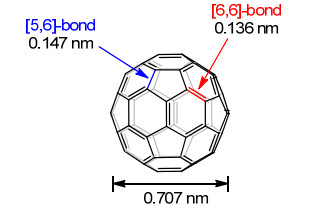
图 1.
C60的分子结构
Figure 1.
The structure of C60

富勒烯(fullerene)作为碳的第三种同素异形体, 不同于已知的石墨和金刚石, 它具有独特的笼状分子结构. Osawa[1]在1970年从理论上预测了[60]富勒烯(C60)具有类似足球的高度对称的Ih结构. 1985年, Smalley、Curl和Kroto等[2]从实验中发现了第一个富勒烯C60.此后C70、C76、C78、C80等一系列类似的笼状分子也相继被发现.这种C60分子, 连同其它一系列结构多样并具有球形结构的碳的同素异形体, 被统称为“富勒烯”.富勒烯的发现使得分子的研究范围从苯的二维平面分子扩展到数目众多的三维笼状分子. 1990年, Krätschmer等[3]首次实现了C60的克级宏量制备.自此, 富勒烯在化学、物理、材料和生命科学等领域的研究进入快速发展期, 在诸多领域里展现出了独特性能, 具有广泛的应用前景.富勒烯分子中, 每个sp2杂化的碳原子与三个相邻的碳原子成键, 构成六边形和五边形的模块.对于一个经典的富勒烯C2n, 根据欧拉定理, 六边形和五边形的数目分别为n-10 (n≠11)和12[4].正是这12个五边形决定了球状分子的曲率, 对富勒烯的稳定性至关重要. C60因其独特的结构和特殊的物理化学性质, 自发现以来一直受到广泛关注.它具有高度对称的Ih结构, 有32个面、12个正五边形、20个正六边形(图 1)[5].其分子中五元环与六元环之间的键称为[5,6]-键, 两个六元环之间的键称为[6,6]-键. C60有60个[5,6]-键和30个[6,6]-键.由于[6,6]-键具有更多的双键性质, 因此反应一般发生在[6,6]-键上. C60的化学性质类似于缺电子的共轭多烯烃, 易于发生环加成、亲核加成、自由基加成和还原反应等多种反应[6].富勒烯因其独特的结构和性质, 在太阳能电池、微电子器件、抗癌药物、光限幅材料、自组装、润滑油、催化剂、化妆品和超导材料等方面具有广泛的应用[7], 吸引了科学家们浓厚的兴趣.
X射线单晶衍射和核磁共振研究表明, C60以及高富勒烯完全共轭[8].在C60被发现后不久, Haddon等[9]通过计算发现, C60的最低未占据分子轨道(LUMO)处于能量较低的三重简并态, 在被还原时最多能够接受6个电子.随后, 循环伏安法(CV)、差分脉冲伏安法(DPV)、Osteryoung方波伏安法(OSWV)和恒电位电解法(CPE)等电分析技术迅速成为研究富勒烯及其衍生物电学性质的重要手段[10].对富勒烯进行衍生化可以改变富勒烯的电学性质, 提高其材料性能以及溶解度.另外, 在富勒烯骨架表面连接不同官能团, 可以实现功能多元化[7].但是运用经典的化学方法对富勒烯衍生物进行表面修饰时, 往往具有反应效率低、反应条件苛刻、反应选择性较差和多加成异构体混合物难以分离等缺点.由于富勒烯独特的电学性质而新发展起来的富勒烯电化学功能化反应, 因其优异的选择性和可控性, 已被认为是制备各种富勒烯衍生物的新颖且高效的策略.近年来, 人们发现富勒烯稠合杂环化合物在恒电位电解获得两个电子后, 富勒烯骨架上的碳-杂原子键会发生断裂开环生成单键连接的富勒烯二负离子, 并在与亲电试剂反应过程中重排成键.因此这种单键连接的富勒烯二负离子已成为构筑新型富勒烯衍生物的重要模块. Echegoyen等[10]在1998年对早期富勒烯及其衍生物的电化学反应进行了系统评述.但由于当时富勒烯电化学发展时间较短, 涵盖的电化学反应较少, 没有涉及到富勒烯稠合杂环化合物的电化学反应研究.本文将近十年来富勒烯稠合杂环化合物的电化学衍生化反应, 按产物加成模式分类进行了综述, 以期为科研工作者了解富勒烯电化学反应新进展提供帮助.
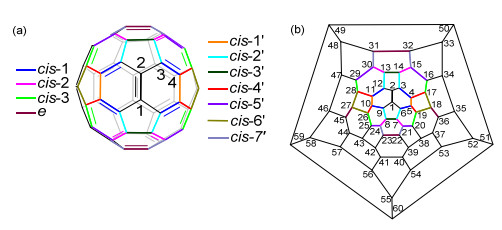
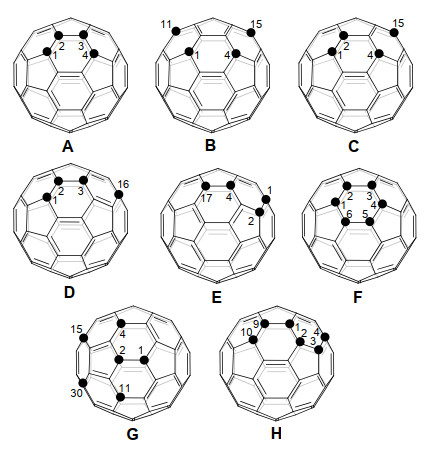
随着加成基团数量的增加, 富勒烯衍生物的区域异构体数量会急剧增加, 从而导致了各异构体繁琐的分离问题.对于C60的[2+1]-、[2+2]-、[2+3]-或[2+4]-环加成反应, 通常会首先得到一个[6,6]-键上的单环加合物, 并可以进一步发生二次环化反应得到8种区域异构体的混合物, 即cis-1、cis-2、cis-3、e等异构体(图 2)[11].理论上, C60的三环加合物和四环加合物分别有46个和262个可能的区域异构体[12].目前已报道的四功能化C60衍生物最常见的是1, 2, 3, 4-加合物(A)[13-14]、1, 4, 11, 15-加合物(B)[15]、1, 2, 4, 15-加合物(C)[16]、1, 2, 3, 16-加合物(D)[17-18]以及1, 2, 4, 17-加合物(E)[19].而六功能化C60衍生物常见的是1, 2, 3, 4, 5, 6-加合物(F)[20]、1, 2, 4, 11, 15, 30-加合物(G)[15, 21]以及1, 2, 3, 4, 9, 10-加合物(H)[19](图 2b, 3).尽管多种多功能化富勒烯衍生物已被报道, 但是多功能化富勒烯衍生物的区域选择性合成仍具有很大的挑战性.
自2011年第一个C60稠合杂环化合物的电化学反应[14a]被报道以来, 越来越多的富勒烯稠合杂环化合物的电化学反应被报道.这些反应按加成模式大致可以分为1, 2, 3, 4-加成、1, 2, 3, 16-加成、1, 4, 9, 25-加成、1, 2, 4, 17-加成、1, 4, 9, 12-加成以及其它加成模式.
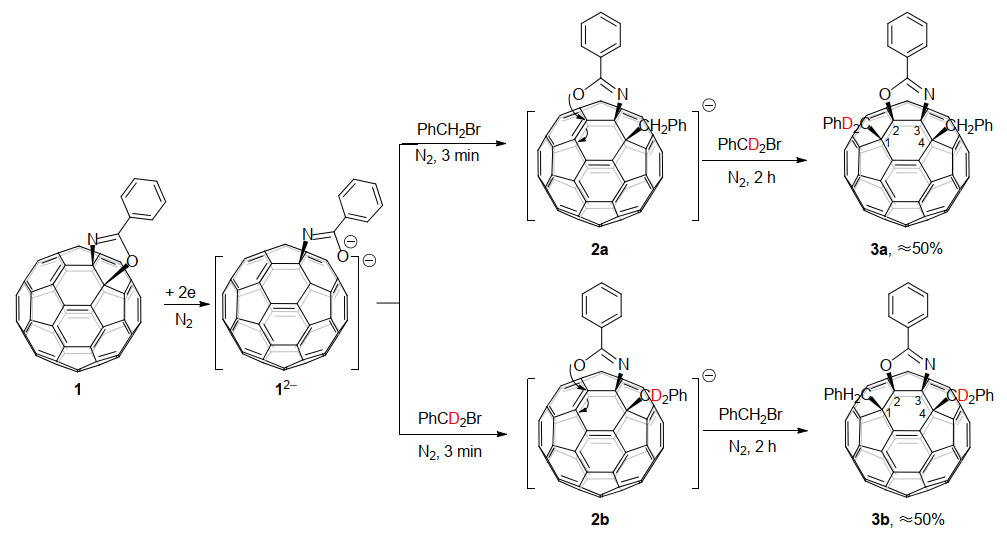
2011年, 高翔课题组[14a]首次报道了富勒烯稠合杂环化合物的电化学衍生化反应.通过电化学方法研究了C60稠合噁唑啉二负离子12-与PhCH2Br的反应, 发现C60稠合噁唑啉得到两个电子后, C60—O键会发生断裂, 在加入PhCH2Br后, 得到了双苄基化的1, 2, 3, 4-C60稠合噁唑啉衍生物3, 产率约为50% (Scheme 1).这个过程伴随着噁唑啉杂环从[6,6]-键到[5,6]-键的断键重排.通过逐步添加PhCH2Br和PhCD2Br, 以区分在每个步骤上加成到C60稠合噁唑啉二负离子的苄基位置, 发现噁唑啉杂环选择性地发生C—O键的断裂开环和重排闭环.另外, 还通过理论计算得到中间体12-和2a的自然键轨道(NBO)电荷密度分布, 证实了苄基进攻位点依次为C(4)和C(1).
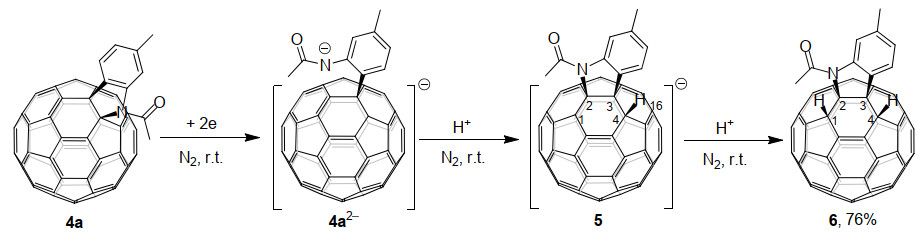
2014年, 王官武课题组[14b]报道了通过电化学方法得到的C60稠合吲哚啉二负离子4a2-的质子化反应.电化学还原使得C60稠合吲哚啉4a得到两个电子后C60—N键发生断裂, 随后用三氟乙酸(TFA)作质子源, 仅需反应20 min, 即可高效选择性地得到二氢化的1, 2, 3, 4-C60稠合吲哚啉衍生物6 (Scheme 2).这个过程也伴随着杂环从[6,6]-键到[5,6]-键的断键与重排.同时, 通过理论计算发现4a2-的富勒烯笼表面电荷最集中的位点为C(16)和C(4), 由此可以得到两种可能的中间体.其中, 更稳定的中间体为进攻C(4)得到的负离子物种5, 继续被质子化后可以得到6.这个结果说明, 产物的区域选择性取决于碳笼表面负电荷的分布、进攻基团的大小和产物的相对稳定性.
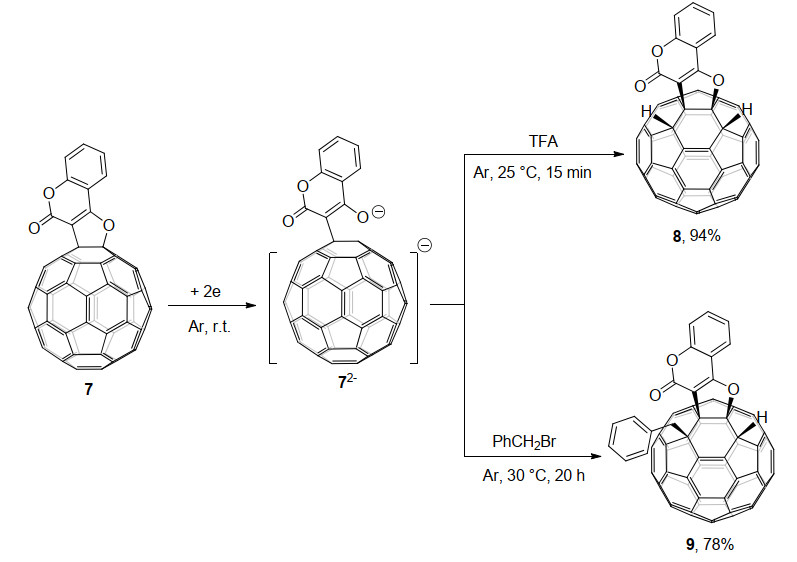
2020年, 该课题组[14c]又报道了电化学得到的C60稠合呋喃香豆素二负离子72-的电化学衍生化反应.他们发现72-可以被TFA质子化, 反应15 min即可高效得到二氢化的1, 2, 3, 4-C60稠合呋喃香豆素衍生物8, 产率为94%.有趣的是, 72-和过量PhCH2Br在30 ℃反应20 h, 可以选择性地得到单苄基化的1, 2, 3, 4-四官能团化富勒烯衍生物9, 产率为78% (Scheme 3).
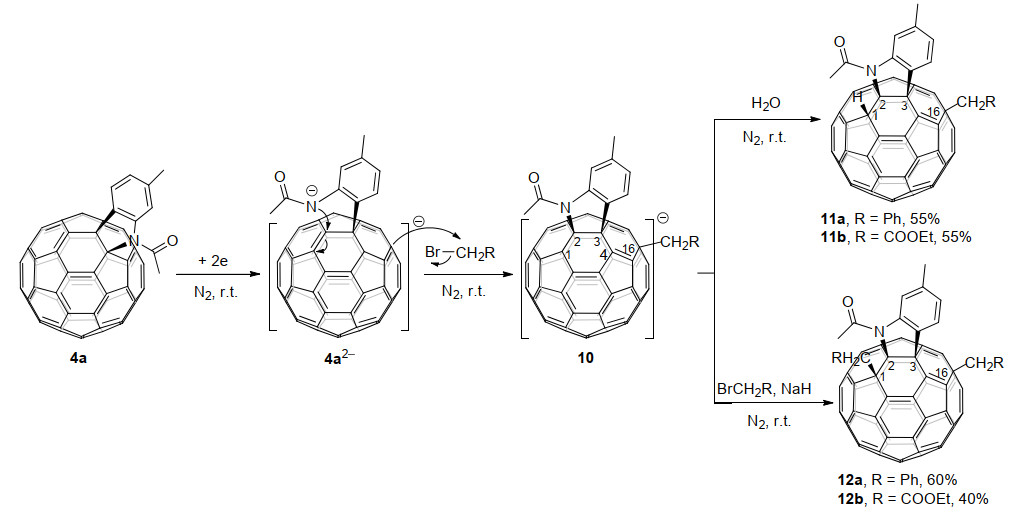
2014年, 王官武课题组[18a]以C60稠合吲哚啉4a为底物, 通过电化学还原得到4a2-, 随后在常温条件下与溴代物反应5 h, 高效选择性地得到了1, 2, 3, 16-加成模式的富勒烯四官能团化衍生物11和12, 并通过11a的单晶X射线衍射证实了产物的分子结构(Scheme 4).有趣的是, 在氢化钠(NaH)不存在或存在的情况下, 富勒烯笼上可以分别区域选择性地加成一个或两个CH2Ph/CH2CO2Et基团.在得到两个电子后, C60稠合吲哚啉发生了C60—N键的断裂, 得到了一个开环的二负离子中间体, 当在C(16)处加成一个CH2Ph/CH2CO2Et基团后, 该中间体从[6,6]-键重排到[5,6]-键生成杂环中间体10.理论计算结果表明, 反应的区域选择性受电荷分布和空间因素的双重控制.
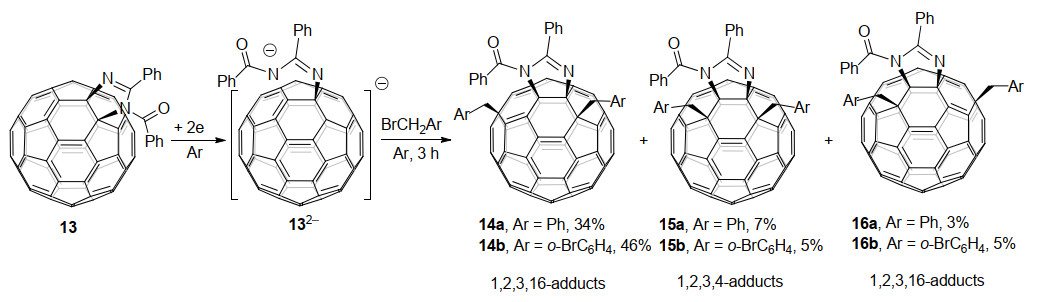
同年, 高翔课题组[18b]也报道了1, 2, 3, 16-四官能团化富勒烯衍生物的电化学合成(Scheme 5).他们用电化学得到的C60稠合咪唑啉二负离子132-与ArCH2Br (Ar=Ph, o-BrPh)反应, 主要得到了四官能团化的1, 2, 3, 16-富勒烯衍生物14, 同时还有少量四官能团化的1, 2, 3, 4-和1, 2, 3, 16-富勒烯衍生物15和16.主产物14b的分子结构也经单晶X射线衍射结构进行了确认.值得一提的是, 这个反应过程中也伴随着C60—N键的断裂与重排, 使杂环从[6,6]-键重排到[5,6]-键.咪唑环上的苯甲酰基团使得反应主要受空间效应影响得到1, 2, 3, 16-加成主产物, 而通过理论计算发现另外两种产物则受电子因素影响生成.但电化学研究表明, 1, 2, 3, 4-加成次要产物的二负离子反而比1, 2, 3, 16-加成主产物的二负离子更稳定, 这也在某种程度上表明, 富勒烯衍生物负离子的稳定性更多地取决于电子因素, 而位阻因素的影响要小得多.此外, 他们还研究了电化学得到的C70稠合咪唑啉二负离子的苄基化反应, 也得到了类似的结果, 但是选择性同样不太理想[22].
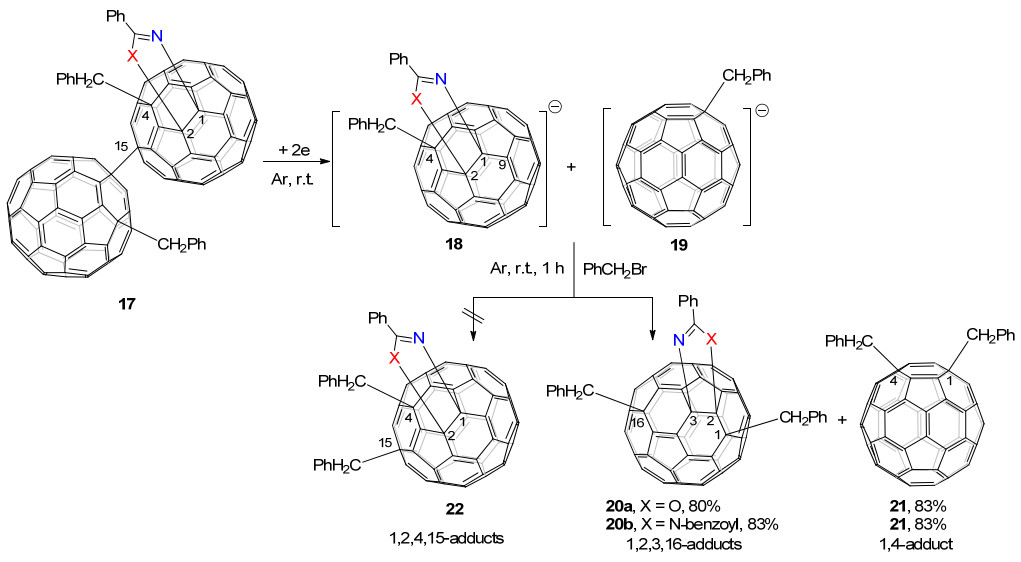
2015年, 该课题组[18c]又发现将C60稠合噁唑啉或咪唑啉单键连接的1, 2, 4, 15-C60二聚体17电还原后, 会解离成1, 2, 4-C60一负离子中间体18以及PhCH2C60一负离子19.之后再与PhCH2Br反应, 出乎意料地也得到了四官能团化的1, 2, 3, 16-C60衍生物19以及双苄基化的1, 4-C60衍生物20, 而不是之前设想的在C(15)位继续发生苄基化反应, 生成四功能化的1, 2, 4, 15-富勒烯衍生物21 (Scheme 6).这说明1, 2, 4-C60一负离子中间体是不稳定的, 会重排为1, 4, 9-C60一负离子.此外, 以二聚体17为底物进行的电化学反应, 得到1, 2, 3, 16-产物的选择性比他们之前报道的C60稠合咪唑啉13为底物的电化学反应[18b]选择性要高, 说明单键连接的富勒烯二聚体也具有作为前体制备新型加成模式富勒烯衍生物的潜力.
2017年, 王官武课题组[18d]报道了一种高区域选择性羰基化的电化学方法, 可以直接得到单羰基化的C60稠合吲哚啉衍生物24 (Scheme 7).通过使用廉价易得的酰氯和氯甲酸酯23与C60稠合吲哚啉二负离子4a2-反应, 酰基和酯基官能团都可以很容易地连接到富勒烯骨架上, 并且选择性地生成四官能团化的1, 2, 3, 16-C60衍生物24.有趣的是, 即使在NaH存在下也只发生了单羰基化反应.另外, 在没有NaH存在下, 则会生成二氢化的1, 2, 3, 4-四官能团化C60衍生物6[14b].
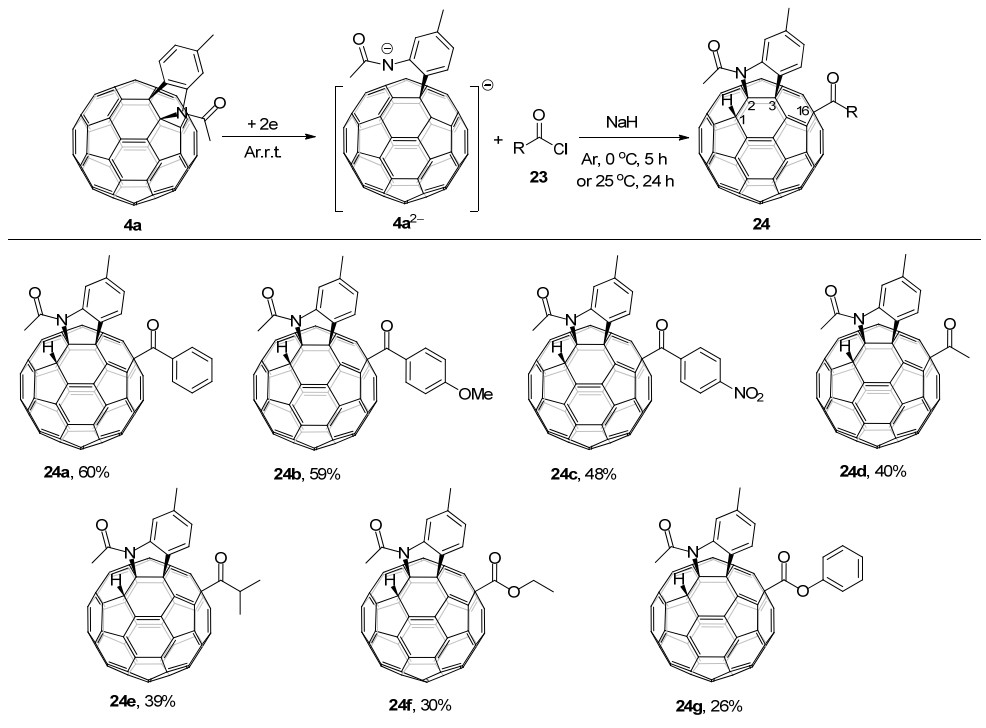
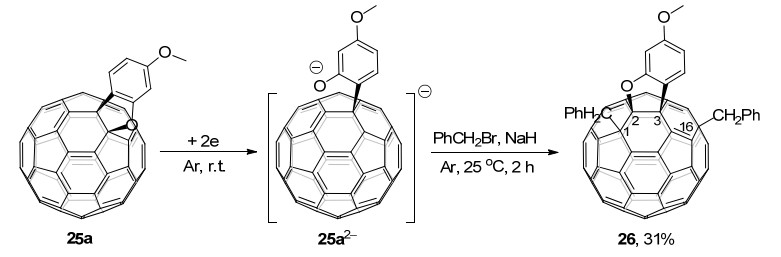
同年, 该课题组[18e]还报道了C60稠合苯并呋喃25a的电化学苄基化反应.电化学还原得到的C60稠合苯并呋喃二负离子25a2-在NaH存在下与PhCH2Br在常温下反应2 h, 可以选择性地得到四官能团化的1, 2, 3, 16-C60衍生物26, 产率为31% (Scheme 8).
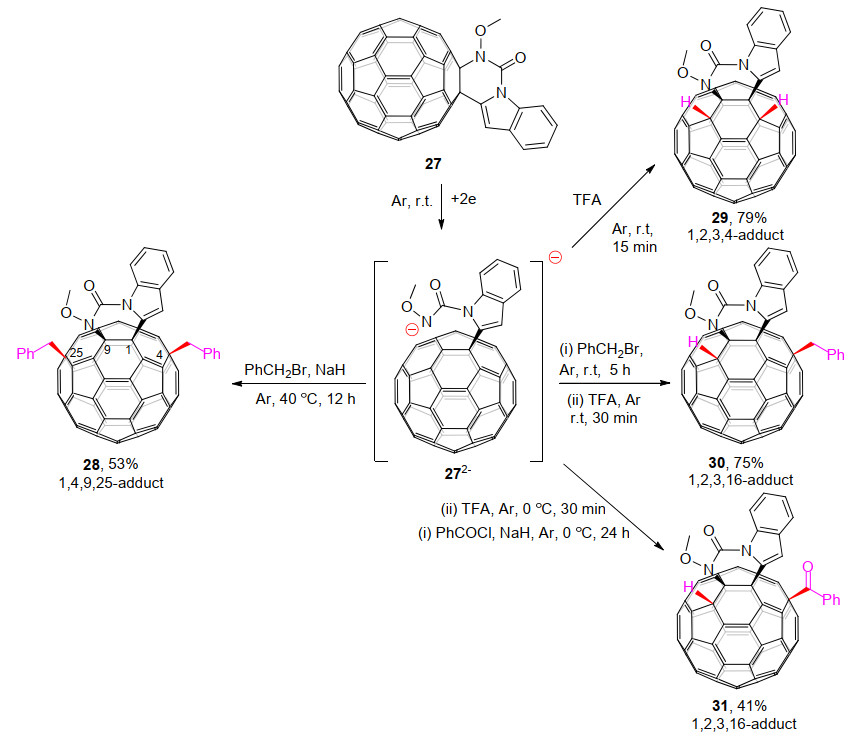
2019年, 王官武课题组[23]报道了C60稠合3, 4-二氢嘧啶并[1, 6-a]吲哚-1(2H)-酮(27)的一系列电化学反应. C60稠合3, 4-二氢嘧啶并[1, 6-a]吲哚-1(2H)-酮二负离子272-与过量PhCH2Br和NaH反应, 可以得到很少被研究[24]的四官能团化的1, 4, 9, 25-C60衍生物28, 并通过X射线单晶衍射确定了其结构(Scheme 9).同时, 272-可以被TFA质子化为二氢化的1, 2, 3, 4-富勒烯衍生物29, 并可与PhCH2Br/PhCOCl和TFA分步反应得到单烷基/羰基化的C60稠合3, 4-二氢嘧啶并[1, 6-a]吲哚-1(2H)-酮衍生物30/31.
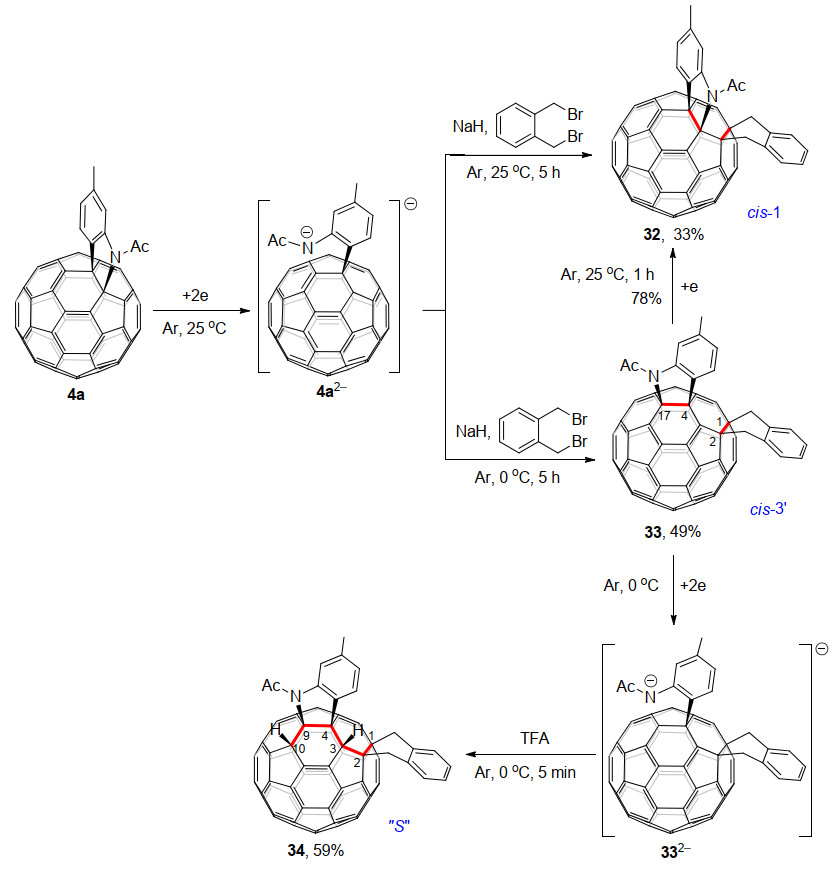
2020年, 王官武课题组[19a]继续对C60稠合吲哚啉4a的电化学反应进行研究, 首次高区域选择性合成了新颖的cis-3'-型四官能团化的1, 2, 4, 17-C60衍生物33和“S”-型六官能团化的1, 2, 3, 4, 9, 10-C60衍生物34, 并且首次得到了这种六加成衍生物的单晶结构(Scheme 10).他们用C60稠合吲哚啉二负离子4a2-与邻二溴苄反应, 在0 ℃时可以选择性地得到之前文献尚未报道的具有新颖加成模式的1, 2, 4, 17-加合物33.而在25 ℃时, 则生成更稳定的1, 2, 3, 4-加合物32.此外, 33得到一个电子后在25 ℃下反应1 h, 杂环会断键重排得到32.进一步将电化学得到的1, 2, 4, 17-加合物二负离子332-质子化, 可以合成具有1, 2, 3, 4, 9, 10-加成模式且显示独特“S”-构型的六功能化C60衍生物34.
同年, 该课题组[19b]还报道了C60稠合吲哚啉4的电化学双羰基化反应, 获得了cis-3' C60衍生物35以及“S”-型C60衍生物39 (Scheme 11).通过电化学方法将C60稠合吲哚啉还原为二价负离子中间体42-后, 再加入亲电试剂邻苯二甲酰氯, 高选择性制备出了cis-3'异构体, 即1, 2, 4, 17-四加成富勒烯衍生物35, 并通过单晶结构证实了这种加成模式.这一四加成富勒烯衍生物进一步在1 equiv.的TFA存在下, 通过逐步电化学还原再质子化转换为“S”-型加成产物, 即1, 2, 3, 4, 9, 10-六加成富勒烯衍生物39.此外, 研究表明这种1, 2, 4, 17-四加成富勒烯衍生物十分稳定.他们将富勒烯四加成产物作为电子传输层, 应用到了钙钛矿太阳能电池上, 得到了与[6,6]-苯基-C61-丁酸甲酯(PC61BM)相近的光电转换效率(PCE).
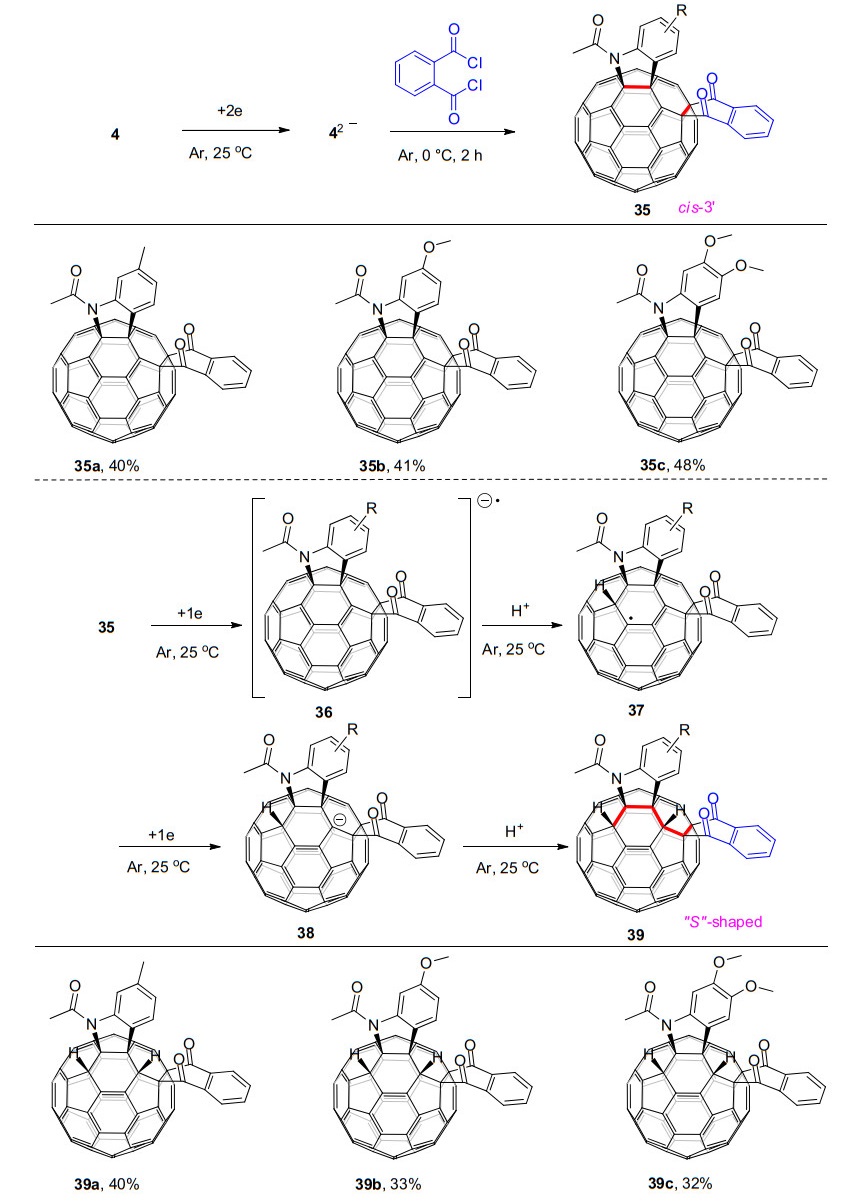
最近, 王官武课题组[25]报道了C60稠合吲哚啉4a与不同大位阻烷基溴代物的电化学烷基化反应, 得到了不同的反应结果(Scheme 12).电化学生成的4a2-与大位阻的2, 4, 6-三溴甲基三甲基苯反应可以得到氢烷基化的1, 2, 3, 16-加合物40a或双烷基化的1, 2, 3, 16-加合物41a.相比之下, 4a2-与大位阻的二苯基溴甲烷的氢烷基化反应仍可得到1, 2, 3, 16-加合物40b, 但相应的双烷基化反应则可以得到较少被报道[24]的1, 4, 9, 12-加成主产物41c以及1, 2, 3, 16-加成次要产物41b.
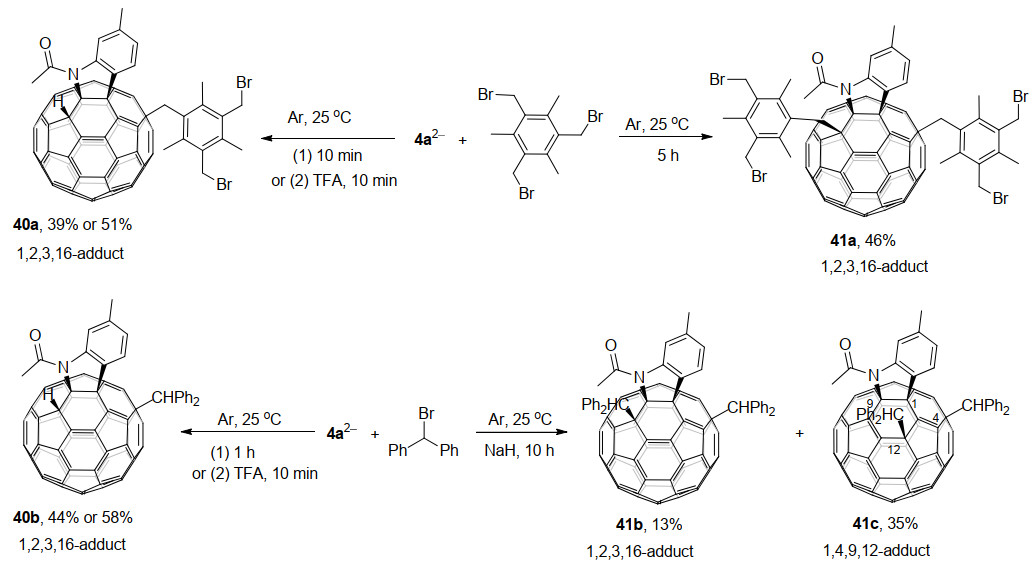
2013年, 王官武课题组和高翔课题组合作[26]研究了以C60稠合磺酸内酯42为底物, 通过电化学还原转化制备具有磺酸基团的单键二聚体43和新型1, 4-C60衍生物44的反应(Scheme 13).他们发现, 原位生成的C60稠合磺酸内酯二价负离子422-与PhCH2Br反应生成的44是通过C60稠合磺酸内酯在接受一个电子后C60—O键开环而引发的.进一步研究发现C60稠合磺酸内酯二价负离子422-通过电化学氧化失去一个电子后, 得到单键连接的二聚体43, 并可通过进一步电化学氧化返回到起始原料C60稠合磺酸内酯42.
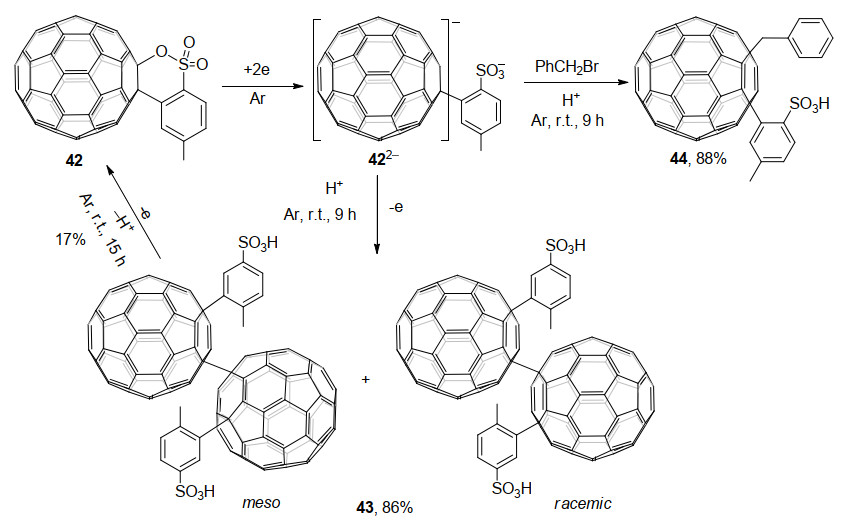
2017年, 王官武课题组[27]报道了电化学生成的C60稠合苯并呋喃二负离子252-与二溴代丙二酸二乙基酯的环丙烷化反应.在252-负离子电荷密度分布和反应物的空间位阻的双重控制下, 主要得到了e异构体45以及少量的两种trans-3异构体46和47, 并且成功培养出了e异构体的单晶(Scheme 14).有趣的是, 当使用空间位阻较大的二溴代丙二酸二乙基酯代替PhCH2Br作为亲电试剂时, C60稠合苯并呋喃二负离子富勒烯表面上的反应位点完全不同, 区域选择性地得到了e和trans-3加合物, 而非1, 2, 3, 16-加成产物[18e].此外, 产物中的稠合苯并呋喃杂环没有从原来的[6,6]-键重排到[5,6]-键, 这与其它C60稠合杂环电化学衍生得到的多加成产物很不相同.
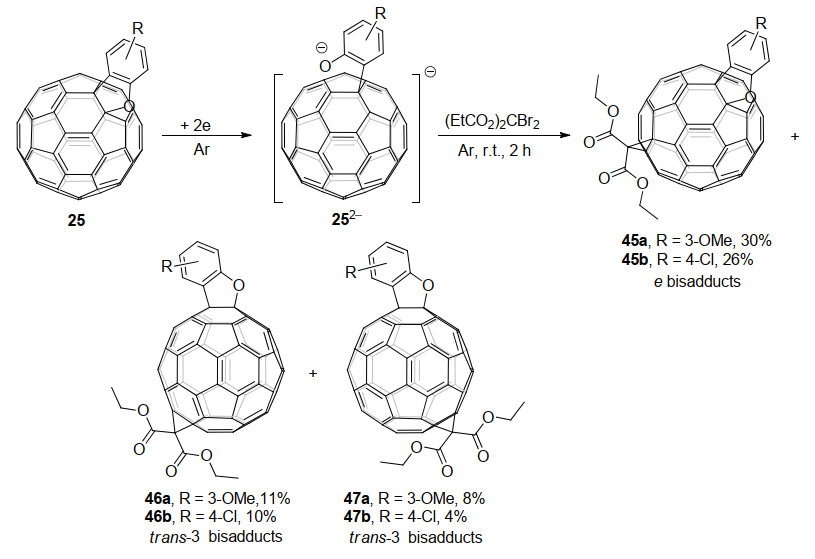
2019年, 该课题组[28]研究了电化学得到的C60稠合内酯二负离子482-的质子化反应.出乎意料的是, 电化学还原生成的C60稠合内酯二负离子中间体在乙酸(AcOH)的作用下, 高效地转化为C60稠合环酮49, 而不是预期的二氢化的1, 2, 3, 4-C60衍生物[14b], 产率为85%~91% (Scheme 15).这是首次报道的内酯直接还原成酮的反应, 从反应式来看也是首次实现Baeyer-Villiger氧化的逆反应.他们通过光谱和X射线单晶衍射对中间体和产物进行了表征, 并且通过对照实验阐明了反应机理:首先, C60稠合内酯49发生电化学还原, C60—O键断裂生成开环的二负离子482-.由于AcOH的酸性较弱, 所以即使在过量的AcOH存在下, 也只有二负离子482-中的羧酸根阴离子被质子化, 得到单负离子50.最后, 中间体50经分子内环化, 同时脱去氢氧根离子(脱去后被另一分子的AcOH中和)得到C60稠合环酮49.
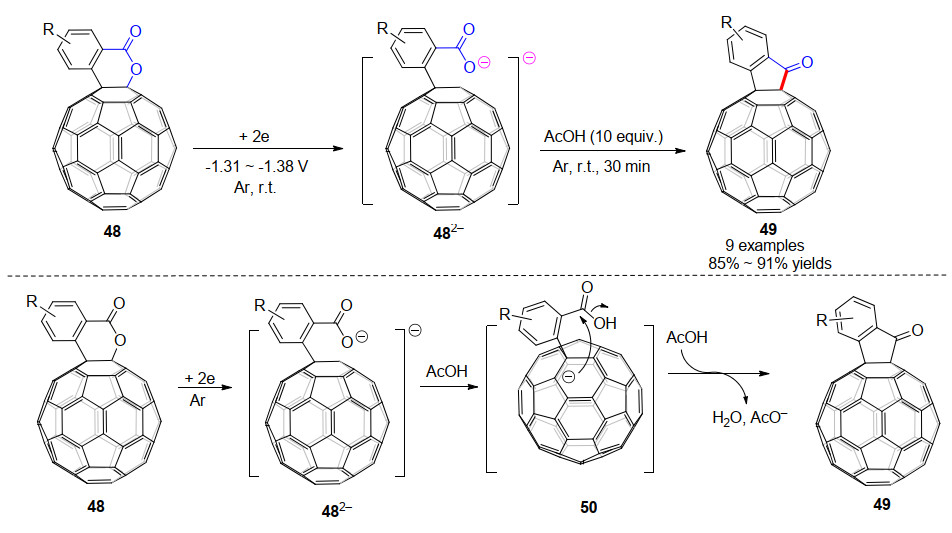
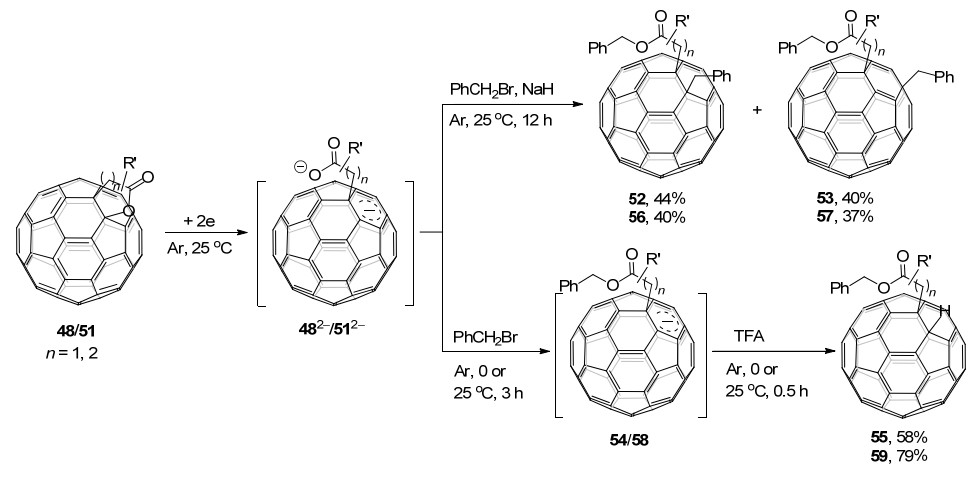
随后, 该课题组[29]继续研究了C60稠合内酯48/51的电化学还原苄基化反应, 得到了三种类型的开环苄基化加合物, 这与之前报道的C60稠合杂环化合物电化学还原苄基化后得到的产物类型有很大不同(Scheme 16).稠合的δ-、γ-内酯二负离子482-和512-均可被苄基化, 并以高收率生成开环的单苄基化和双苄基化的加合物.根据实验结果及理论计算, 他们给出了可能的反应机理: C60稠合内酯48/51接受两个电子后, C60—O键断裂开环生成二负离子中间体482-/512-, 它们的一个负电荷分布在C60笼上, 另一个负电荷分布在羧酸根的氧原子上; 随后, 第一个苄基加成在羧酸根的氧原子上, 得到单负离子; 之后, 通过电子和空间效应影响, 单负离子与第二分子PhCH2Br发生SN2反应, 生成1, 2-加合物52/56和1, 4-加合物53/57.如果采用TFA进行质子化, 则得到1, 2-氢化富勒烯55/59.另外, 代表性的富勒烯产物被用作钙钛矿太阳能电池的电子传输材料, 得到了比PC61BM略高的PCE.
富勒烯衍生物在生命科学和材料科学等领域具有广泛的应用前景, 合成新颖的富勒烯衍生物是富勒烯表面修饰领域的重要研究方向.电化学还原使得富勒烯的碳笼由原来的亲电性变为亲核性, 从而能与亲电试剂发生反应.这种极性反转的策略极大地拓宽了富勒烯化学的反应范围, 对通过电化学合成独特富勒烯多功能化衍生物具有深远的意义, 同时也将极大地拓展了富勒烯功能材料的应用.富勒烯及其衍生物通过电化学还原, 可以改变其富勒烯笼表面的电荷分布, 使得某些特定位点的电荷密度增加, 更易于发生亲电反应, 从而可以得到一些经典化学方法难以得到的新型加成模式的产物.
对于C60稠合杂环化合物, 它们得到两个电子后杂环中的碳-杂原子键会发生断裂, 此时富勒烯笼上非官能团化碳原子中电荷最集中的位点一般是杂环与碳笼相连碳的邻位或对位, 这也是与亲电试剂反应的第一个位点.若与此富勒烯化合物二负离子反应的亲电试剂进攻基团较小, 随着碳-杂键进一步重排, 则会生成1, 2, 3, 4-、1, 2, 3, 16-加成产物; 若亲电试剂位阻较大, 则会生成1, 4, 9, 12-、1, 4, 9, 25-加成产物.此外, 生成产物的相对稳定性也是影响产物选择性的重要因素, 即较稳定结构的产物更容易生成.利用电化学得到的C60稠合杂环化合物二负离子与亲电试剂反应, 为富勒烯衍生物的加成提供了一系列新的模式.但是目前研究的C60稠合杂环化合物的电化学反应较为有限, 仍有许多C60稠合杂环化合物的电化学反应未被研究.相信未来C60稠合杂环化合物的电化学反应将会是合成富勒烯多官能团化衍生物的重要手段.预期新颖的C60稠合六元杂环化合物、七元杂环化合物或是C70稠合杂环化合物的电化学反应将会是以后富勒烯电化学研究的重要方向.
Osawa, E. Kagaku (Kyoto) 1970, 25, 854. http://ci.nii.ac.jp/naid/10021261422
Kroto, H. W.; Heath, J. R.; O'brien, S. O.; Curl, R. F.; Smalley, R. E. Nature 1985, 318, 162. doi: 10.1038/318162a0
Krätschmer, W.; Lamb, L. D.; Fostiropoulos, K.; Huffman, D. R. Nature 1990, 347, 354. doi: 10.1038/347354a0
Ceulemans, A.; Fowler, P. W. Nature 1991, 353, 52. doi: 10.1038/353052a0
Liu, S.; Lu, Y. J.; Kappes, M. M.; Ibers, J. A. Science 1991, 254, 408. doi: 10.1126/science.254.5030.408
For selected reviews, see:
(a) Murata, M.; Murata, Y.; Komatsu, K. Chem. Commun. 2008, 6083.
(b) Vougioukalakis, G. C.; Roubelakis, M. M.; Orfanopoulos, M. Chem. Soc. Rev. 2010, 39, 817.
(c) Itami, K.; Chem. Rec. 2011, 11, 226.
(d) Wang, G.-W.; Li, F.-B. Curr. Org. Chem. 2012, 16, 1109.
(e) Maroto, E. E.; Izquierdo, M.; Reboredo, S.; Marco-Martínez, J.; Filippone, S.; Martín, N. Acc. Chem. Res. 2014, 47, 2660.
(f) Gan, L. Chin. J. Chem. 2018, 36, 991.
(g) Lin, H.-S.; Matsuo, Y. Chem. Commun. 2018, 54, 11244.
For selected reviews, see:
(a) Nakamura, E.; Isobe, H. Acc. Chem. Res. 2003, 36, 807.
(b) Giacalone, F.; Martín, N. Chem. Rev. 2006, 106, 5136.
(c) Thompson, B. C.; Fréchet, J. M. J. Angew. Chem., Int. Ed. 2008, 47, 58.
(d) Guldi, D. M.; Illescas, B. M.; Atienza, C. M.; Wielopolski, M.; Martín, N. Chem. Soc. Rev. 2009, 38, 1587.
(e) Balch, A. L.; Winkler, K. Chem. Rev. 2016, 116, 3812.
(f) Illescas, B. M.; Rojo, J.; Delgado, R.; Martín, N. J. Am. Chem. Soc. 2017, 139, 6018.
(g) Wang, Y.; Zheng, L.; Li, J.; Liu, C.; Yao, J. Chin. J. Org. Chem. 2018, 38, 3143(in Chinese).
(王宇飞, 郑丽萍, 李靖靖, 刘超, 姚建华, 有机化学, 2018, 38, 3143.)
(h) Umeyama, T.; Imahori, H. Acc. Chem. Res. 2019, 52, 2046. For recent examples, see:
(i) Sun, Y.; Gao, H.; Zhang, Y.; Wang, Y.; Kan, B.; Wan, X.; Zhang, H.; Chen, Y. Chin. J. Org. Chem. 2018, 38, 228(in Chinese).
(孙延娜, 高欢欢, 张雅敏, 王云闯, 阚斌, 万相见, 张洪涛, 陈永胜, 有机化学, 2018, 38, 228.)
(j) Li, W.; Yan, D.; Liu, F.; Russell, T.; Zhan C.; Yao, J. Sci. China: Chem. 2018, 61, 1609.
(k) Sun, W.; Ye, L.; Liu, J.; Zheng, L.; Guo, W.; Han, S.; Shao, C.; Jiang, H. Chin. J. Org. Chem. 2019, 39, 2867(in Chinese).
(孙卫东, 叶琳, 刘佳, 郑璐, 郭文彩, 韩森凯, 邵成园, 江华, 有机化学, 2019, 39, 2867.)
(a) Haddon, R. C. Acc. Chem. Res. 1992, 25, 127.
(b) Johnson, R. D.; Bethune, D. S.; Yannoni, C. S. Acc. Chem. Res. 1992, 25, 169.
Haddon, R. C.; Brus, L. E.; Raghavachari, K. Chem. Phys. Lett. 1986, 131, 165. doi: 10.1016/0009-2614(86)80538-3
Echegoyen, L.; Echegoyen, L. E. Acc. Chem. Res. 1998, 31, 593. doi: 10.1021/ar970138v
Nakamura, Y.; O-kawa, K.; Nishimura, J. Bull. Chem. Soc. Jpn. 2003, 76, 865. doi: 10.1246/bcsj.76.865
Hirsch, A.; Brettreich, M. Fullerenes:Chemistry and Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005. http://www.researchgate.net/publication/285260113_Fullerenes_Chemistry_and_Reactions
(a) Jensen, A. W.; Khong, A.; Saunders, M.; Wilson, S. R.; Schuster, D. I. J. Am. Chem. Soc. 1997, 119, 7303.
(b) Yamada, M.; Schweizer, W. B.; Schoenebeck, F.; Diederich, F. Chem. Commun. 2010, 46, 5334.
(c) He, C.-L.; Liu, R.; Li, D.-D.; Zhu, S.-E.; Wang, G.-W. Org. Lett. 2013, 15, 1532.
(d) Chen, M.; Bao, L.; Peng, P.; Zheng, S.; Xie, Y.; Lu, X. Angew. Chem., Int. Ed. 2016, 55, 11887.
(e) Jiang, S.-P.; Zhang, M.; Wang, C.-Y.; Yang, S.; Wang, G.-W. Org. Lett. 2017, 19, 5110.
(a) Yang, W.-W.; Li, Z.-J.; Li, F.-F.; Gao, X. J. Org. Chem. 2011, 76, 1384.
(b) Xiao Y.; Wang, G. Chin. J. Chem. 2014, 32, 699.
(c) Majid, H.; Niu, C.; Wang, G.-W. Org. Chem. Front. 2020, 7, 1249.
(a) Schick, G.; Kampe, K.-D.; Hirsch, A. J. Chem. Soc., Chem. Commun. 1995, 2023.
(b) Murata, Y.; Shiro, M.; Komatsu, K. J. Am. Chem. Soc. 1997, 119, 8117.
(c) Deng, L.-L.; Xie, S.-L.; Yuan, C.; Liu, R.-F.; Feng, J.; Sun, L.-C.; Lu, X.; Xie, S.-Y.; Huang, R.-B.; Zheng, L.-S. Sol. Energy Mater. Sol. Cells 2013, 111, 193.
(d) Clikeman, T. T.; Deng, S. H. M.; Avdoshenko, S.; Wang, X.-B.; Popov, A. A.; Strauss, S. H.; Boltalina, O. V. Chem.-Eur. J. 2013, 19, 15404.
(a) Kadish, K. M.; Gao, X.; Caemelbecke, E. V.; Suenobu, T.; Fukuzumi, S. J. Am. Chem. Soc. 2000, 122, 563.
(b) Matsuo, Y.; Iwashita, A.; Abe, Y.; Li, C.-Z.; Matsuo, K.; Hashiguchi, M.; Nakamura, E. J. Am. Chem. Soc. 2008, 130, 15429.
(c) Nambo, M.; Wakamiya, A.; Yamaguchi, S.; Itami, K. J. Am. Chem. Soc. 2009, 131, 15112.
(d) Kuvychko, I. V.; Streletskii, A. V.; Shustova, N. B.; Seppelt, K.; Drewello, T.; Popov, A. A.; Strauss, S. H.; Boltalina, O. V. J. Am. Chem. Soc. 2010, 132, 6443.
(e) Chang, W.-W.; Li, Z.-J.; Yang, W.-W.; Gao, X. Org. Lett. 2012, 14, 2386.
Rubin, Y.; Ganapathi, P. S.; Franz, A.; An, Y.-Z.; Qian, W.; Neier, R. Chem.-Eur. J. 1999, 5, 3162. doi: 10.1002/(SICI)1521-3765(19991105)5:11<3162::AID-CHEM3162>3.0.CO;2-H
(a) Xiao, Y.; Zhu, S.-E.; Liu, D.-J.; Suzuki, M.; Lu, X.; Wang, G.-W. Angew. Chem., Int. Ed. 2014, 53, 3006.
(b) Hou, H.-L.; Li, Z.-J.; Gao, X. Org. Lett. 2014, 16, 712.
(c) Li, Z.-J.; Li, S.-H.; Sun, T.; Hou, H.-L.; Gao, X. J. Org. Chem. 2015, 80, 3566.
(d) Lin, H.-S.; Matsuo, Y.; Wang, J.-J.; Wang, G.-W. Org. Chem. Front. 2017, 4, 603.
(e) Li, F.; Wang, J.-J.; Wang, G.-W. Chem. Commun. 2017, 53, 1852.
(a) Liu, K.-Q.; Wang, J.-J.; Yan, X.-X.; Niu, C.; Wang, G.-W. Chem. Sci. 2020, 11, 384.
(b) Yan, X.-X.; Li, B.; Lin, H.-S.; Jin, F.; Niu, C.; Liu, K.-Q.; Wang, G.-W.; Yang, S. Research 2020, 2020, 2059190.
(a) Hsu, H.-F.; Shapley, J. R. J. Am. Chem. Soc. 1996, 118, 9192.
(b) Tajima, Y.; Takeuchi, K. J. Org. Chem. 2002, 67, 1696.
(c) Chuang, S.-C.; Clemente, F. R.; Khan, S. I.; Houk, K. N.; Rubin, Y. Org. Lett. 2006, 8, 4525.
(a) Birkett, P. R.; Hitchcock, P. B.; Kroto, H. W.; Taylor, R.; Walton, D. R. M. Nature 1992, 357, 479.
(b) Gan, L.; Huang, S.; Zhang, X.; Zhang, A.; Cheng, B.; Cheng, H.; Li, X.; Shang, G. J. Am. Chem. Soc. 2002, 124, 13384.
Hou, H.-L.; Li, Z.-J.; Wang, Y.; Gao, X. J. Org. Chem. 2014, 79, 8865. doi: 10.1021/jo5019238
Majid, H.; Chen, M.; Yang, S.; Wang, G.-W. Org. Lett. 2019, 21, 8568. doi: 10.1021/acs.orglett.9b03112
Chen, S.; Li, Z.-J.; Li, S.-H.; Gao, X. Org. Lett. 2015, 17, 5192. doi: 10.1021/acs.orglett.5b02528
Yang, Y.; Niu, C.; Chen, M.; Yang, S.; Wang, G.-W. Org. Biomol. Chem. 2020, 18, 4783. doi: 10.1039/D0OB00876A
Liu, R.; Li, F.; Xiao, Y.; Li, D.-D.; He, C.-L.; Yang, W.-W.; Gao, X.; Wang, G.-W. J. Org. Chem. 2013, 78, 7093. doi: 10.1021/jo400920f
Wang, J.-J.; Lin, H.-S.; Niu, C.; Wang, G.-W. Org. Biomol. Chem. 2017, 15, 3248. doi: 10.1039/C7OB00463J
Niu, C.; Zhou, D.-B.; Yang, Y.; Yin, Z.-C.; Wang, G.-W. Chem. Sci. 2019, 10, 3012. doi: 10.1039/C8SC05089A
Niu, C.; Li, B.; Yin, Z.-C.; Yang S.; Wang, G.-W. Org. Lett. 2019, 21, 7346. doi: 10.1021/acs.orglett.9b02635

图 2 (a) 加成于两个[6,6]-键的cis和e异构体及加成于一个[6,6]-键和一个[5,6]-键的cis'异构体的C60的双环化衍生物和(b)碳原子编号的C60 Schlegel图
Figure 2 (a) cis and e isomers fused to two [6,6]-junctions and cis' isomers fixed to a [6,6]-junction and a [5,6]-junction for bis- cycloadducts of C60, and (b) Schlegel diagram of C60 with numbering of the C-atoms

图 3 常见四功能化和六功能化C60衍生物的加成模式
Figure 3 Common addition patterns of tetra- and hexa- functionalized [60]fullerene derivatives

图式 1 C60稠合噁唑啉的电化学苄基化反应
Scheme 1 Electrochemical benzylation of the C60-fused oxazoline

图式 3 C60稠合呋喃香豆素的电化学功能化反应
Scheme 3 Electrochemical functionalizations of the C60-fused furochromenone

图式 5 C60稠合咪唑啉的电化学苄基化反应
Scheme 5 Electrochemical benzylation of the C60-fused imidazoline

图式 6 1, 2, 4, 15-C60单键连接二聚体的电化学苄基化反应
Scheme 6 Electrochemical benzylation of the singly-bonded 1, 2, 4, 15-C60 dimers

图式 7 C60稠合吲哚啉的电化学单羰基化反应
Scheme 7 Electrochemical monoacylation of the C60-fused indoline

图式 8 C60稠合苯并呋喃的电化学苄基化反应
Scheme 8 Electrochemical benzylation of the C60-fused benzofuran

图式 9 C60稠合3, 4-二氢嘧啶并[1, 6-a]吲哚-1(2H)-酮的电化学功能化反应
Scheme 9 Electrochemical functionalizations of the C60-fused 3, 4-dihydropyrimido[1, 6-a]indol-1(2H)-one

图式 10 1, 2, 4, 17-及1, 2, 3, 4, 9, 10-功能化C60衍生物的电化学合成
Scheme 10 Electrosynthesis of 1, 2, 3, 16- and 1, 2, 3, 4, 9, 10-functionalized C60 derivatives

图式 11 C60稠合吲哚啉的电化学双羰基化以及进一步电化学质子化反应
Scheme 11 Electrochemical diacylation of the C60-fused indolines and further electrochemical protonation

图式 12 C60稠合吲哚啉与大体积烷基溴代物的电化学烷基化反应
Scheme 12 Electrochemical alkylations of the [60]fulleroindoline with bulky alkyl bromides

图式 13 电化学还原C60稠合磺酸内酯为C60稠合磺酸
Scheme 13 Electrochemical reduction of the C60-fused sultone to the C60-fused sulfonic acids

图式 14 C60稠合苯并呋喃的电化学环丙烷化反应
Scheme 14 Electrochemical cyclopropanation of the C60-fused benzofurans

图式 15 C60稠合内酯的电化学逆Baeyer-Villiger反应
Scheme 15 Retro Baeyer-Villiger reactions of the C60-fused lactones

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:


