-
[1]
(a) Petasis, N. A.; Patane, M. A. Tetrahedron 1992, 48, 5757.
(b) Mehta, G.; Singh, V. Chem. Rev. 1999, 99, 881.
(c) Yet, L. Chem. Rev. 2000, 100, 2963.
-
[2]
Suffness, M. Taxol:Science and Applications, CRC Press, Boca Raton, FL, 1995. http://www.cabdirect.org/abstracts/19950316216.html
-
[3]
(a) Tang, Y.-Z.; Liu, Y.-H.; Chen, J.-X. Mini-Rev. Med. Chem. 2012, 12, 53.
(b) Shang, R.; Wang, J.; Guo, W.; Liang, J. Curr. Top. Med. Chem. 2013, 13, 3013.
(c) Goethe, O.; Heuer, A.; Ma, X.; Wang, Z.; Herzon, S. B. Nat. Prod. Rep. 2019, 36, 220.
-
[4]
(a) Armanino, N.; Charpentier, J.; Flachsmann, F.; Goeke, A.; Liniger, M.; Kraft, P. Angew. Chem., Int. Ed. 2020, 59, 16310.
(b) Kraft, P.; Bajgrowicz, J. A.; Denis, C.; Fráter, G. Angew. Chem., Int. Ed. 2000, 39, 2980.
(c) Birkbeck, A. A. Challenges in the Synthesis of Natural and Non-Natural Volatiles. In The Chemistry and Biology of Volatiles, Ed.: Herrmann, A., John Wiley & Sons, Ltd., New York, 2010, pp. 173~193.
(d) Vesley, J. A.; Massie, S. N. US 3985769, 1976.
(e) Markert, T. WO 99/54430, 1998.
(f) Fráter, G.; Bajgrowicz, J. A.; Kraft, P. Tetrahedron 1998, 54, 7633.
(g) Granier, T.; Bajgrowicz, J. A.; Hanhart, A. US 7888309, 2011.
-
[5]
(a) Martinez, H.; Ren, N.; Matta, M. E.; Hillmyer, M. A. Polym. Chem. 2014, 5, 3507.
(b) Hill, A. R.; Balogh, J.; Moncho, S.; Su, H.-L.; Tuba, R.; Brothers, E. N.; Al-Hashimi, M.; Bazzi, H. S. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 3137.
-
[6]
For selected reviews for Diels-Alder reaction in synthesis, see:
(a) Nicolaou, K. C.; Snyder, S. A.; Montagnon, T. Vassilikogiannakis. G. Angew. Chem., Int. Ed. 2002, 41, 1668.
(b) Takao, K.-I.; Munakata, R.; Tadano, K.-I. Chem. Rev. 2005, 105, 4779.
(c) Wessig, P.; Müller, G. Chem. Rev. 2008, 108, 2051.
(d) Funel, J.-A.; Abele, S. Angew. Chem., Int. Ed. 2013, 52, 3822.
(e) Jiang, X.; Wang, R. Chem. Rev. 2013, 113, 5515.
(f) Heravi, M. M.; Vavsari, V. F. RSC Adv. 2015, 5, 50890.
(g) Yang, B.; Gao, S. Chem. Soc. Rev. 2018, 47, 7926.
(h) Tasdelen, M. A. Polym. Chem. 2011, 2, 2133.
-
[7]
Selected reviews for metal-catalyzed[4+2] reactions:
(a) Reymond, S.; Cossy, J. Chem. Rev. 2008, 108, 5359.
(b) Carmona, D.; Lamata, M. P.; Oro, L. A. Coord. Chem. Rev. 2000, 200~202, 717.
(c) Wender, P. A.; Smith, T. E. Tetrahedron 1998, 54, 1255.
(d) Frühauf, H.-W. Chem. Rev. 1997, 97, 523.
(e) Kagan, H. B.; Riant, O. Chem. Rev. 1992, 92, 1007.
(f) Robinson, J. E. Modern Rhodium-Catalyzed Organic Reactions; Ed.: Evans, P. A., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005, pp. 241~262.
-
[8]
(a) Ben-Shoshan, R.; Sarel, S. J. Chem. Soc. D 1969, 883.
(b) Victor, R.; Ben-Shoshan, R.; Sarel, S. Tetrahedron Lett. 1970, 4253.
(c) Sarel, S. Acc. Chem. Res. 1978, 11, 204.
(d) Aumann, R. J. Am. Chem. Soc. 1974, 96, 2631.
(e) Taber, D. F.; Kanai, K.; Jiang, Q.; Bui, G. J. Am. Chem. Soc. 2000, 122, 6807.
(f) Taber, D. F.; Joshi, P. V.; Kanai, K. J. Org. Chem. 2004, 69, 2268.
(g) Kurahashi, T.; de Meijere, A. Synlett 2005, 2619.
(h) Iwasawa, N.; Owada, Y.; Matsuo, T. Chem. Lett. 1995, 115.
(i) Owada, Y.; Matsuo, T.; Iwasawa, N. Tetrahedron 1997, 53, 11069.
(j) Murakami, M.; Itami, K.; Ubukata, M.; Tsuji, I.; Ito, Y. J. Org. Chem. 1998, 63, 4.
(k) Shu, D.; Li, X.; Zhang, M.; Robichaux, P. J.; Tang, W. Angew. Chem., Int. Ed. 2011, 50, 1346.
(l) Grabowski, N. A.; Hughes, R. P.; Jaynes, B. S.; Rheingold, A. L. J. Chem. Soc., Chem. Commun. 1986, 1694.
(m) Cho, S. H.; Liebeskind, L. S. J. Org. Chem. 1987, 52, 2631.
(n) Semmelhack, M. F.; Ho, S.; Steigerwald, M.; Lee, M. C. J. Am. Chem. Soc. 1987, 109, 4397.
(o) Brancour, C.; Fukuyama, T.; Ohta, Y.; Ryu, I.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. Chem. Commun. 2010, 46, 5470.
(p) Jiang, G.-J.; Fu, X.-F.; Li, Q.; Yu, Z.-X. Org. Lett. 2012, 14, 692.
(q) Li, X.; Song, W.; Tang, W. J. Am. Chem. Soc. 2013, 135, 16797.
(r) Fukuyama, T.; Ohta, Y.; Brancour, C.; Miyagawa, K.; Ryu, I.; Dhimane, A.-L.; Fensterbank, L.; Malacria, M. Chem.-Eur. J. 2012, 18, 7243.
(s) Farley, C. M.; Sasakura, K.; Zhou, Y.-Y.; Kanale, V. V.; Uyeda, C. J. Am. Chem. Soc. 2020, 142, 4598.
-
[9]
(a) Jiao, L.; Lin, M.; Zhuo, L.-G.; Yu, Z.-X. Org. Lett. 2010, 12, 2528.
(b) Mazumder, S.; Shang, D.; Negru, D. E.; Baik, M.-H.; Evans, P. A. J. Am. Chem. Soc. 2012, 134, 20569.
(c) Kim, S.; Chung, Y. K. Org. Lett. 2014, 16, 4352.
(d) Wang, J.; Hong, B.; Hu, D.; Kadonaga, Y.; Tang, R.; Lei, X. J. Am. Chem. Soc. 2020, 142, 2238.
-
[10]
Selected reviews for metal-catalyzed[2+2+2] reactions:
(a) Vollhardt, K. P. C. Angew. Chem., Int. Ed. 1984, 23, 539.
(b) Chopade, P. R.; Louie, J. Adv. Synth. Catal. 2006, 348, 2307.
(c) Kotha, S.; Brahmachary, E.; Lahiri, K. Eur. J. Org. Chem. 2005, 4741.
(d) Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2011, 40, 3430.
(e) Shibata, T.; Tsuchikama, K. Org. Biomol. Chem. 2008, 6, 1317.
(f) Li, C.; Zhang, H.; Feng, J.; Zhang, Y.; Wang, J. Org. Lett. 2010, 12, 3082.
(g) Shaw, M. H.; Melikhova, E. Y.; Kloer, D. P.; Whittingham, W. G.; Bower, J. F. J. Am. Chem. Soc. 2013, 135, 4992.
-
[11]
(a) Deiters, A.; Martin, S. F. Chem. Rev. 2004, 104, 2199.
(b) Grubbs, R. H.; Miller, S. J.; Fu, G. C. Acc. Chem. Res. 1995, 28, 446.
(c) Fürstner, A. Top. Catal. 1997, 4, 285.
(d) Donohoe, T. J.; Orr, A. J.; Bingham, M. Angew. Chem., Int. Ed. 2006, 45, 2664.
(e) Maier, M. E. Angew. Chem., Int. Ed. 2000, 39, 2073.
(f) Michaut, A.; Rodriguez, J. Angew. Chem., Int. Ed. 2006, 45, 5740.
-
[12]
Hu, Y.-J.; Li, L.-X.; Han, J.-C.; Min, L.; Li, C.-C. Chem. Rev. 2020, 120, 5910. doi: 10.1021/acs.chemrev.0c00045
-
[13]
(a) Liang, Y.; Jiang, X.; Yu, Z.-X. Chem. Commun. 2011, 47, 6659.
(b) Liang, Y.; Jiang, X.; Fu, X.-F.; Ye, S.; Wang, T.; Yuan, J.; Wang, Y.; Yu, Z.-X. Chem.-Asian J. 2012, 7, 593.
-
[14]
(a) Illuminati, G.; Mandolini, L. Acc. Chem. Res. 1981, 14, 95.
(b) Galli, C.; Mandolini, L. Eur. J. Org. Chem. 2000, 2000, 3117.
-
[15]
Lautens, M.; Klute, W.; Tam, W. Chem. Rev. 1996, 96, 49. doi: 10.1021/cr950016l
-
[16]
Yu, Z.-X.; Wang, Y.; Wang, Y. Chem.-Asian J. 2010, 5, 1072. doi: 10.1002/asia.200900712
-
[17]
(a) Reed, H. W. B. J. Chem. Soc. 1954, 1931.
(b) Ziegler, K.; Holzkamp, E.; Breil, H.; Martin, H. Angew. Chem. 1955, 67, 426.
-
[18]
Wender, P. A.; Ihle, N. C. J. Am. Chem. Soc. 1986, 108, 4678. doi: 10.1021/ja00275a085
-
[19]
Park, J. W.; Park, J. E.; Park, J. H.; Hong, M. R.; Kim, S. M.; Chung, Y. K.; Kim, C. H. Synlett 2016, 27, 455.
-
[20]
Llorente, N.; Fernández-Pérez, H.; Bauzá, A.; Frontera, A.; Vidal-Ferran, A. Catal. Sci. Technol. 2018, 8, 5251. doi: 10.1039/C8CY00684A
-
[21]
(a) tom Dieck, H.; Dietrich, J. Chem. Ber. 1984, 117, 694.
(b) tom Dieck, H.; Dietrich, J. Angew.Chem., Int. Ed. 1985, 24, 781.
(c) Mallien, M.; Haupt, E. T. K.; tom Dieck, H. Angew. Chem., Int. Ed. 1988, 27, 1062.
-
[22]
Lee, H.; Campbell, M. G.; Sánchez, R. H.; Börgel, J.; Raynaud, J.; Parker, S. E.; Ritter, T. Organometallics 2016, 35, 2923. doi: 10.1021/acs.organomet.6b00474
-
[23]
Kennedy, C. R.; Zhong, H. Y.; Macaulay, R. L.; Chirik, P. J. J. Am. Chem. Soc. 2019, 141, 8557. http://www.researchgate.net/publication/332911264_Regio-_and_Diastereoselective_Iron-Catalyzed_44-Cycloaddition_of_13-Dienes
-
[24]
(a) Braconi, E.; Götzinger, A. C.; Cramer, N. J. Am. Chem. Soc. 2020, 142, 19819.
(b) Baldenius, K.-U.; tom Dieck, H.; König, W. A.; Icheln, D.; Runge, T. Angew. Chem., Int. Ed. 1992, 31, 305.
-
[25]
Selected reviews for metal-catalyzed C-C bond activation of strained rings:
(a) Murakami, M.; Matsuda, T. Chem. Commun. 2011, 47, 1100.
(b) Ruhland, K. Eur. J. Org. Chem. 2012, 2012, 2683.
(c) Souillart, L.; Cramer, N. Chem. Rev. 2015, 115, 9410.
(d) Chen, F.; Wang, T.; Jiao, N. Chem. Rev. 2014, 114, 8613.
(e) Cleavage of Carbon-Carbon Single Bonds by Transition Metals, Eds.: Murakami, M.; Chatani, N., Wiley-VCH, Weinheim, Germany, 2016.
(f) Dong, G. C-C Bond Activation, In Topics in Current Chemistry, Eds.: Bayley, H.; Houk, K. N.; Hughes, G.; Hunter, C. A.; Ishihara, K.; Krische, M. J.; Lehn, J.-M.; Luque, R.; Olivucci, M.; Siegel, J. S.; Thiem, J.; Venturi, M.; Wong, C.-H.; Wong, H. N. C; You, S.-L.; Yam, V. W.-W.; Yan, C. Springer Verlag, Berlin and Heidelberg, Germany, 2014, DOI: 10.1007/978-3-642-55055-3.
(g)Rubin,M.;Rubina,M.;Gevorgyan,V.Chem.Rev.2007,107,3117.
(h)Fumagalli,G.;Stanton,S.;Bower,J.F.Chem.Rev.2017,117,9404.
(i)Dai,H.;Wu,F.;Bai,D.Chin.J.Org.Chem.2020,40,1423(inChinese).
(代洪雪,吴芬,白大昌,有机化学,2020,40,1423.
-
[26]
(a) Evans, J. A.; Everitt, G. F.; Kemmitt, R. D. W.; Russell, D. R. J. Chem. Soc., Chem. Commun. 1973, 158.
(b) Liebeskind, L. S.; Baysdon, S. L.; South, M. S.; Blount, J. F. J. Organomet. Chem. 1980, 202, C73.
(c) Liebeskind, L. S.; Baysdon, S. L.; South, M. S.; Iyer, S.; Leeds, J. P. Tetrahedron 1985, 41, 5839.
(d) Huffman, M. A.; Liebeskind, L. S.; Pennington, W. T. Organometallics 1990, 9, 2194.
(e) Masuda, Y.; Hasegawa, M.; Yamashita, M.; Nozaki, K.; Ishida, N.; Murakami, M. J. Am. Chem. Soc. 2013, 135, 7142.
(f) Okumura, S.; Sun, F.; Ishida, N.; Murakami, M. J. Am. Chem. Soc. 2017, 139, 12414.
(g) Xu, T.; Dong, G. Angew. Chem., Int. Ed. 2012, 51, 7567.
(h) Xu, T.; Ko, H. M.; Savage, N. A.; Dong, G. J. Am. Chem. Soc. 2012, 134, 20005.
(i) Deng, L.; Chen, M.; Dong, G. J. Am. Chem. Soc. 2018, 140, 9652.
(j) Lu, G.; Fang, C.; Xu, T.; Dong, G.; Liu, P. J. Am. Chem. Soc. 2015, 137, 8274.
(k) Xu, T.; Savage, N. A.; Dong, G. Angew. Chem., Int. Ed. 2014, 53, 1891.
(l) Chen, P.-H.; Xu, T.; Dong, G. Angew. Chem., Int. Ed. 2014, 53, 1674.
(m) Sun, T. W.; Zhang, Y.; Qiu, B.; Wang, Y.; Qin, Y.; Dong, G.; Xu, T. Angew. Chem., Int. Ed. 2018, 57, 2859.
(n) Deng, L.; Xu, T.; Li, H.; Dong, G. J. Am. Chem. Soc. 2016, 138, 369.
(o) Chen, P.-H.; Sieber, J.; Senanayake, C. H.; Dong, G. Chem. Sci. 2015, 6, 5440.
(p) Zhu, Z.; Li, X.; Chen, S.; Chen, P.-H.; Billett, B. A.; Huang, Z.; Dong, G. ACS Catal. 2018, 8, 845.
(q) Xu, Z.-Y.; Zhang, S.-Q.; Liu, J.-R.; Chen, P.-P.; Li, X.; Yu, H.-Z.; Hong, X.; Fu, Y. Organometallics 2018, 37, 592.
(r) Bender, M.; Turnbull, B. W. H.; Ambler, B. R.; Krische, M. J. Science 2017, 357, 779.
(s) Ambler, B. R.; Turnbull, B. W. H.; Suravarapu, S. R.; Uteuliyev, M. M.; Huynh, N. O.; Krische, M. J. J. Am. Chem. Soc. 2018, 140, 9091.
(t) Deng, L.; Dong, G. Trends in Chem. 2020, 2, 183.
-
[27]
Juliá-Hernández, F.; Ziadi, A.; Nishimura, A.; Martin, R. Angew. Chem., Int. Ed. 2015, 54, 9537. doi: 10.1002/anie.201503461
-
[28]
Yang, S.; Xu, Y.; Li, J. Org. Lett. 2016, 18, 6244. doi: 10.1021/acs.orglett.6b02943
-
[29]
Zou, H.; Wang, Z.-L.; Huang G. Chem.-Eur J. 2017, 23, 12593. doi: 10.1002/chem.201702316
-
[30]
Reppe, W.; Schlichting, O.; Klager, K.; Toepel, T. Liebigs Ann. Chem. 1948, 560, 1. doi: 10.1002/jlac.19485600102
-
[31]
(a) Wender, P. A.; Christy, J. P. J. Am. Chem. Soc. 2007, 129, 13402.
(b) Wender, P. A.; Christy, J. P.; Lesser, A. B.; Gieseler, M. T. Angew. Chem., Int. Ed. 2009, 48, 7687.
-
[32]
Chai, Z.; Wang, H.-F.; Zhao, G. Synlett 2009, 11, 1785. http://www.researchgate.net/publication/239238177_Ni-Catalyzed_Carbocyclization_of_16-Enynes_Mediated_by_Dialkylzinc_Reagents_Me2Zn_or_Et2Zn_Makes_a_Difference
-
[33]
Nasrallah, D. J.; Croatt, M. P. Eur. J. Org. Chem. 2014, 2014, 3767. doi: 10.1002/ejoc.201402109
-
[34]
Greco, A.; Carbonar, A.; Dall'Asta, G. J. Org. Chem. 1970, 35, 271. doi: 10.1021/jo00826a067
-
[35]
Murakami, M.; Ashida, S.; Matsuda, T. J. Am. Chem. Soc. 2006, 128, 2166. doi: 10.1021/ja0552895
-
[36]
Tao, J.-Y.; Fang, D.-C.; Chass, G. A. Phys. Chem. Chem. Phys. 2012, 14, 6937. doi: 10.1039/c2cp40067g
-
[37]
Lainhart, B. C.; Alexanian, E. J. Org. Lett. 2015, 17, 1284. doi: 10.1021/acs.orglett.5b00267
-
[38]
Gilbertson, S. R.; DeBoef, B. J. Am. Chem. Soc. 2002, 124, 8784. doi: 10.1021/ja026536x
-
[39]
DeBoef, B.; Counts, W. R.; Gilbertson, S. R. J. Org. Chem. 2007, 72, 799. doi: 10.1021/jo0620462
-
[40]
Canlas, G. M. R.; Gilbertson, S. R. Chem. Commun. 2014, 50, 5007. doi: 10.1039/C4CC01320D
-
[41]
(a) Evans, P. A.; Robinson, J. E.; Baum, E. W.; Fazal, A. N. J. Am. Chem. Soc. 2002, 124, 8782.
(b) Evans, P. A.; Baum, E. W. J. Am. Chem. Soc. 2004, 126, 11150.
(c) Evans, P. A.; Baum, E. W.; Fazal, A. N.; Pink, M. Chem. Commun. 2005, 63.
-
[42]
Wender, P. A.; Christy, J. P. J. Am. Chem. Soc. 2006, 128, 5354. doi: 10.1021/ja060878b
-
[43]
(a) Hilt, G.; Janikowski, J. Angew. Chem., Int. Ed. 2008, 47, 5243.
(b) Varela, J. A.; Castedo, L.; Saá, C. Org. Lett. 2003, 5, 2841.
-
[44]
Yamasaki, R.; Ohashi, M.; Maeda, K.; Kitamura, T.; Nakagawa, M.; Kato, K.; Fujita, T.; Kamura, R.; Kinoshita, K.; Masu, H.; Azumaya, I.; Ogoshi, S.; Saito, S. Chem.-Eur J. 2013, 19, 3415. doi: 10.1002/chem.201204087
-
[45]
Jiménez, T.; Carreras, J.; Ceccon, J.; Echavarren, A. M. Org. Lett. 2016, 18, 1410. doi: 10.1021/acs.orglett.6b00342
-
[46]
Davis, R. E.; Dodds, T. A.; Hseu, T. H.; Wagnon, J. C.; Devon, T.; Tancrede, J.; McKennis, J. S.; Pettit, R. J. Am. Chem. Soc. 1974, 96, 7562. doi: 10.1021/ja00831a034
-
[47]
D'yakonov, V. A.; Kadikova, G. N.; Dzhemilev, U. M. Russ. Chem. Rev. 2018, 87, 797. doi: 10.1070/RCR4793
-
[48]
(a) Mach, K.; Antropiusová, H.; Sedmera, P.; Hanuš, V.; Tureček, F. J. Chem. Soc., Chem. Commun. 1983, 805.
(b) Mach, K.; Antropiusová, H.; Petrusová, L.; Hanuš, V.; Tureček, F.; Sedmera, P. Tetrahedron 1984, 40, 3295.
-
[49]
D'yakonov, V. A.; Kadikova, G. N.; Dzhemilev, U. M. Tetrahedron Lett. 2011, 52, 2780. doi: 10.1016/j.tetlet.2011.03.131
-
[50]
D'yakonov, V. A.; Kadikova, G. N.; Khalilov, L. M.; Dzhemilev, U. M. Russ. J. Org. Chem. 2013, 49, 1139. doi: 10.1134/S1070428013080071
-
[51]
Dzhemilev, U. M.; Kadikova, G. N.; Kolokol'tsev, D. I.; D'yakonov, V. A. Tetrahedron 2013, 69, 4609. doi: 10.1016/j.tet.2013.04.019
-
[52]
(a) D'yakonov, V. A.; Kadikova, G. N.; Kolokol'tsev, D. I.; Ramazanov, I. R.; Dzhemilev, U. M. J. Organomet. Chem. 2015, 794, 23.
(b) D'yakonov, V. A.; Kadikova, G. N.; Kolokol'tsev, D. I.; Ramazanov, I. R.; Dzhemilev, U. M. Eur. J. Org. Chem. 2015, 2015, 4464.
(c) D'yakonov, V. A.; Kadikova, G. N.; Nasretdinov, R. N.; Kolokol'tsev, D. I.; Dzhemilev, U. M. Tetrahedron Lett. 2017, 58, 1714.
(d) D'yakonov, V. A.; Kadikova, G. N.; Khalilov, L. M.; Dzhemilev, U. M. Russ. J. Org. Chem. 2018, 54, 832.
-
[53]
Achard, M.; Tenaglia, A.; Buono, G. Org. Lett. 2005, 7, 2353. doi: 10.1021/ol050618j
-
[54]
Achard, M.; Mosrin, M.; Tenaglia, A.; Buono, G. J. Org. Chem. 2006, 71, 2907. doi: 10.1021/jo052630v
-
[55]
Clavier, H.; Le Jeune, K.; de Riggi, I.; Tenaglia, A.; Buono, G. Org. Lett. 2011, 13, 308. doi: 10.1021/ol102783x
-
[56]
(a) D'yakonov, V. A.; Kadikova, G. N.; Gazizullina, G. F.; Khalilov, L. M.; Dzhemilev, U. M. Tetrahedron Lett. 2015, 56, 2005.
(b) D'yakonov, V. A.; Kadikova, G. N.; Gazizullina, G. F.; Dzhemilev, U. M. Russ. Chem. Bull. 2016, 65, 200.
-
[57]
D'yakonov, V. A.; Kadikova, G. N.; Gazizullina, G. F.; Dzhemilev, U. M. ChemistrySelect 2018, 3, 6221. doi: 10.1002/slct.201801028
-
[58]
(a) D'yakonov, V. A.; Kadikova, G. N.; Nasretdinov, R. N.; Dzhemileva, L. U.; Dzhemilev, U. M. Eur. J. Org. Chem. 2020, 623.
(b) Kadikova, G. N.; D'yakonov, V. A.; Nasretdinov, R. N.; Dzhemileva, L. U.; Dzhemilev, U. M. Mendeleev Commun. 2020, 30, 318.
-
[59]
Oonishi, Y.; Hosotani, A.; Sato, Y. J. Am. Chem. Soc. 2011, 133, 10386. doi: 10.1021/ja203824v
-
[60]
Oonishi, Y.; Hosotani, A.; Sato, Y. Angew. Chem., Int. Ed. 2012, 51, 11548. doi: 10.1002/anie.201206508
-
[61]
Liu, T.; Han, L.; Han, S.; Bi, S. Organometallics 2015, 34, 280. doi: 10.1021/om501118e
-
[62]
(a) Xia, Y.; Liang, Y.; Chen, Y.; Wang, M.; Jiao, L.; Huang, F.; Liu, S.; Li, Y.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 3470.
(b) Shi, F.-Q.; Li, X.; Xia, Y.; Zhang, L.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 15503.
(c) Liang, Y.; Liu, S.; Xia, Y.; Li, Y.; Yu, Z.-X. Chem.-Eur J. 2008, 14, 4361.
(d) Liang, Y.; Zhou, H.; Yu, Z.-X. J. Am. Chem. Soc. 2009, 131, 17783.
(e) Liang, Y.; Liu, S.; Yu, Z.-X. Synlett 2009, 905.
(f) Mercier, E.; Fonovic, B.; Henry, C.; Kwon, O.; Dudding, T. Tetrahedron Lett. 2007, 48, 3617.
(g) González, I.; Pla-Quintana, A.; Roglans, A.; Dachs, A.; Solà, M.; Parella, T.; Farjas, J.; Roura, P.; Lloveras, V.; Vidal-Gancedo, J. Chem. Commun. 2010, 46, 2944.
(h) Zhao, L.; Wen, M.; Wang, Z.-X. Eur. J. Org. Chem. 2012, 19, 3587.
-
[63]
Faustino, H.; Alonso, I.; Mascareñas, J. L.; López, F. Angew. Chem., Int. Ed. 2013, 52, 6526. doi: 10.1002/anie.201302713
-
[64]
Faustino, H.; Bernal, P.; Castedo, L.; López, F.; Mascareñas, J. L. Adv. Synth. Catal. 2012, 354, 1658. doi: 10.1002/adsc.201200047
-
[65]
(a) Rigby, J. H.; Henshilwood, J. A. J. Am. Chem. Soc. 1991, 113, 5122.
(b) Rigby, J. H.; Ateeq, H. S.; Charles, N. R.; Henshilwood, J. A.; Short, K. M.; Sugathapala, P. M. Tetrahedron 1993, 49, 5495.
(c) Rigby, J. H.; Ahmed, G.; Ferguson, M. D. Tetrahedron Lett. 1993, 34, 5397.
(d) Rigby, J. H.; Sandanayaka, V. P. Tetrahedron Lett. 1993, 34, 935.
(e) Rigby, J. H.; Pigge, F. C.; Ferguson, M. D. Tetrahedron Lett. 1994, 35, 8131.
(f) Rigby, J. H.; Sugathapala, P.; Heeg, M. J. J. Am. Chem. Soc. 1995, 117, 8851.
(g) Rigby, J. H.; Kondratenko, M. A.; Fiedler, C. Org. Lett. 2000, 2, 3917.
(h) Rigby, J. H.; Laurent, S. B.; Kamal, Z.; Heeg, M. J. Org. Lett. 2008, 10, 5609.
-
[66]
Rigby, J. H.; Kirova-Snover, M. Tetrahedron Lett. 1997, 38, 8153. doi: 10.1016/S0040-4039(97)10221-0
-
[67]
De, S.; Misra, S.; Rigby, J. H. Org. Lett. 2015, 17, 3230. doi: 10.1021/acs.orglett.5b01326
-
[68]
Magauer, T.; Mulzer, J.; Tiefenbacher, K. Org. Lett. 2009, 11, 5306. doi: 10.1021/ol902263k
-
[69]
Yao, Z.-K.; Li, J.; Yu, Z.-X. Org. Lett. 2011, 13, 134. doi: 10.1021/ol102700m
-
[70]
Wang, Y.; Wang, J.; Su, J.; Huang, F.; Jiao, L.; Liang, Y.; Yang, D.; Zhang, S.; Wender, P. A.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 10060. doi: 10.1021/ja072505w
-
[71]
Jiang, G.-J.; Fu, X.-F.; Li, Q.; Yu, Z.-X. Org. Lett. 2012, 14, 692. doi: 10.1021/ol2031526
-
[72]
Fu, X.-F.; Xiang, Y.; Yu, Z.-X. Chem.-Eur J. 2015, 21, 4242. doi: 10.1002/chem.201405712
-
[73]
Wender, P. A.; Gamber, G. G.; Hubbard, R. D.; Zhang, L. J. Am. Chem. Soc. 2002, 124, 2876. doi: 10.1021/ja0176301
-
[74]
Wang, Y.; Yu, Z.-X. Acc. Chem. Res. 2015, 48, 2288. doi: 10.1021/acs.accounts.5b00037
-
[75]
Fan, X.; Zhuo, L.-G.; Tu, Y. Q.; Yu, Z.-X. Tetrahedron 2009, 65, 4709. doi: 10.1016/j.tet.2009.04.020
-
[76]
Jiao, L.; Yuan, C.; Yu, Z.-X. J. Am. Chem. Soc. 2008, 130, 4421. doi: 10.1021/ja7100449
-
[77]
Yuan, C.; Jiao, L.; Yu, Z.-X. Tetrahedron Lett. 2010, 51, 5674. doi: 10.1016/j.tetlet.2010.08.028
-
[78]
(a) Schuda, P. F.; Phillips, J. L.; Morgan, T. M. J. Org. Chem. 1986, 51, 2742.
(b) Nishida, M.; Iseki, K.; Shibasaki, M.; Ikegami, S. Chem. Pharm. Bull. 1990, 38, 3230.
(c) Banwell, M. G.; Austin, K. A. B.; Willis, A. C. Tetrahedron 2007, 63, 6388.
-
[79]
Fan, X.; Liu, C.-H.; Yu, Z.-X. Rhodium(Ⅰ)-Catalyzed Cycloadditions Involving Vinylcyclopropanes and Their Derivatives. In Rhodium Catalysis in Organic Synthesis, Ed.:Tanaka, K., Wiley-VCH, Weinheim, Germany, 2019, pp. 229~276.


 下载:
下载:


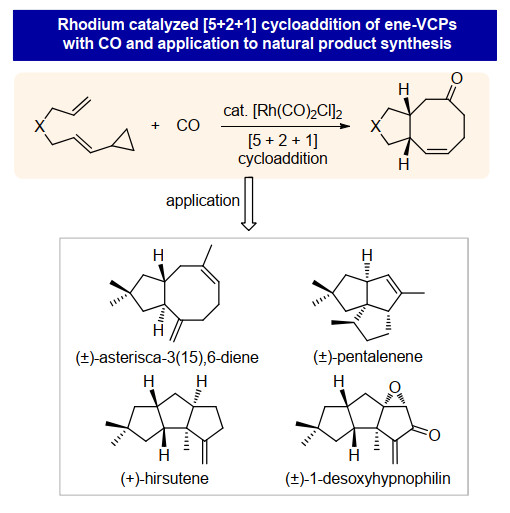
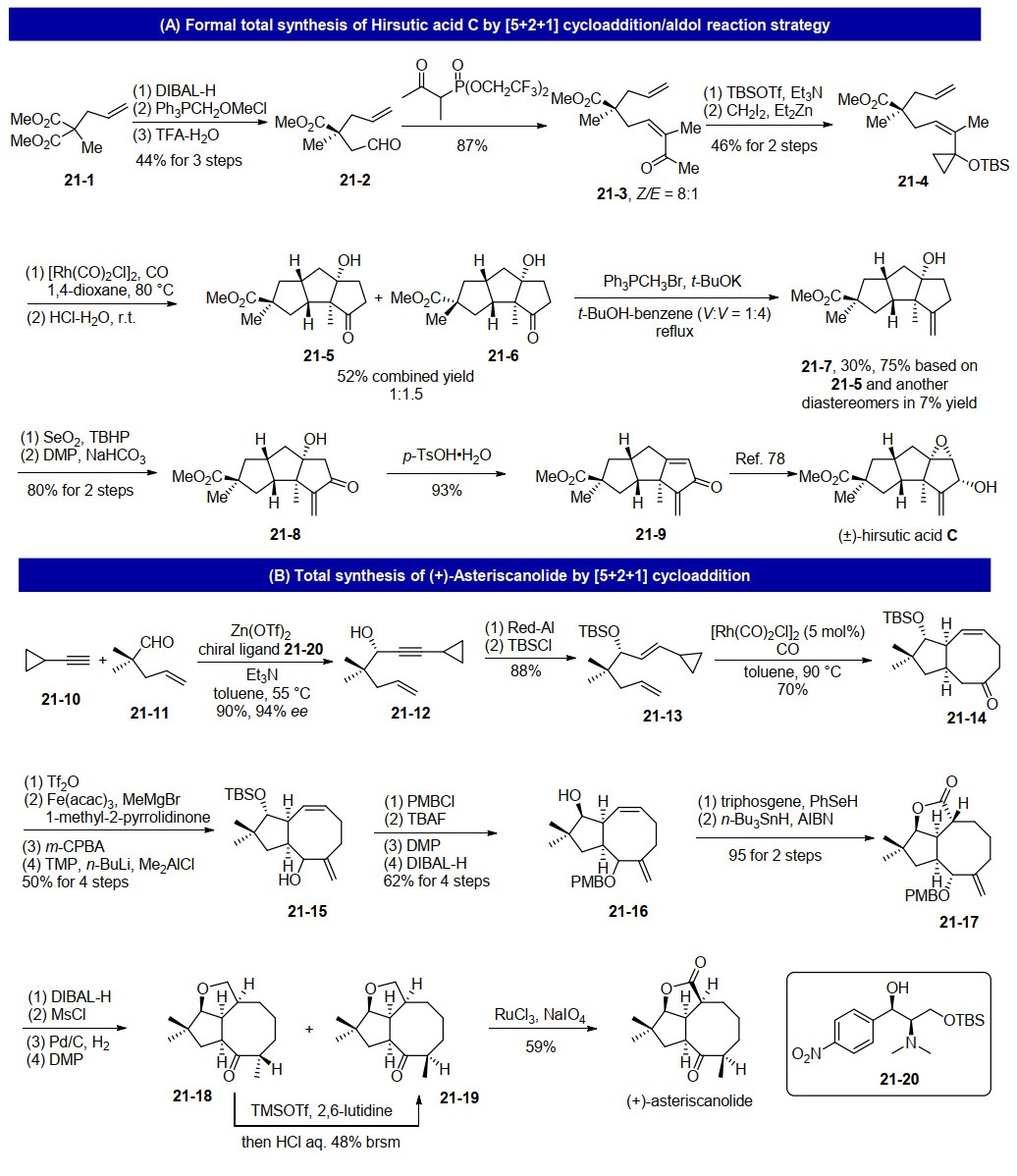
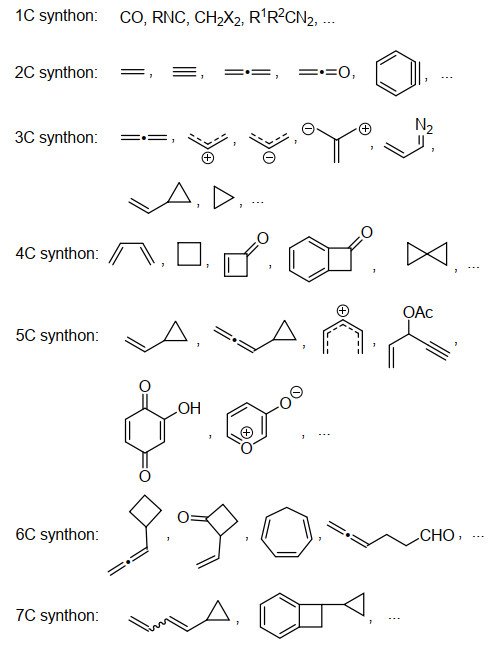

 下载:
下载:























