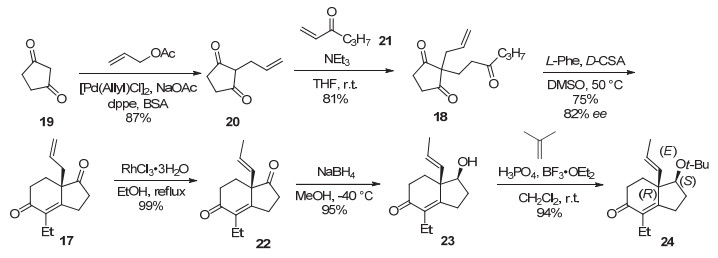
图 1.
(-)-BTX, homoBTX及(-)-BTX-A的结构
Figure 1.
Structures of (-)-BTX, homoBTX and (-)-BTX-A

箭毒蛙毒素(-)-batrachotoxin [(-)-BTX (1), 图 1]是20世纪60年代从哥伦比亚北部雨林箭毒蛙(Phyllobates aurotaenia)的皮肤中提取到的甾体类生物碱[1], 后来在巴布亚新几内亚Pitohui鸟的羽毛中也被发现[2]. (-)-BTX是目前已知毒性最强的非肽类神经毒素之一, 其LD50仅为2 μg/kg.除了其极强的毒性之外, 它还是一种非常重要的钠离子通道(Navs)特异性激活剂, 对研究细胞离子通道的结构和功能具有重要价值[3].在分离(-)-BTX的同时还得到(-)-batrachotoxinin A [(-)-BTX-A, 2]和(-)-homobatrachotoxin (3)]. (-)-BTX与(-)-BTX-A具有相同的骨架结构, (-)-BTX的C-20位为羟基的吡咯甲酸酯, 而(-)-BTX-A C-20位是一个游离的羟基.后者的毒性要低1000倍, 可以通过酯化反应转化为(-)-BTX. (-)-BTX以及(-)-BTX-A的分子结构包括绝对和相对立体化学, 系通过单晶X射线衍射法以及化学转化得到确定[4].
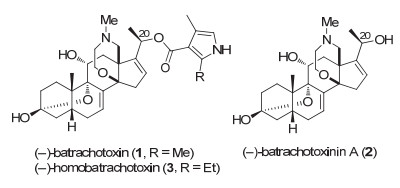
(-)-BTX所表现的出色而重要生理活性, 使之极具生物、医药、毒理研究价值.然而其在自然界中的含量极低, 无法满足生物、生化和药化研究的需要.加上该分子具有挑战性的复杂结构, 已成为全合成的重要目标分子.近几十年来, 诸多课题组对此开展了合成研究[5-9].然而除了Imhof小组[6]于1972年从11α-乙酰氧基孕酮出发实现了其半合成外, 迄今仅有三个小组完成其全合成. 1998年, Kishi课题组[7]从(±)-Wieland-Miesher酮(4)出发, 通过分子内Diels-Alder反应和氧杂Michael加成等反应, 首次完成了BTX-A外消旋体(±)-2的全合成(Scheme 1, a). 2016年, Du Bois课题组[8]从(+)-Hajos-Parrish酮(7, 也称Hajos-Wiechert酮)出发, 通过自由基串联环化等方法, 首次完成了(-)-BTX (1)及其对映体的不对称全合成(Scheme 1, b).最近, 罗陀平课题组[9]同样从(+)-Hajos-Parrish酮(7)出发, 通过光氧化还原偶联和去对称化等关键反应, 完成了(-)-BTX-A (2)的不对称全合成(Scheme 1, c).此外, 2018年, Inoue小组[5a]也从(±)-Wieland-Miesher酮(4)出发, 通过桥头自由基偶联及Pd/Ni催化的Ullmann反应等关键反应, 实现了BTX四环核心骨架的构筑.
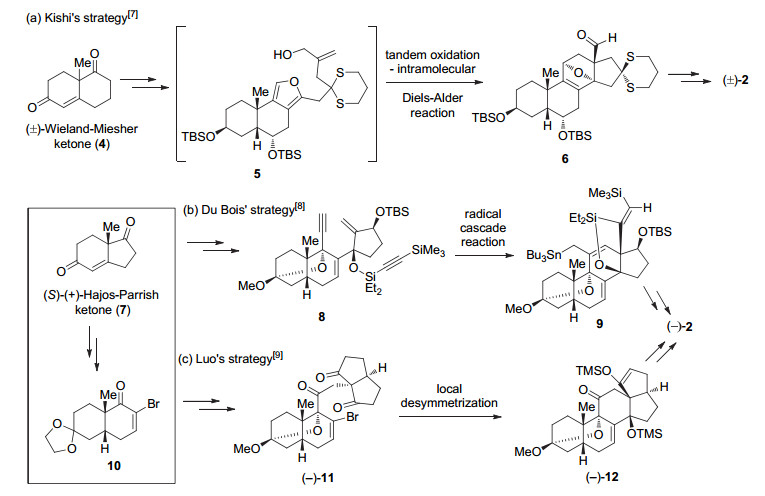
基于本课题组对箭毒蛙生物碱[10a- 10b]、生物活性复杂天然产物[10c- 10h]全合成以及相关高效合成方法学[11]的兴趣, 我们开展了BTX的全合成研究.本文将报道batrachotoxin不对称全合成的一种新策略及合成探索的初步结果, 即官能化的CD环的构筑.
结构上, (-)-BTXs为甾体生物碱, 以高氧代甾体四环为核心骨架, 包含一个分子内的3β-半缩酮亚结构和一个氮杂氧杂七元桥环, 由此构成该天然产物的六环结构.如Scheme 1所示, 上述Kishi、Du Bois和罗陀平小组的全合成均以AB环为出发点, 依次引入D环和C环, 即采用AB+D+C的合成策略.其他研究组的合成研究大多集中于ABC环的构筑[5].我们对(-)-BTX (1)进行的逆合成分析见Scheme 2, 采用的是以A环与CDE环通过Diels-Alder反应构筑B环的汇聚合成策略.由于亲双烯体A环13为已知化合物[12], 因此, 研究的重点是双烯CDE骨架14的构筑. 14可由15经脱氢生成.而后者有望通过对C环共轭烯酮的分子内氧杂共轭加成构造氮杂氧杂七元桥环.在Kishi的全合成中[7], 该七元桥环系通过对D环共轭烯酮的分子内氧杂共轭加成构筑. CD环16中官能化的角甲基可由烯丙基转化而得.最后, Hajos-Wiechert酮类似物17计划由三酮18通过Hajos-Wiechert型反应[13](Hajos-Wiechert反应亦称Hajos-Parrish-Eder-Sauer-Wiechert反应)合成[14].
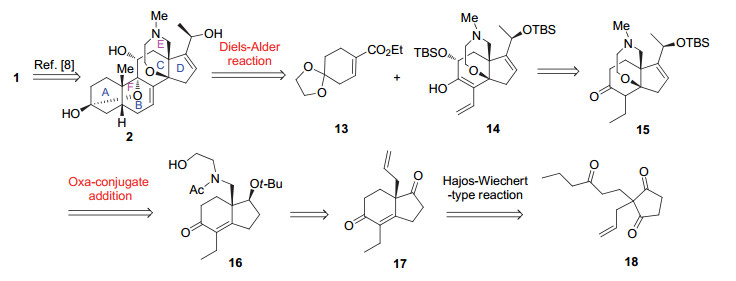
合成始于商品化试剂1, 3-环戊二酮19.首先依Williams等[15]报道的步骤, 在[Pd(Allyl)Cl]2催化下与烯丙基醋酸酯进行Tsuji-Trost烯丙基化[16], 以87%的产率得到2-烯丙基1, 3-环戊二酮(20)[17].随后, 20与1-己烯-3-酮(21)在NEt3作用下进行Michael加成反应[18].当按文献方法以乙酸乙酯为溶剂, 顺利得到了三羰基化合物18, 但产率仅为37%.反应产率太低可归因于化合物20在乙酸乙酯中溶解性较差.将溶剂换成四氢呋喃后产率提高到81% (Scheme 1).
接下来, 重点研究了三酮18的Hajos-Wiechert型反应[13], 即有机小分子催化的分子内的去对称化以构筑含手性季碳的CD片段17.经典的Hajos-Wiechert环化反应底物在L-脯氨酸催化下可以获得较高的产率(71%)和对映选择性(99.5% ee)[14c].对非经典底物18的反应, 需要重新筛选催化剂及反应条件, 这方面已有许多报道[13].参照Ando等[13b- 13c]的方法, 以L-苯丙氨酸为有机手性催化剂, D-10-樟脑磺酸(D-CSA)作添加剂, 在乙腈中反应, 仅以53%的产率和64%的ee值得到环化产物(+)-17.为此, 对反应的溶剂进行了筛选.以D-10-樟脑磺酸为添加剂, 分别在N, N-二甲基甲酰胺(DMF)、1, 4-二氧六环、甲苯和二甲基亚砜中反应.实验结果表明, 溶剂对反应的影响较大(表 1).其中, 以DMF作溶剂, 产率为83%, 但ee值只有73%(表 1, Entry 2).而在1, 4-二氧六环和甲苯中反应, 产率和ee值均不理想(ee值分别为63%和53%, 表 1, Entries 3, 4).以二甲基亚砜为溶剂, 可以获得较高的产率和ee值(表 1, Entry 5, 产率75%, ee值为81%).还探索了对甲苯磺酸(p-TSA)和对甲苯磺酸吡啶盐(PPTS)作为添加剂对反应的影响.结果显示, 反应产率分别下降13%和14%, 对ee值基本没有影响(表 1, Entries 6, 7).将催化剂L-苯丙氨酸的用量从100 mol%降为30 mol%, ee值随之降为15% (Entry 8).最终确定了以二甲亚砜作溶剂, D-10-樟脑磺酸作添加剂的最佳反应条件(表 1, Entry 5).在该条件下, 以75%的产率和81%的ee合成了烯丙基化的CD环.通过后续化合物24的衍生化及与已知化合物比对, 推定化合物17季碳中心的绝对构型为R.
 下载:
导出CSV
下载:
导出CSV
 |
||||||
| Entry | Additive | Solvent | Temp./℃ | Time | Yieldb/% | eec/% |
| 1 | D-CSA | MeCN | 30~70 | 7 d | 53 | 64 |
| 2 | D-CSA | DMF | 50 | 24 h | 83 | 73 |
| 3 | D-CSA | 1, 4-dioxane | 50 | 7 d | 51 | 63 |
| 4 | D-CSA | Toluene | 50 | 7 d | 45 | 53 |
| 5 | D-CSA | DMSO | 50 | 24 h | 75 | 81 |
| 6 | p-TSA | DMSO | 50 | 24 h | 61 | 81 |
| 7 | PPTS | DMSO | 50 | 24 h | 62 | 81 |
| 8d | D-CSA | DMSO | 50 | 24 h | — | 15 |
| a Reaction condition: To a solution of the compound 18 (1.0 mmol) in solvent (2 mL) was added successively L-Phe (1.0 mmol, 1.0 equiv.) and additive (0.5 mmol, 0.5 equiv.). The reaction was monitored by TLC until the compound 18 consumed completely. b Isolated yield; c The ee value was detected by chiral HPLC; d L-Phe (0.3 mmol, 0.3 equiv.). | ||||||
在合成了光学活性的CD环(+)-17后, 接着研究了其向CDE环的转化.首先是末端烯烃双键的迁移.根据Benn等[19]报道的方法, 烯丙基化合物17在三氯化钌水合物催化下, 几乎定量地被转化为内烯烃22.根据烯烃双键上邻位两个氢之间偶合常数(J=15.5 Hz), 确定了其构型为E型.随后, 化合物22在-40 ℃下经硼氢化钠(0.5 equiv.)还原, 选择性地将饱和酮羰基还原[20], 高产率、高立体选择性地得到单一构型的化合物23 (95%).在H3PO4/BF3•OEt2催化下23与异丁烯反应[21], 以94%的产率得到叔丁基保护的产物24.化合物24的相对和绝对立体化学通过与从已知化合物[22]转化得到的化合物的比旋光、1H NMR谱和13C NMR谱数据比对确定.
接下来, 探索了化合物24中非共轭烯烃双键的选择性氧化切除.遗憾的是, 尝试多种氧化方法(m-CPBA/ NaIO4[23], OsO4/NMO/NaIO4[24], K2OsO4/NMO, RuCl3• 3H2O/NaIO4[25])均未获成功.推测可能是相邻季碳位阻的影响导致反应无法进行.随后, 尝试了臭氧化断裂双键的方法.然而, 当按照常规的臭氧解操作[26], 以反应液呈淡蓝色作为反应终止的标志, 没有分离到预期的产物25.我们推测, 可能化合物24中的共轭烯烃双键也被氧化[27].为避免过度氧化, 通过加入显色剂(油溶紫)来控制臭氧化反应的时间, 以选择性地氧化非共轭双键, 实验结果见表 2.当通入臭氧100和150 s时, 分别以38%和47%的产率得到目标产物25, 并回收61%和50%的原料(表 1, Entries 1, 2).将反应时间延长至200 s时, 产率提高到68%(表 1, Entry 3).而当反应时间为250 s时, 原料全部转化, 产率达到82%(表 1, Entry 4).继续延长反应时间, 产率开始降低(71%, 表 1, Entry 5).由此, 确定烯烃24臭氧化反应的时间是250 s.
下载:
导出CSV
 |
|||
| Entry | Time/s | Yield a/% of 25 | Recovery a/% of 24 |
| 1 | 100 | 38 | 61 |
| 2 | 150 | 47 | 50 |
| 3 | 200 | 68 | 10 |
| 4 | 250 | 82 | 0 |
| 5 | 300 | 71 | 0 |
| a isolated yield. | |||
得到化合物25后, 25与O-TBS保护的乙醇胺26经还原胺化[NaBH(OAc)3]得化合物27.后者经乙酰化(AcCl/NEt3)得酰胺28, 两步总产率77%.化合物28在四丁基氟化铵作用下顺利脱除TBS保护基, 以85%的产率生成CDE环前体16 (Scheme 4).
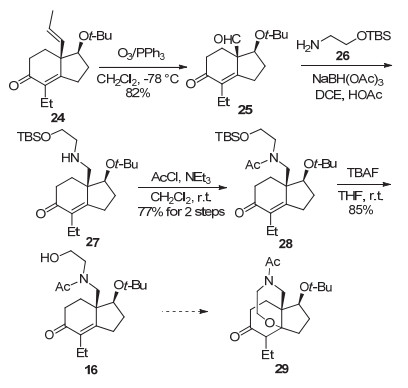
最后, 对化合物16分子内氧杂Michael加成反应以构建E环的条件进行初步探索.遗憾的是, 尝试了多种碱性[1, 8-二氮杂二环十一碳-7-烯(DBU)、NaH、t-BuOK]及酸性条件(Tf2NH)[28], 均未得到期望的环化产物29.
设计了(-)-batrachotoxin (1)的不对称全合成新策略: A环与CDE环通过Diels-Alder反应构建B环和整个分子中的五环骨架, 并探索了CDE骨架的构筑.从1, 3-环戊二酮出发, 经L-苯丙氨酸催化的Hajos-Wiechert型反应, 以81%的ee合成了Hajos-Wiechert酮类似物17.随后通过臭氧氧化及还原胺化等七步反应合成了(-)-BTX分子中官能化的CD环, 即CDE骨架的前体16.进一步的工作正在进行中, 结果将另文报道.
旋光用Perkin-Elmer 341型旋光仪测定; 1H NMR和13C NMR谱用Bruker公司AvanceII-400/500型核磁共振仪(400 MHz/100 MHz、500 MHz/125 MHz)测定, 以Me4Si为内标测定.红外谱图由Nicolet Avatar 360型FTIR仪测定(KBr盐片涂膜); 高分辨质谱由Finnigan公司Mat-LCQ型质谱仪测定; 熔点由Yanaco MP-500型熔点仪测定(温度计未校正).柱色谱采用300~400目硅胶(烟台江友硅胶开发有限公司); 洗脱剂采用乙酸乙酯和石油醚(b.p. 60~90 ℃); ee值由Agilent 1260型高效液相色谱测定; 所用其他溶剂均用标准方法纯化后使用.
将[PdCl(Allyl)]2 (91.5 mg, 0.25 mmol)和1, 2-双(二苯基膦)乙烷(400 mg, 1.0 mmol)溶于无水四氢呋喃(THF) (25 mL)中, 然后依次加入1, 3-环戊二酮(1.47 g, 15.0 mmol)、烯丙基醋酸酯(1.00 g, 10.0 mmol)、N, O-双三甲硅基乙酰胺(BSA, 3.05 g, 15.0 mmol)和醋酸钠(34.0 mg, 0.25 mmol).混合物在氮气保护下加热回流反应18 h后, 减压蒸馏除去溶剂.残留物重新溶于二氯甲烷(50 mL), 并用盐酸(1.0 mol/L, 15 mL)洗涤.水相用二氯甲烷萃取(15 mL×2).合并有机相, 无水硫酸钠干燥, 过滤, 减压浓缩得1.20 g淡黄色粗产物2-烯丙基-1, 3-环戊二酮(20), 产率87%.该粗产物不经纯化直接用于下一步反应.
将化合物20 (1.10 g, 7.96 mmol)和1-己烯-3-酮21 (1.16 mL, 9.95 mmol)溶于THF (50 mL)中, 然后加入三乙胺(1.66 mL, 11.9 mmol).混合物在室温下反应24 h.待原料反应完全后, 减压蒸馏除去大部分溶剂.加入20 mL二氯甲烷稀释, 用10 mL水洗.水相用二氯甲烷萃取(10 mL×3).合并有机相, 用2 mL饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物用硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:10]分离得到1.52 g 2-烯丙基-2-(3-氧己基)环戊烷-1, 3-二酮(18), 产率81%.无色液体. 1H NMR (500 MHz, CDCl3) δ: 5.63~5.51 (m, 1H), 5.12~5.02 (m, 2H), 2.86~2.74 (m, 2H), 2.70~2.58 (m, 2H), 2.40 (t, J=7.2 Hz, 2H), 2.33 (t, J=6.6 Hz, 4H), 1.90 (t, J=7.2 Hz, 2H), 1.59~1.49 (m, 2H), 0.88 (t, J=7.6 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 215.7 (2C), 210.2, 131.1, 120.0, 59.3, 44.7, 39.4, 36.5, 35.7 (2C), 27.0, 17.1, 13.6; IR (film) vmax: 2962, 2933, 2875, 1721, 1417, 993, 924 cm-1; HRMS-ESI calcd for C14H20O3Na [M+Na]+ 259.1305, found 259.1309.
将化合物18 (1.76 g, 7.45 mmol)溶于无水二甲基亚砜(DMSO) (15 mL)中, 然后加入L-苯丙氨酸(1.23 g, 7.45 mmol)和D-10-樟脑磺酸(0.87 g, 3.72 mmol).混合物在50 ℃下搅拌24 h后, 加入50 mL乙酸乙酯和50 mL水稀释, 水相用乙酸乙酯萃取(50 mL×3).合并有机相, 用2 mL饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物用硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:10]分离得1.22 g化合物17, 产率75%.淡黄色油状液体.产物对映体过量(ee)通过手性HPLC (Chiralcel AS-H)测定, ee值为81%[色谱条件为: V(hexane):V(IPA)=99:1, 1.0 mL/min, λ=254 nm].保留时间分别为: tR(minor)=18.2 min, tR(major)=20.4 min.[α]D20+234.9 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.78~5.62 (m, 1H), 5.19~5.02 (m, 2H), 2.96~2.85 (m, 1H), 2.85~2.72 (m, 1H), 2.72~2.60 (m, 1H), 2.57~2.33 (m, 5H), 2.31~2.21 (m, 2H), 2.16 (ddd, J=13.7, 5.5, 1.8 Hz, 1H), 1.73 (ddd, J=13.7, 13.7, 5.8 Hz, 1H), 0.94 (t, J=7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 216.5, 197.1, 161.4, 136.5, 131.9, 119.2, 52.4, 39.4, 35.7, 32.4, 26.5, 24.3, 18.5, 13.4; IR (film) vmax: 2964, 2932, 2872, 1743, 1663, 1447, 1358, 1159, 1119, 919 cm-1; HRMS-ESI calcd for C14H18O2Na [M+Na]+ 241.1199, found 241.1196.
将化合物17 (1.91 g, 8.76 mmol)溶于乙醇(35 mL)中, 加入RhCl3•3H2O (0.12 g, 0.44 mmol), 反应液成棕色.加热回流反应1 h后, 补加RhCl3•3H2O (0.12 g, 0.44 mmol), 继续回流反应1 h.降至室温, 减压蒸馏除去溶剂.将浓缩液直接用硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:10]分离得1.90 g化合物22, 产率99%.淡黄色油状液体. [α]D20+66.9 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.48 (dq, J=15.5, 6.5 Hz, 1H), 5.22 (dq, J=15.5, 1.5 Hz, 1H), 2.94~2.81 (m, 1H), 2.78~2.63 (m, 2H), 2.40~2.25 (m, 5H), 2.08 (ddd, J=13.2, 4.6, 2.6 Hz, 1H), 1.77 (ddd, J=5.7, 13.2, 13.2 Hz, 1H), 1.68 (dd, J=6.5, 1.5 Hz, 3H), 0.97 (t, J=7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 214.5, 197.8, 159.0, 137.7, 130.0, 128.2, 56.5, 35.5, 32.9, 28.9, 24.1, 18.4, 17.9, 13.6; IR (film) vmax: 2963, 2933, 2872, 1746, 1714, 1667, 1449, 1349, 1155, 1119, 974 cm-1; HRMS-ESI calcd for C14H18O2Na [M+Na]+ 241.1199, found 241.1196.
将化合物22 (7.21 g, 33 mmol)溶于甲醇(132 mL)中, 在-40 ℃下分批加入NaBH4 (0.63 g, 16.5 mmol).加完后在该温度下继续反应1 h.待原料反应完全后, 依次加入2 mL丙酮和饱和碳酸氢钠(10 mL)淬灭反应.减压蒸馏除去溶剂, 然后加入100 mL二氯甲烷和50 mL水溶解.水相用二氯甲烷萃取(30 mL×3).合并有机相, 用5 mL饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物用硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:2]分离得6.88 g化合物23, 产率95%.粘稠液体. [α]D20+100.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.55 (dq, J=15.8, 1.6 Hz, 1H), 5.30 (dq, J=15.8, 6.4 Hz, 1H), 3.77 (dd, J=11.1, 6.7 Hz, 1H), 2.78 (br s, 1H), 2.52~2.31 (m, 3H), 2.28~2.08 (m, 4H), 2.03~1.94 (m, 1H), 1.79~1.65 (m, 5H), 0.92 (t, J=7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 199.24, 163.91, 136.89, 128.54, 127.76, 80.65, 51.89, 33.62, 32.26, 29.83, 24.99, 18.71, 18.31, 13.09; IR (film) vmax: 3424, 2931, 2870, 1644, 1448, 1352, 1258, 1103, 977 cm-1; HRMS-ESI calcd for C14H20O2Na [M+Na]+ 243.1356, found 243.1357.
将化合物23 (5.79 g, 26.3 mmol)溶于异丁烯的二氯甲烷溶液(2.0 mol/L, 132 mL)中, 然后加入磷酸(0.88 g, 8.95 mmol).氮气保护后, 在-78 ℃下滴加三氟化硼乙醚(1.10 mL, 8.95 mmol).在-78 ℃下搅拌2 h后在室温下继续反应18 h.待原料反应完全后, 在冰浴下加入氨水(20 mL)淬灭反应.水相用二氯甲烷萃取(30 mL×3).合并有机相, 用2 mL饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物用硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:10]分离得6.85 g化合物24, 产率94%.淡黄色蜡状固体产物. m.p. 81~83 ℃; [α]D20+55.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.56 (dq, J=15.8, 1.6 Hz, 1H), 5.16 (dq, J=15.8, 6.5 Hz, 1H), 3.50 (dd, J=10.7, 6.6 Hz, 1H), 2.45~2.24 (m, 3 H), 2.24~2.05 (m, 3H), 1.94 (ddd, J=12.7, 5.0, 2.2 Hz, 1H), 1.84~1.75 (m, 1H), 1.69~1.57 (m, 5H), 1.14 (s, 9H), 0.91 (t, J=7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 198.9, 163.0, 136.4, 128.8, 126.6, 79.9, 72.9, 51.2, 33.4, 32.4, 29.9, 28.4 (3C), 24.9, 18.5, 18.0, 13.0; IR (film) vmax: 2970, 2871, 1667, 1453, 1362, 1191, 1105, 977, 907 cm-1; HRMS-ESI calcd for C18H28O2Na [M+Na]+ 299.1982, found 299.1985.
于25 mL Schlenk管中将化合物24 (250 mg, 0.90 mmol)溶于16 mL二氯甲烷中, 然后加入油溶紫(1.0 mg).在-78 ℃下搅拌后通入臭氧250 s.加入PPh3 (285 mg, 1.09 mmol), 在室温下搅拌2 h后.减压蒸馏除去溶剂, 所得残留物过硅胶柱纯化[V(乙酸乙酯):V(石油醚)=1:10], 得196 mg化合物25, 产率82%.白色固体. m.p. 98~100 ℃; [α]D20+84.6 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 9.78 (s, 1H), 3.90 (dd, J=10.0, 7.3 Hz, 1H), 2.81~2.66 (m, 2H), 2.59~2.47 (m, 1H), 2.41 (ddd, J=17.8, 5.2, 2.0 Hz, 1H), 2.35~2.12 (m, 4H), 1.94~1.75 (m, 2H), 1.19 (s, 9H), 0.59 (t, J=7.6 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ: 201.7, 197.2, 157.5, 138.1, 81.1, 74.0, 60.8, 34.0, 31.4, 28.3 (3C), 28.0, 26.3, 19.0, 12.9; IR (film) vmax: 2971, 2932, 2873, 1709, 1654, 1463, 1349, 1257, 1194, 1088, 797 cm-1; HRMS-ESI calcd for C16H24O3Na [M+Na]+ 287.1618, found 287.1621.
将化合物25 (594 mg, 2.25 mmol)溶于1, 2-二氯乙烷(10 mL), 然后依次加入化合物26 (1.18 g, 6.74 mmol)和NaBH(OAc)3 (1.43 g, 6.74 mmol).室温下搅拌12 h后, 在冰浴下加入5 mL饱和碳酸氢钠淬灭反应.加入15 mL二氯甲烷稀释, 水相用二氯甲烷萃取(15 mL×3).合并有机相, 用2 mL饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩得淡黄色油状液体.将粗产物溶于20 mL干燥的二氯甲烷中, 在冰浴下依次加入三乙胺(1.60 mL, 11.2 mmol)和乙酰氯(0.48 ml, 6.74 mmol).加完后在室温下反应4 h.待原料反应完后, 在冰浴下加入5 mL饱和碳酸氢钠溶液淬灭反应.水相用二氯甲烷萃取(15 mL×3).合并有机相, 用2 mL饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物用硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:5]分离得0.81 g化合物28, 两步总产率77%.淡黄色油状液体. [α]D20+26.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 4.00~3.52 (m, 5H), 3.51~3.31 (m, 2H), 2.78~2.06 (m, 8H), 1.99 (s, 3H), 1.95~1.63 (m, 2H), 1.19 (s, 9H, minor), 1.16 (s, 9H, major), 0.92 (t, J=7.7 Hz, 3H), 0.86 (s, 9H, major), 0.86 (s, 9H, minor), 0.03 (s, 6H, major), 0.00 (s, 6H, minor); 13C NMR (100 MHz, CDCl3) δ: 198.3 (major), 197.4 (minor), 172.0 (major), 171.7 (minor), 162.9, 137.1 (minor), 136.5 (major), 81.8 (major), 80.7 (minor), 73.8 (minor), 73.3 (major), 61.2, 52.5 (major), 51.1 (minor), 49.6 (minor), 49.0 (major), 48.7 (minor), 47.8 (major), 33.6 (major), 33.4 (minor), 32.8 (major), 32.5 (minor), 30.3 (major), 30.2 (minor), 28.7 (minor), 28.6 (major), 26.3 (minor), 26.2 (major), 25.9 (minor), 25.8 (major), 22.0 (minor), 21.9 (major), 19.0 (major), 18.9 (minor), 18.15 (major), 18.10 (minor), 12.4 (major), 12.3 (minor), -5.5; IR (film) vmax: 2959, 2930, 2858, 1659, 1463, 1362, 1257, 1193, 1104, 927, 836, 778 cm-1; HRMS-ESI calcd for C26H47NO4SiNa [M+Na]+ 488.3167, found 488.3165.
将化合物28 (751 mg, 1.61 mmol)溶于THF (8 mL)中, 在冰浴下滴加四丁基氟化铵的THF溶液(1.0 mol/L, 3.20 mL, 3.20 mmol).加完后在室温下反应4 h.加入饱和NaHCO3淬灭反应.减压蒸馏除去溶剂, 然后加入乙酸乙酯(30 mL)和水(20 mL)溶解, 水相用乙酸乙酯萃取(20 mL×3).合并有机相, 用2 mL饱和食盐水洗涤, 无水硫酸钠干燥, 过滤, 减压浓缩.粗产物用硅胶柱层析[V(乙酸乙酯):V(石油醚)=1:5]分离得482 mg化合物16, 产率85%.淡黄色油状液体. [α]D20+51.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 3.93~3.63 (m, 4H), 3.62~3.32 (m, 4H), 2.72~2.06 (m, 8H), 2.01 (s, 3H), 1.94~1.60 (m, 2H), 1.18 (s, 9H, minor), 1.16 (s, 9H, major), 0.92 (t, J=7.5 Hz, 3H, minor), 0.89 (t, J=7.5 Hz, 3H, major); 13C NMR (100 MHz, CDCl3) δ: 198.4 (major), 197.5 (minor), 173.7 (minor), 172.8 (major), 163.2 (major), 162.8 (minor), 137.3 (minor), 136.5 (major), 81.7 (major), 80.6 (minor), 74.0 (minor), 73.4 (major), 61.2 (minor), 60.0 (major), 52.7, 50.5 (major), 50.1 (minor), 48.9 (major), 48.6 (minor), 47.5, 33.6 (major), 33.3 (minor), 32.7 (major), 32.5 (minor), 30.3 (major), 30.2 (minor), 28.64 (minor), 28.60 (major), 26.4 (minor), 26.1 (major), 21.9, 19.0 (major), 18.9 (minor), 12.3; IR (film) vmax: 3452, 2971, 2932, 2873, 1644, 1420, 1363, 1258, 1193, 1088, 753 cm-1; HRMS-ESI calcd for C20H33NO4Na [M+Na]+ 374.2302, found 374.2297.
辅助材料(Supporting Information) 化合物24绝对和相对立体化学的确定, 化合物16~18, 22~25, 28的1H NMR和13C NMR碳谱以及化合物17的手性HPLC谱图.这些材料可以免费从本刊网站(http://sioc-jour-nal.cn/)上下载.
(a) Märki, F.; Witkop, B. Experientia 1963, 19, 329.
(b) Daly, J. W.; Witkop, B.; Bommer, P.; Biemann, K. J. Am. Chem. Soc. 1965, 87, 124.
Dumbacher, J. P.; Beehler, B. M.; Spande, T. F.; Garraffo, H. M.; Daly, J. W. Science 1992, 258, 799. doi: 10.1126/science.1439786
(a) Linford, N. J.; Cantrell, A. R.; Qu, Y.; Scheuer, T.; Catterall, W. A. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 13947.
(b) Li, H.-L.; Hadid, D.; Ragsdale, D. S. Mol. Pharmacol. 2002, 61, 905.
(c) Wang, S.-Y; Mitchell, J.; Tikhonov, D. B.; Zhorov, B. S.; Wang, G. K. Mol. Pharmacol. 2006, 69, 788.
(d) Du, Y.; Garden, D. P.; Wang, L.; Zhorov, B. S.; Dong, K. J. Biol. Chem. 2011, 286, 13151.
(e) Toma, T.; Logan, M. M.; Menard, F.; Devlin, A. S; Du Bois, J. ACS Chem. Neurosci. 2016, 7, 1463.
(f) Ayvazyan, N. M.; O'Leary, V. B.; Dolly, J. O.; Ovsepian, S. V. Drug Discovery Today 2019, 24, 1968.
(a) Tokuyama, T.; Daly, J.; Witkop, B.; Karle, I. L.; Karle, J. J. Am. Chem. Soc. 1968, 90, 1917.
(b) Tokuyama, T.; Daly, J.; Witkop, B. J. Am. Chem. Soc. 1969, 91, 3931.
(a) Sakata, K.; Wang, Y.-H.; Urabe, D.; Inoue, M. Org. Lett. 2018, 20, 130.
(b) Sloan Devlin, A.; Du Bois, J. Chem. Sci. 2013, 4, 1059.
(c) Lacrouts, P.; Parsons, P. J.; Penkett, C. S.; Raza, A. R. Synlett 2005, 2767.
(d) Trudeau, S.; Deslongchamps, P. J. Org. Chem. 2004, 69, 832.
(e) Sultan, A.; Raza, A. R.; Khan, K. M. J. Chem. Soc. Pak. 2013, 35, 1369.
(f) Kurosu, M.; Kishi, Y. Tetrahedron Lett. 1998, 39, 8929.
(g) Grinsteiner, T. J.; Kishi, Y. Tetrahedron Lett. 1994, 35, 8337.
(h) Grinsteiner, T. J.; Kishi, Y. Tetrahedron Lett. 1994, 35, 8333.
(i) Hudson, P.; Pairaudeau, G.; Parsons, P. J.; Jahans, A. W.; Drew, M. G. B. Tetrahedron Lett. 1993, 34, 7295.
(j) Magnus, P.; Leapheart, T.; Walker, C. J. Chem. Soc., Chem. Commun. 1985, 1185.
(k) Keana, J. F. W.; Schumaker, R. R. J. Org. Chem. 1976, 41, 3840.
(l) Schumaker, R. R.; Keana, J. F. W. J. Chem. Soc., Chem. Commun. 1972, 622.
(a) Imhof, R.; Gössinger, E.; Graf, W.; Berner, H.; Berner-Fenz, L.; Wehrli, H. Helv. Chim. Acta 1972, 55, 1151.
(b) Imhof, R.; Gössinger, E.; Graf, W.; Berner-Fenz, L.; Berner, H.; Schaufelberger, R.; Wehrli, H. Helv. Chim. Acta 1973, 56, 139.
Kurosu, M.; Marcin, L. R.; Grinsteiner, T. J.; Kishi, Y. J. Am. Chem. Soc. 1998, 120, 6627. doi: 10.1021/ja981258g
Logan, M. M.; Toma, T.; Thomas-Tran, R.; Du Bois, J. Science 2016, 354, 865. doi: 10.1126/science.aag2981
Guo, Y.-L.; Guo, Z.-X.; Lu, J.-T.; Fang, R.-T.; Chen, S.-C.; Luo, T.-P. J. Am. Chem. Soc. 2020, 142, 3675. doi: 10.1021/jacs.9b12882
(a) Deng, H.-Q.; Qian, X.-Y.; Li, Y.-X.; Zheng, J.-F.; Xie, L.-F.; Huang, P.-Q. Org. Chem. Front. 2014, 1, 258.
(b) Liu, X.-K.; Zheng, X.; Ruan, Y.-P.; Ma, J.; Huang, P.-Q. Org. Biomol. Chem. 2012, 10, 1275.
(c) Yang, R.-F.; Huang, P.-Q. Chem.-Eur. J. 2010, 16, 10319.
(d) Huang, P.-Q.; Huang, S.-Y.; Gao, L.-H.; Mao, Z.-Y.; Chang, Z.; Wang, A.-E Chem. Commun. 2015, 51, 4576.
(e) Guo, L.-D.; Huang, X.-Z.; Luo, S.-P.; Cao, W.-S.; Ruan, Y.-P.; Ye, J.-L.; Huang, P.-Q. Angew. Chem., Int. Ed. 2016, 55, 4064.
(f) Geng, H.; Huang, P.-Q. Chem. Rec. 2019, 19, 523.
(g) Huang, P.-Q. Chin. J. Org. Chem. 2018, 38, 2461(in Chinese). (黄培强, 有机化学, 2018, 38, 2461.)
(h) Liu, Z.-J.; Huang, P.-Q. J. Org. Chem. 2019, 84, 5627.
(a) Liu, Y.-P.; Zhu, C.-J.; Yu, C.-C.; Wang, A.-E; Huang, P.-Q. Eur. J. Org. Chem. 2019, 7169.
(b) Xu, Z.; Wang, X.-G.; Wei, Y.-H.; Ji, K.-L.; Zheng, J.-F.; Ye, J.-L.; Huang, P.-Q. Org. Lett. 2019, 21, 7587.
(c) Wang, S.-R.; Huang P.-Q. Chin. J. Chem. 2019, 37, 887.
(d) Wu, D.-P.; He, Q.; Chen, D.-H.; Ye, J.-L.; Huang, P.-Q. Chin. J. Chem. 2019, 37, 315.
(e) Ye, J.-L.; Zhu, Y.-N.; Geng, H.; Huang, P.-Q. Sci. China: Chem. 2018, 61, 687.
(f) Ou, W.; Han, F.; Hu, X.-N.; Chen, H.; Huang, P.-Q. Angew. Chem., Int. Ed. 2018, 57, 11354.
Danishefsky, S.; Kitahara, T.; Yan, C.-F.; Morris, J. J. Am. Chem. Soc. 1979, 101, 6996. doi: 10.1021/ja00517a036
(a) Hagiwara, H.; Uda, J. J. Org. Chem. 1988, 53, 2308.
(b) Hagiwara, H.; Sakai, H.; Uchiyama, T.; Ito, Y.; Morita, N.; Hoshi, T.; Suzuki, T.; Ando, M. J. Chem. Soc., Perkin Trans. 12002, 583.
(c) Sakai, H.; Hagiwara, H.; Ito, Y.; Hoshi, T.; Suzuki, T.; Ando, M. Tetrahedron Lett. 1999, 40, 2965.
(d) Przeździecka, A.; Stepanenko, W.; Wicha, J. Tetrahedron: Asymmetry 1999, 10, 1589.
(e) Shigehisa, H.; Mizutani, T.; Tosaki, S. Y.; Ohshima, T.; Shibasaki, M. Tetrahedron 2005, 61, 5057.
(f) Zhang, X.-M.; Wang, M.; Tu, Y.-Q.; Fan, C.-A.; Jiang, Y.-J.; Zhang, S.-Y.; Zhang, F.-M. Synlett 2008, 2831.
(a) Hajos, Z. G.; Parrish, D. R. DE 2102623, 1971.
(b) Eder, U.; Sauer, G.; Wiechert, R. Angew. Chem., Int. Ed. Engl. 1971, 10, 496.
(c) Hajos, Z. G.; Parrish, D. R. J. Org. Chem. 1974, 39, 1615. For an essay, see:
(d) Barbas III, C. F. Angew. Chem., Int. Ed. 2008, 47, 42.
Ruprah, P. K.; Cros, J. P.; Pease, J. E.; Whittingham, W. G.; Williams, J. M. J. Eur. J. Org. Chem. 2002, 3145.
(a) Tsuji, J.; Takahashi H.; Morikawa, M. Tetrahedron Lett. 1965, 6, 4387.
(b) Trost, B. M.; Fullerton, T. J. J. Am. Chem. Soc. 1973, 95, 292. For a review, see:
(c) Trost, B. M. Angew. Chem., Int. Ed. Engl. 1989, 28, 1173.
Schmiedel, V. M.; Hong, Y. J.; Lentz, D.; Tantillo, D. J.; Christmann, M. Angew. Chem., Int. Ed. 2018, 57, 2419. doi: 10.1002/anie.201711766
Peixoto, P. A.; Jean, A.; Maddaluno, J.; De Paolis, M. Angew. Chem., Int. Ed. 2013, 52, 6971. doi: 10.1002/anie.201301465
(a) Hanselmann, R.; Benn, M. Synth. Commun. 1996, 26, 945.
(b) Corbu, A.; Gauron, G.; Castro, J. M.; Dakir, M.; Arseniyadis, S. Org. Lett. 2007, 9, 4745.
Brady, T. P.; Kim, S. H.; Wen, K.; Theodorakis, E. A. Angew. Chem., Int. Ed. 2004, 43, 739. doi: 10.1002/anie.200352868
Hajos, Z. G.; Mitcheli, R. A.; Parrish, D. R.; Oliveto, E. P. J. Org. Chem. 1967, 32, 3008.
(a) Zeng, C.; Zheng, C.; Zhao, J.; Zhao, G. Org. Lett. 2013, 15, 5846.
(b) Corbu, A.; Gauron, G.; Castro, J. M.; Dakir, M.; Arseniyadis, S. Org. Lett. 2007, 9, 4745.
(a) Schroeter, M.; Weyerstahl, P. Synth. Commun. 2004, 34, 1535.
(b) Sridhar, Y.; Srihari, P. Org. Biomol. Chem. 2013, 11, 4640.
(a) Takikawa, H.; Hosoe, A.; Ueda, K.; Sasaki, M. Biosci. Biotechnol. Biochem. 2004, 68, 1961.
(b) Butera, J.; Bagli, J.; Doubleday, W.; Humber, L.; Treasurywala, A.; Loughney, D.; Sestanj, K.; Millen, J.; Sredy, J. J. Med. Chem. 1989, 32, 757.
Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J. Org. Chem. 1981, 46, 3936. doi: 10.1021/jo00332a045
Hanselmann, R.; Benn, M. Synth. Commun. 1996, 26, 945. doi: 10.1080/00397919608003699
Letort, A.; Long, D.-L.; Prunet, J. J. Org. Chem. 2016, 81, 12318. doi: 10.1021/acs.joc.6b02264
Wabnitz, T. C.; Spencer, J. B. Org. Lett. 2003, 5, 2141. doi: 10.1021/ol034596h

图 1 (-)-BTX, homoBTX及(-)-BTX-A的结构
Figure 1 Structures of (-)-BTX, homoBTX and (-)-BTX-A

图式 1 已报道(±)-BTX-A和(-)-BTX-A全合成路线的主要特征
Scheme 1 Key features of the reported total syntheses of (±)-BTX-A and (-)-BTX-A

图式 2 (-)-Batrachotoxin (1)的逆合成分析
Scheme 2 Retrosynthetic analysis of (-)-batrachotoxin (1)

图式 3 (-)-BTX官能化的CD片段16的合成
Scheme 3 Synthesis of functionalized CD segment 16 of (-)-BTX
表 1 L-苯丙氨酸催化的18的Hajos-Wiechert型反应的探索a
Table 1. Investigation on the L-phenylalanine-catalyzed Hajos-Wiechert-type reaction of 18
|
||||||
| Entry | Additive | Solvent | Temp./℃ | Time | Yieldb/% | eec/% |
| 1 | D-CSA | MeCN | 30~70 | 7 d | 53 | 64 |
| 2 | D-CSA | DMF | 50 | 24 h | 83 | 73 |
| 3 | D-CSA | 1, 4-dioxane | 50 | 7 d | 51 | 63 |
| 4 | D-CSA | Toluene | 50 | 7 d | 45 | 53 |
| 5 | D-CSA | DMSO | 50 | 24 h | 75 | 81 |
| 6 | p-TSA | DMSO | 50 | 24 h | 61 | 81 |
| 7 | PPTS | DMSO | 50 | 24 h | 62 | 81 |
| 8d | D-CSA | DMSO | 50 | 24 h | — | 15 |
| a Reaction condition: To a solution of the compound 18 (1.0 mmol) in solvent (2 mL) was added successively L-Phe (1.0 mmol, 1.0 equiv.) and additive (0.5 mmol, 0.5 equiv.). The reaction was monitored by TLC until the compound 18 consumed completely. b Isolated yield; c The ee value was detected by chiral HPLC; d L-Phe (0.3 mmol, 0.3 equiv.). | ||||||
 下载: 导出CSV
下载: 导出CSV
表 2 臭氧化反应时间的探索
Table 2. Investigation on reaction time of the ozonolysis
|
|||
| Entry | Time/s | Yield a/% of 25 | Recovery a/% of 24 |
| 1 | 100 | 38 | 61 |
| 2 | 150 | 47 | 50 |
| 3 | 200 | 68 | 10 |
| 4 | 250 | 82 | 0 |
| 5 | 300 | 71 | 0 |
| a isolated yield. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们