Received Date:
22 July 2020 Revised Date:
29 July 2020 Available Online:
25 November 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21532003, 21871152, 21790332)
Abstract:
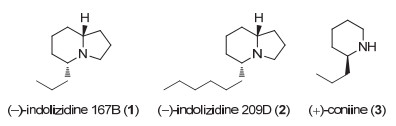
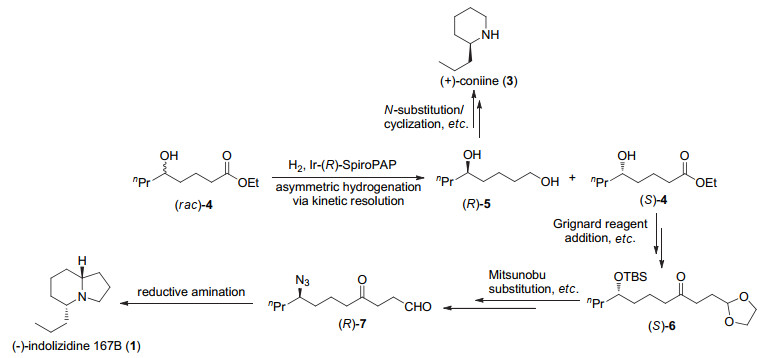
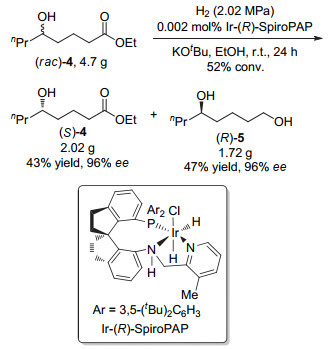
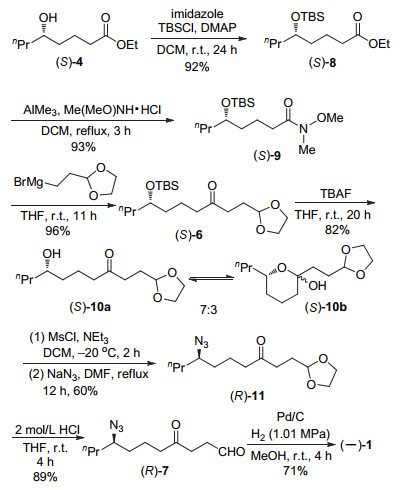
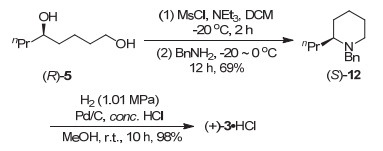
The enantioselective syntheses of (-)-indolizidine 167B and (+)-coniine were described based on the asymmetric hydrogenation of racemic δ-hydroxy esters via kinetic resolution. With optically active chiral δ-hydroxy ester (S)-4 and chiral 1, 5-diol (R)-5 obtained by asymmetric hydrogenation of racemic ethyl 5-hydroxyoctanoate (rac-4) with chiral spiro iridium catalyst Ir-(R)-SpiroPAP as chiral starting materials, the efficient enantioselective syntheses of (-)-indolizidine 167B and (+)-coniine were achieved by using intramolecular reductive amination and N-substitution/cyclization, respectively, as a key step to construct the chiral aza-bicyclic[4.3.0]nonane skeleton and chiral piperidine ring. This provides new efficient methods for enantioselective syntheses of indolizidine and piperidine alkaloids.
Dedicated to the 40th anniversary of Chinese Journal of Organic Chemistry.
[1]
(a) Daly, J. W.; Spande, T. F. In Alkaloids: Chemical and Biological Perspectives, Ed.: Pelletier, S. W., Wiley, New York, 1986, Vol. 4, p. 1. (b) Daly, J. W.; Nishizawa, Y.; Padgett, W. L.; Tokuyama, T. Smith, A. L.; Holmes, A. B.; Kibayashi, C.; Arostam, R. S. Neurochem. Res. 1991, 16, 1213. (c) Michael, J. P. Nat. Prod. Rep. 2007, 24, 191. (d) Michael, J. P. Nat. Prod. Rep. 2008, 25, 139. (e) Ratmanova, N. K.; Andreev, I. A.; Leontiev, A.; Momotova, D.; Novoselov, A. M.; Ivanova, O. A.; Trushkov, I. V. Tetrahedron2020, 76, 131031.
[2]
(a) Daly, J. W. Fortschr. Chem. Org. Naturst. 1982, 41, 205. (b) Aronstam, R. S.; Daly, J. W.; Spande, T. F.; Narayanan, T. K.; Albuquerque, E. X. Neurochem. Res. 1986, 11, 1227.
[3]
For selected recent papers for enantioselective synthesis of indolizidine 167B and 209D, see: (a) Sun, Z.; Yu, S.; Ding, Z.; Ma, D. J. Am. Chem. Soc. 2007, 129, 9300. (b) Yu, R. T.; Lee, E. E.; Malik, G.; Rovis, T. Angew. Chem., Int. Ed. 2009, 48, 2379. (c) Kapat, A.; Nyfeler, E.; Giuffredi, G. T.; Renaud, P. J. Am. Chem. Soc. 2009, 131, 17746. (d) Liu, H.; Su, D.; Cheng, G.; Xu, J.; Wang, X.; Hu, Y. Org. Biomol. Chem. 2010, 8, 1899. (e) Abels, F.; Lindemann, C.; Schneider, C. Chem.-Eur. J. 2014, 20, 1964. (f) Li, Y.-J.; Hou, C.-C.; Chang, K.-C. Eur. J. Org. Chem. 2015, 1659. (g) Chiou, W.-H.; Chen, H.-Y. RSC Adv. 2017, 7, 684. (h) Koo, S. M.; Vendola, A. J.; Momm, S. N.; Morken, J. P. Org. Lett. 2020, 22, 666.
[4]
For selected recent papers for enantioselective synthesis of coniine, see: (a) Sattely, E. S.; Cortez, G. A.; Moebius, D. C.; Schrock, R. R.; Hoveyda, A. H. J. Am. Chem. Soc. 2005, 127, 8526. (b) Beng, T. K.; Gawley, R. E. J. Am. Chem. Soc. 2010, 132, 12216. (c) Ren, H.; Wulff, W. D. J. Am. Chem. Soc. 2011, 133, 5656. (d) Arena, G.; Zill, N.; Salvadori, J.; Girard, N.; Mann, A.; Taddei, M. Org. Lett. 2011, 13, 2294. (e) Damodar, K.; Lingaiah, M.; Bhunia, N.; Das, B. Synthesis2011, 2478. (f) Bosque, I.; Gonzá-Gómez, J. C.; Foubelo, F.; Yus, M. J. Org. Chem. 2012, 77, 780. (g) Ren, H.; Wulff, W. D. Org. Lett. 2013, 15, 242. (h) Berthold, D.; Geissler, A. G. A.; Giofré, S.; Breit, B. Angew. Chem. Int. Ed. 2019, 58, 9994.
[5]
Mody, N. V.; Henson, R.; Hedin, P. A.; Kokpol, U.; Miles, D. H. Experientia1976, 32, 829. doi: 10.1007/BF02003710

 下载:
下载:

 下载:
下载:






