现了量子点/金属纳米粒子在有机相和水相之间的转移。实验过程中,向体系中加入谷胱甘肽四甲基铵盐(GTMA)的甲醇溶液后,GTMA通过配体交换作用取代了包覆在CdSe@CdZnS量子点/金属纳米粒子(Au、Ag、CdS和PbS)表面的油酸,导致粒子从有机溶剂转移到水相(图 9)。随后,向上述体系中加入十六烷基三甲基溴化铵(CTAB),由于CTA+与纳米粒子表面的阴离子通过静电作用结合并形成络合物,使纳米粒子表面疏水,从水相转移到有机相。之后,向体系中加入癸酸四甲基铵盐(TMAD),其中的D-(癸酸根)与CTA+形成了更为疏水的(CTA)+D-并溶于有机相,而负电性的纳米粒子与四甲基铵根离子(TMA+)则以一种亲水性盐的形式转移到水中。可以看出,在整个相转移过程中,甲醇被用作有效的介导溶剂,以改善量子点/金属纳米粒子与有机或水不溶性配体之间的界面接触。
图 9

Sastry等[45, 46]首先利用α-环糊精(α-CD)和十八硫醇(ODT)形成的包合物α-CD-ODT与羧酸之间的配体交换制备了α-CD-ODT稳定的AuNPs;然后向该AuNPs的水溶胶中加入等体积的氯仿,并剧烈摇动,AuNPs几乎完全从水相转移到氯仿相。进一步机理研究表明,在两相混合物的晃动过程中,结合在AuNPs表面的α-CD-ODT中的α-CD分子被移动并从包合物中分离出来,而疏水性的ODT依旧通过硫醇键结合在AuNPs表面,使得AuNPs更具疏水性,从而由水相转移到氯仿相。
Caruso等[10]报道了一种基于配体交换的金和钯纳米粒子相转移的方法。通过向金/钯纳米粒子的甲苯分散液中加入4-N,N-二甲基氨基吡啶(DMAP)的水溶液,纳米粒子能够跨越液-液界面从甲苯相直接转移到水中。然而,吡啶或4-氨基吡啶的加入则会导致粒子聚集并在甲苯/水界面处沉淀。从这一结果可以发现,相转移过程中叔胺(强碱性)与供电子基团(弱碱性)结合的必要性。进一步机理研究表明,将DMAP的水溶液加入到含有金/钯纳米粒子的甲苯分散液中,导致DMAP在甲苯/水相边界的分配,并物理吸附到粒子表面。其中DMAP分子取代纳米粒子表面的四烷基铵,并通过环内氮原子与粒子的表面原子形成一个不稳定的供体-受体复合物而物理吸附到纳米粒子表面,环外氮原子部分质子化产生的表面电荷阻止了金或钯纳米粒子的团聚,这促使纳米粒子从甲苯相转移到水相。该相转移方法不需要沉淀过程就能够产生具有高纳米粒子浓度的水凝胶。与硫醇稳定的粒子相比,DMAP与金属纳米粒子表面没有形成强共价键,容易从粒子表面除去,因此在催化领域具有重要的应用前景。
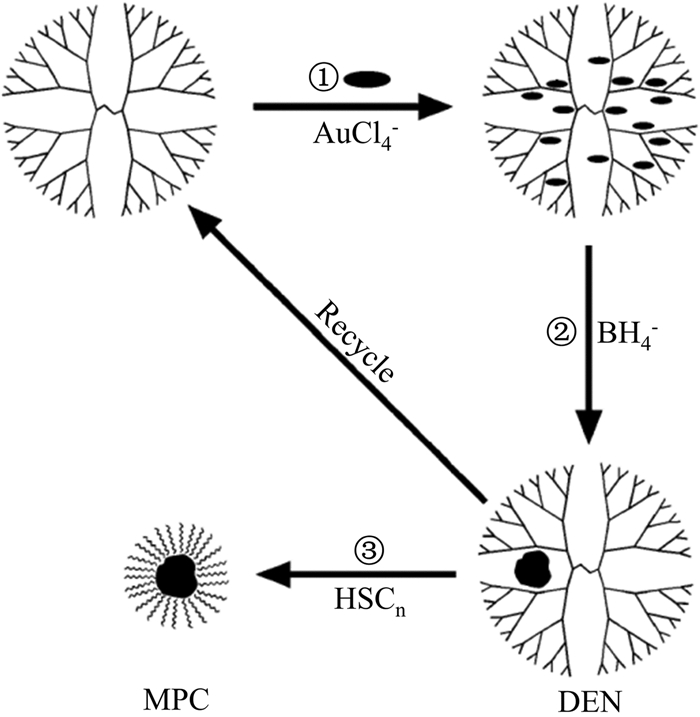
Garcia-Martinez等[47]使用正构烷硫醇完全地把单分散AuNPs从其水溶液中的树状聚合物模板上提取到有机相。该过程首先通过混合聚酰胺-胺(PAMAM)树状分子和HAuCl4水溶液将AuCl4-螯合在树状分子中。然后把所得复合物通过NaBH4还原,得到树状分子包裹的AuNPs。之后,为了将AuNPs从树状分子中提取出来,则向该体系中加入过量的NaBH4(以提供足够高的离子强度)及含有适量烷硫醇的甲苯溶液。通过振荡上述体系,正构烷硫醇与树状分子之间实现了完全的配体交换而自组装在AuNPs表面,将其从树状大分子中提取出来并转运到甲苯相(图 10)。这种相转移方法实现了在不损坏纳米粒子和模板的情况下二者的分离,为制备高度分散的单层保护簇提供了方法。
图 10

Roberts等[48]提出了一种直接的β-D-葡萄糖辅助水相合成AuNPs的方法,并利用配体交换将AuNPs从水相萃取到有机相。通过将所制备的β-D-葡萄糖稳定的AuNPs水溶胶与十二烷硫醇的正己烷溶液混合,并剧烈搅拌,由于十二烷硫醇完全取代了AuNPs表面的β-D-葡萄糖而自组装在粒子表面,使得AuNPs由亲水性变为疏水性,并从水相转移到甲苯相。同时,还发现大量的还原剂NaBH4和强碱性条件对该过程有不利影响,即BH4-通过暂时地稳定纳米粒子来阻止烷硫醇进入纳米粒子表面[49],这不利于纳米粒子表面配体交换反应的进行,因此不利于相转移的发生。这种方法可有效地将单分散的AuNPs萃取到有机相,并可采用精确控制的CO2膨胀液相粒子沉积技术制备广域的、局部有序的纳米粒子阵列和薄膜。
此外,他们[50]以十二烷硫醇作为萃取剂,在浓盐酸辅助下,将羧甲基纤维素钠(CMC)稳定的铂纳米粒子从水相萃取到己烷相,成功地制备了十二烷硫醇包覆的铂纳米粒子。最初由于CMC和铂纳米粒子在水中的强相互作用[51, 52],抑制了十二烷硫醇与CMC在粒子表面的交换,所以铂纳米粒子不能直接从水相转移到含有十二烷硫醇的己烷溶液中。然而,浓盐酸的加入使CMC结构中的COOH占主导地位,明显削弱了CMC与铂纳米粒子之间的相互作用,使得十二烷硫醇可以有效地与铂纳米粒子产生更强的相互作用。十二烷硫醇取代铂纳米粒子表面的CMC,导致铂纳米粒子具有强的疏水性,因此从水相转移到己烷相,并可得到十二烷硫醇包覆的铂纳米粒子的有机分散液,且该过程不可逆。这项研究为金属纳米粒子的可控合成和高效构建提供了机会。
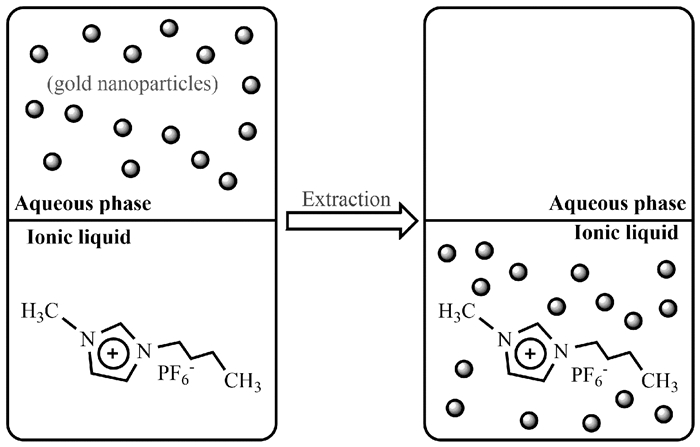
Wei等[53]报道了一种不需要使用烷硫醇就可以将AuNPs从水相转移到离子液体的方法。该方法首先以柠檬酸盐为还原剂,通过在水溶液中还原HAuCl4制备了柠檬酸盐稳定的AuNPs。之后向上述体系中加入与水不互溶的离子液体六氟磷酸1-丁基-3-甲基咪唑鎓([C4mim][PF6]),形成两相体系。当剧烈摇动两相混合物时,AuNPs虽然能够从水层完全转移到离子液体层,但由于严重的聚集,离子液体中出现了AuNPs的沉淀。然而,通过向水相中加入十四烷基三甲基溴化铵(TTAB)可以解决AuNPs沉淀的问题。这主要是因为TTAB与柠檬酸盐通过配体交换作用包覆在AuNPs表面,粒子的类醇极性及其离子性质有效促进了其从水相到[C4mim][PF6]的转移(图 11)。这一发现为进一步研究不同尺寸和形状的金属纳米粒子在离子液体中的修饰和应用开辟了新的途径。
图 11

1.7 离子交换响应型
纳米粒子表面通常包覆有由阴、阳离子组成的有机盐(如离子液体等)。当其与不同阴阳离子的无机盐混合时,通过离子交换,纳米粒子倾向于形成更稳定或更具疏水性的状态。因此,通过加入适当的盐来调控纳米粒子的亲疏水性,可以有效促使纳米粒子的相转移。
Li等[54, 55]以柠檬酸为碳源、室温咪唑离子液体为修饰剂,采用一锅法制备了高电荷密度的亲水性碳量子点Imi-CQDs-Br,研究了Imi-CQDs-Br在水/乙酸乙酯和水/疏水性离子液体之间的相转移。研究发现,当向乙酸乙酯与Imi-CQDs-Br水分散液两相体系中加入双三氟甲磺酰亚胺锂(LiNTf2),由于离子交换作用,水溶性的Imi-CQDs-Br转变为油溶性的Imi-CQDs-NTf2,导致碳量子点从水相转移到乙酸乙酯中。之后,向该体系中加入NaBr,碳量子点又从乙酸乙酯回到水相,这可能与溴代咪唑离子液体在水中有较高溶解度有关。对于Imi-CQDs-Br在水和疏水性离子液体1-丁基-3-甲基咪唑双三氟甲磺酸盐[C4mim][NTf2]之间的相转移,通过简单的振荡,体系中Br-与NTf2-发生交换,Imi-CQDs+和NTf2-结合形成了疏水性的Imi-CQDs-NTf2,使得碳量子点从水相转移到离子液体中。该相转移过程避免了添加剂的加入,不但简化了工艺而且有利于其在实际过程中的应用。
Naka等[56]通过阴离子交换的方法实现了AuNPs从水相到离子液体相的高效转移。实验表明,在搅拌作用下,向咪唑氯离子液体改性的AuNPs水分散液中加入六氟磷酸后,由于AuNPs表面的氯离子与六氟磷酸根进行交换,使得粒子由亲水性转变为疏水性,并从水相转移到疏水性离子液体六氟磷酸1-甲基-3-己基咪唑鎓中。该体系中离子液体改性的AuNPs在可循环双相催化过程中具有潜在的应用前景。
2. 结语
本文综述了环境响应型纳米粒子在不互溶两相间相转移的研究进展,重点分析了纳米粒子的界面效应、自组装行为及溶剂化效应等在相转移过程中的作用,这将有助于我们进一步加深对纳米粒子在两相界面行为的认识。通过纳米粒子的相转移,人们能够在水或有机溶剂中合成纳米粒子,并随后将纳米粒子转移到不同的环境中进一步应用。该方法不仅节省了不同极性纳米粒子的制备成本,而且实现了在温和条件下对纳米粒子极性的调控,这使得纳米粒子的合成、纯化及循环再利用等过程变得更加简洁、高效。
虽然目前对环境响应型纳米粒子相转移的研究有了较大的进展,但该领域的研究还处于基础研究的起步阶段,面临的挑战仍然很多,至少包括以下几个方面:(1)相转移机制需进一步阐明,以便于准确把握环境因素的影响,从而获得理想的相转移体系;(2)将相转移策略用于合成具有良好尺寸、形貌和表面功能化的复合纳米粒子,这种独特的纳米结构有望在光电子、能源和环境等领域的应用取得突破;(3)设计开发新型纳米粒子可逆相转移体系,使相转移过程更为绿色和高效;(4)纳米粒子相转移的研究对于合理设计纳米粒子并用于相转移催化、生物探针或药物载体等具有重要意义,因此可进一步拓展纳米粒子相转移的应用范围。
-
-
[1]
Jiang Z, Le N D B, Gupta A, et al. Chem. Soc. Rev., 2015, 44(13): 4264~4274. doi: 10.1039/C4CS00387J
-
[2]
周希, 刘美红, 王春鹏, 等. 化学通报, 2018, 81(1): 65~69. http://www.ccspublishing.org.cn/article/id/a4b8703c-b893-4193-9618-37ed7b9e4378
-
[3]
Ryu J, Jung N, Lim D, et al. Chem. Commun., 2014, 50(100): 15940~15943. doi: 10.1039/C4CC07143C
-
[4]
Teixeira I F, Barbosa E C M, Tsang S C E, et al. Chem. Soc. Rev., 2018, 47(20): 7783~7817. doi: 10.1039/C8CS00479J
-
[5]
Cao L, Wang X, Meziani M J, et al. J. Am. Chem. Soc., 2007, 129(37): 11318~11319. doi: 10.1021/ja073527l
-
[6]
Michalet X, Pinaud F F, Bentolila L A, et al. Science, 2005, 307(5709): 538~544. doi: 10.1126/science.1104274
-
[7]
Jung K, Song H, Lee G, et al. ACS Nano, 2014, 8(3): 2590~2601. doi: 10.1021/nn500276n
-
[8]
Ueno K, Oshikiri T, Sun Q, et al. Chem. Rev., 2018, 118(6): 2955~2993. doi: 10.1021/acs.chemrev.7b00235
-
[9]
Yang J, Lee J Y, Ying J Y. Chem. Soc. Rev., 2011, 40(3): 1672~1696. doi: 10.1039/B916790K
-
[10]
Gittins D I, Caruso F. Angew. Chem. Int. Ed., 2001, 40(16): 3001~3004. doi: 10.1002/1521-3773(20010817)40:16<3001::AID-ANIE3001>3.0.CO;2-5
-
[11]
Jessop P G, Mercer S M, Heldebrant D J. Energy Environ. Sci., 2012, 5(6): 7240~7253. doi: 10.1039/c2ee02912j
-
[12]
Cole-Hamilton D J. Science, 2003, 299(5613): 1702~1706. doi: 10.1126/science.1081881
-
[13]
Russell T P. Science, 2002, 297(5583): 964~967. doi: 10.1126/science.1075997
-
[14]
Yang J, Sargent E H, Kelley S O, et al. Nat. Mater., 2009, 8(8): 683~689. doi: 10.1038/nmat2490
-
[15]
Zhou K, Wang Y, Huang X, et al. Angew. Chem. Int. Ed., 2011, 50(27): 6109~6114. doi: 10.1002/anie.201100884
-
[16]
周迪, 孟哲一, 张明辉, 等. 化学学报, 2015, 73: 716~722. https://www.cnki.com.cn/Article/CJFDTOTAL-XDYC201502002.htm
-
[17]
Wang H, Yang H, Liu H, et al. Langmuir, 2013, 29(22): 6687~6696. doi: 10.1021/la4003093
-
[18]
Ansar S M, Chakraborty S, Kitchens C L. Nanomaterials, 2018, 8(5): 339~411. doi: 10.3390/nano8050339
-
[19]
Chakraborty S, Kitchens C L. J. Phys. Chem. C, 2019, 123: 26450~26460. doi: 10.1021/acs.jpcc.9b08352
-
[20]
Imura Y, Morita C, Endo H, et al. Chem. Commun., 2010, 46(48): 9206~9208. doi: 10.1039/c0cc03194a
-
[21]
Zhao S, Qiao R, Zhang X L, et al. J. Phys. Chem. C, 2007, 111(22): 7875~7878. doi: 10.1021/jp070457w
-
[22]
Gui R, Wan A, Jin H, et al. Mater. Lett., 2013, 96: 20~23. doi: 10.1016/j.matlet.2013.01.005
-
[23]
Peng Z, Walther T, Kleinermanns K. J. Phys. Chem. B, 2005, 109(33): 15735~15740. doi: 10.1021/jp051849a
-
[24]
Xiong D, Cui G, Wang J, et al. Angew. Chem. Int. Ed., 2015, 54(25): 7265~7269. doi: 10.1002/anie.201500695
-
[25]
Pei X, Xiong D, Wang H, et al. Angew. Chem. Int. Ed., 2018, 57(14): 3687~3691. doi: 10.1002/anie.201800037
-
[26]
Xiong R, Chen M, Cui X, et al. ACS Appl. Mater. Interf., 2019, 11(25): 22851~22857. doi: 10.1021/acsami.9b05421
-
[27]
Pocoví-Martínez S, Francés-Soriano L, Zaballos-García E, et al. RSC Adv., 2013, 3(15): 4867~4871. doi: 10.1039/c3ra23212c
-
[28]
Kim H, Lee T S. Mol. Cryst. Liq. Cryst., 2019, 685(1): 78~86. doi: 10.1080/15421406.2019.1645464
-
[29]
Li D, Zhao B. Langmuir, 2007, 23(4): 2208~2217. doi: 10.1021/la0628165
-
[30]
Horton J M, Bai Z, Jiang X, et al. Langmuir, 2011, 27(5): 2019~2027. doi: 10.1021/la1044706
-
[31]
Qin B, Zhao Z, Song R, et al. Angew. Chem. Int. Ed., 2008, 47(51): 9875~9878. doi: 10.1002/anie.200803582
-
[32]
Feng X, Ma H, Huang S, et al. J. Phys. Chem. B, 2006, 110(25): 12311~12317. doi: 10.1021/jp0609885
-
[33]
Li Z, Yuan X, Feng Y, et al. Phys. Chem. Chem. Phys., 2018, 20(18): 12808~12816. doi: 10.1039/C8CP01617H
-
[34]
Li Z, Feng Y, Yuan X, et al. Int. J. Mol. Sci., 2019, 20(7): 1685~1700. doi: 10.3390/ijms20071685
-
[35]
Yuan X, Li Z, Feng Y, et al. J. Mol. Liq., 2019, 277: 805~811. doi: 10.1016/j.molliq.2019.01.017
-
[36]
Li Z, Li R, Yuan X, et al. Green Energy Environ., 2019, 4(2): 131~138. doi: 10.1016/j.gee.2019.01.011
-
[37]
李志勇, 冯莹, 王慧勇, 等. 化学进展, 2019, 31(11): 1550~1559. https://www.cnki.com.cn/Article/CJFDTOTAL-JFJY202001002.htm
-
[38]
江金强, 代鹏, 宗奕吾, 等. 物理化学学报, 2009, 25(11): 2285~2290. doi: 10.3866/PKU.WHXB20091027
-
[39]
Peng L, You M, Wu C, et al. ACS Nano, 2014, 8(3): 2555~2561. doi: 10.1021/nn4061385
-
[40]
Wu Y, Zhang C, Qu X, et al. Langmuir, 2010, 26(12): 9442~9448. doi: 10.1021/la100458j
-
[41]
Edwards E W, Chanana M, Wang D, et al. Angew. Chem. Int. Ed., 2008, 47(2): 320~323. doi: 10.1002/anie.200702597
-
[42]
Stocco A, Chanana M, Su G, et al. Angew. Chem. Int. Ed., 2012, 51(38): 9647~9651. doi: 10.1002/anie.201203493
-
[43]
Dorokhin D, Tomczak N, Han M, et al. ACS Nano, 2009, 3(3): 661~667. doi: 10.1021/nn8006515
-
[44]
Wei Y, Yang J, Ying J Y. Chem. Commun., 2010, 46(18): 3179~3181. doi: 10.1039/b926194j
-
[45]
Lala N, Lalbegi S P, Adyanthaya S D, et al. Langmuir, 2001, 17(12): 3766~3768. doi: 10.1021/la0015765
-
[46]
Patil V, Malvankar R B, Sastry M. Langmuir, 1999, 15(23): 8197~8206. doi: 10.1021/la990170t
-
[47]
Garcia-Martinez J C, Crooks R M. J. Am. Chem. Soc., 2004, 126(49): 16170~16178. doi: 10.1021/ja046567n
-
[48]
Liu J, Anand M, Roberts C B. Langmuir, 2006, 22(9): 3964~3971. doi: 10.1021/la060450q
-
[49]
Yang J, Lee J Y, Deivaraj T C, et al. Langmuir, 2003, 19(24): 10361~10365. doi: 10.1021/la034596q
-
[50]
Liu J, Sutton J, Roberts C B. J. Phys. Chem. C, 2007, 111(31): 11566~11576. doi: 10.1021/jp071967t
-
[51]
Wu N, Fu L, Su M, et al. Nano Lett., 2004, 4(2): 383~386. doi: 10.1021/nl035139x
-
[52]
Anand M, Bell P W, Fan X, et al. J. Phys. Chem. B, 2006, 110(30): 14693~14701. doi: 10.1021/jp0614401
-
[53]
Wei G, Yang Z, Lee C, et al. J. Am. Chem. Soc., 2004, 126(16): 5036~5037. doi: 10.1021/ja039874m
-
[54]
Wang B, Song A, Feng L, et al. ACS Appl. Mater. Inter., 2015, 7(12): 6919~6925. doi: 10.1021/acsami.5b00758
-
[55]
Sun X, Yin K, Liu B, et al. J. Mater. Chem. C, 2017, 5(20): 4951~4958. doi: 10.1039/C7TC00543A
-
[56]
Itoh H, Naka K, Chujo Y. J. Am. Chem. Soc., 2004, 126(10): 3026~3027. doi: 10.1021/ja039895g
-
[1]
-

-

 下载:
下载:



 下载:
下载:










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 17
- 文章访问数: 2238
- HTML全文浏览量: 444
