Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences(CAS), Beijing 100190
b.
School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049
Received Date:
30 June 2020 Revised Date:
03 August 2020 Available Online:
25 November 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21772204, 21521002), and the Key Research Program of Frontier Sciences, Chinese Academy of Sciences (No. QYZDJ-SSW-SLH023)
Abstract:
Inspired by enzyme allosteric catalysis, the study on artificial stimuli-responsive asymmetric catalytic systems has attracted more and more attentions in recent years. In order to precisely control the catalytic activity and stereoselectivity, stimuli-responsive functionalities have been introduced into the catalyst design. A variety of asymmetric reactions featuring on/off-switchable catalysis and/or stereodivergent catalysis have been successfully achieved by using light-, coordination-, pH-and redox-driven chiral switchable catalysts. By selecting representative examples, the catalyst design principles, allosteric mechanism and their applications in switchable asymmetric reactions sre mainly introduced. At the same time, advantages and limitations of this emerging field are summarized, and perspectives for its future development are given.
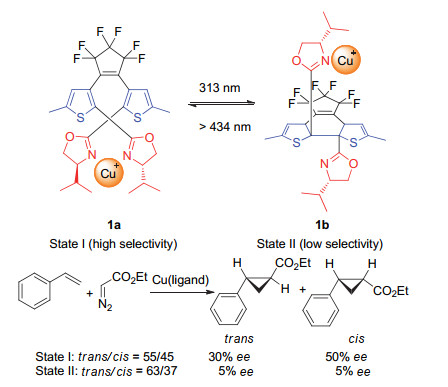
Scheme 1.
A photo-driven catalyst based on 1, 2-dithienyl- ethylene and its application in switchable asymmetric cyclopropanation reactions developed by Branda group
Scheme 2.
A bifunctional organic catalyst based on the first generation molecular motor and its application in switchable asymmetric Michael addition reaction developed by Feringa group
Scheme 3.
Highly active bifunctional organic catalyst based on the first generation molecular motor and its application in switchable asymmetric Henry reactions developed by Feringa group
Scheme 4.
A Pd/bisphosphine catalyst based on the first generation molecular motor and its application in switchable asymmetric reactions developed by Feringa group
Scheme 5.
An anion-binding catalyst based on the first generation molecular motor and its application in switchable asymmetric reactions developed by Feringa group
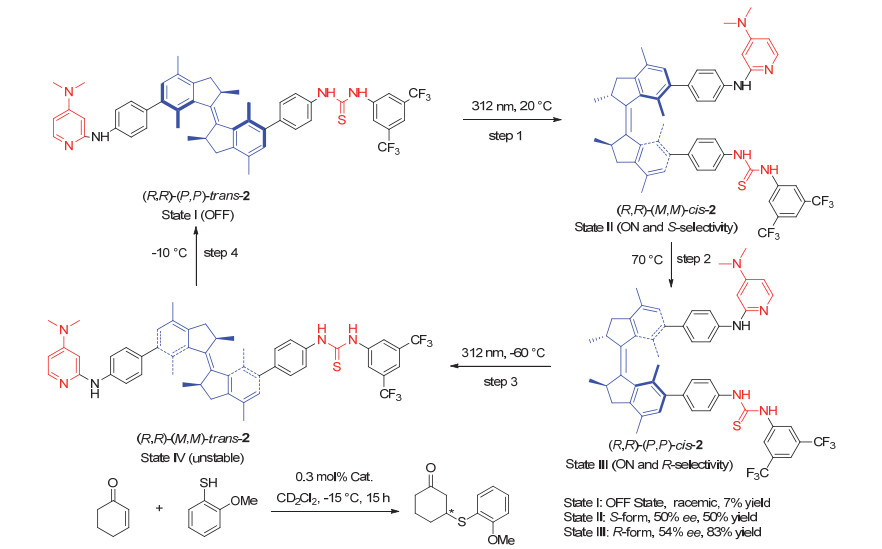
Scheme 6.
Chiral diphenol ligand based on the second generation molecular motor and its applicaton in switchable asymmetric reactions developed by Feringa group
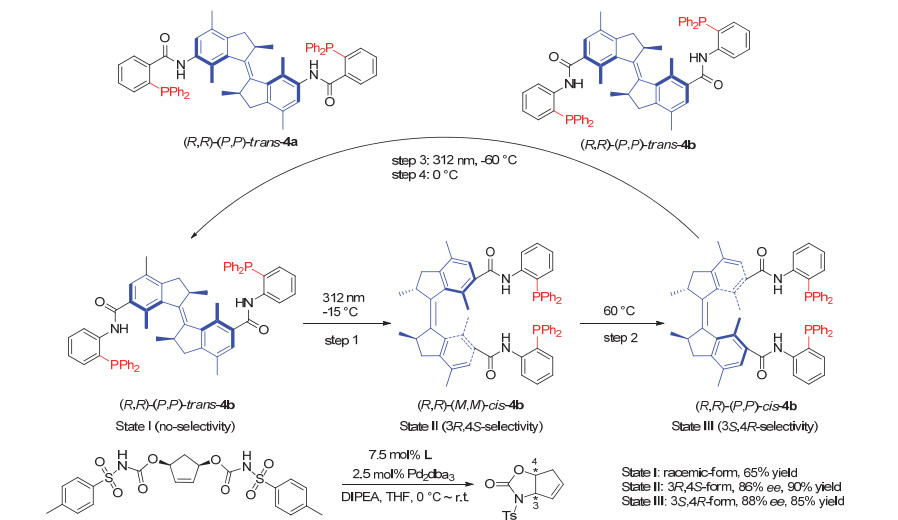
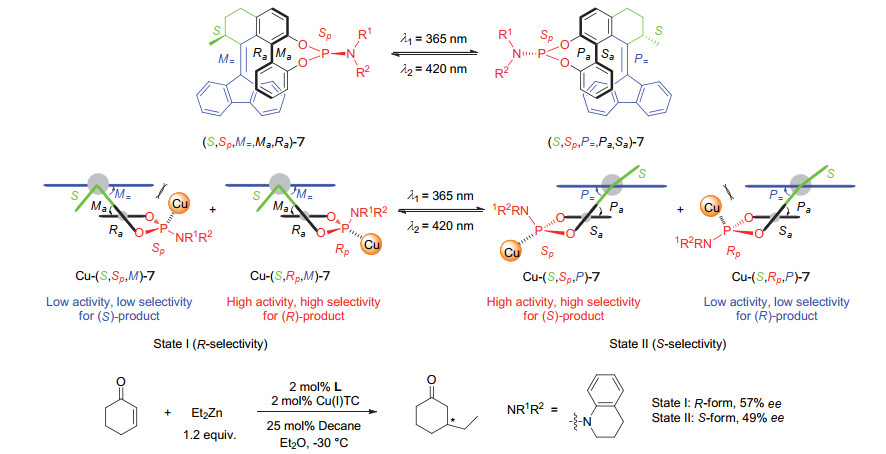
Scheme 7.
Chiral phosphoramidite ligands based on the second generation molecular motor and their application in switchable asymmetric reactions developed by Feringa group
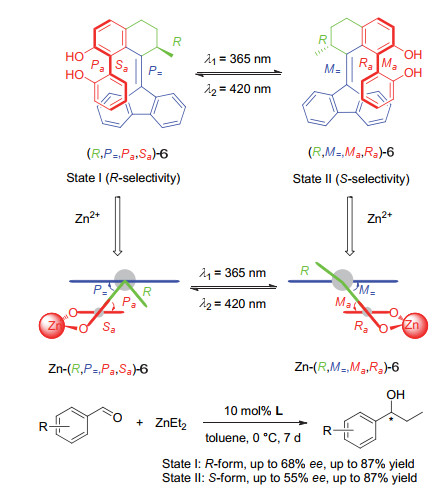
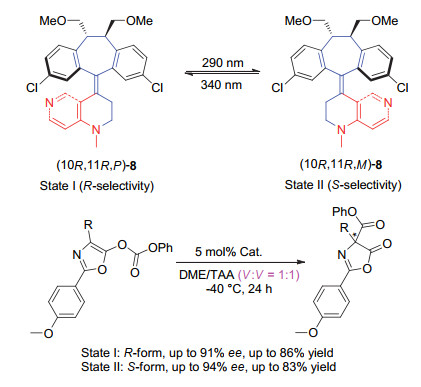
Scheme 8.
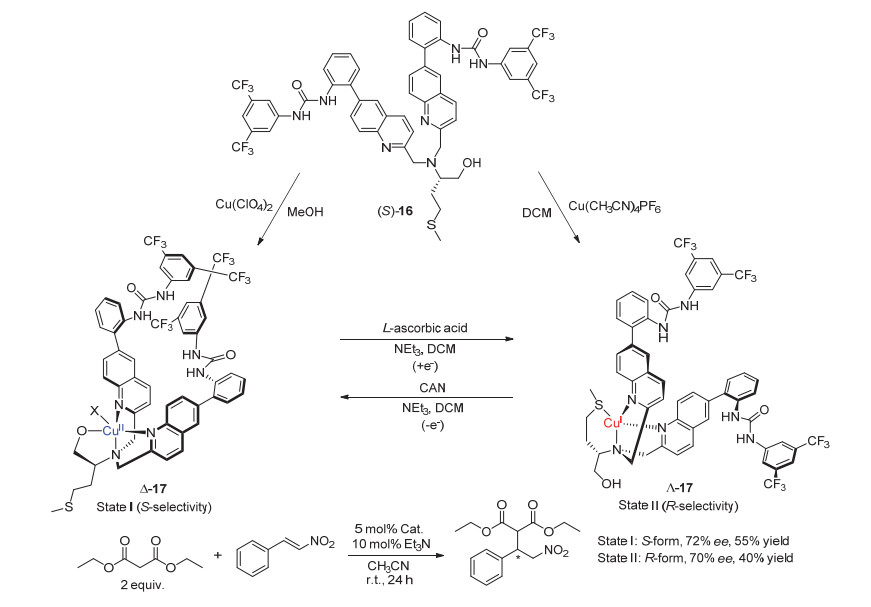
A switchable chiral helicene catalyst and its application in switchable asymmetric Steglich rearrangement reactions developed by Chen and Yu et al.
Dedicated to the 40th anniversary of Chinese Journal of Organic Chemistry.
[1]
(a) Traut, T. Enzyme Activity: Allosteric Regulation, John Wiley & Sons Ltd, Chichester, 2014. (b) Traut, T. Allosteric Regulatory Enzymes, Springer, New York, 2008.
[2]
van Leeuwen, P. W. N. M. Homogeneous Catalysis: Understanding the Art, Springer, Dordrecht, 2004.
[3]
van Leeuwen, P. W. N. M. Supramolecular Catalysis, Wiley-VCH, Weinheim, 2008.
[4]
(a) Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev.2014, 43, 1660. (b) Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev.2014, 43, 1734. (c) Wang, Q.-Q. In Handbook of Macrocyclic Supramolecular Assembly, Eds.: Liu, Y.; Chen, Y.; Zhang, H.-Y., Springer, Singapore, 2019, pp. 1~124. (d) Tang, Y.; He, Y.; Feng, Y.; Fan. Q. Prog. Chem.2018, 30, 476 (in Chinese). (唐雨平, 何艳梅, 冯宇, 范青华, 化学进展, 2018, 30, 476.
[5]
(a) Lüning, U. Angew. Chem., Int. Ed.2012, 51, 8163. (b) Blanco, V.; Leigh, D. A.; Marcos, V. Chem. Soc. Rev.2015, 44, 5341. (c) Vlatković, M.; Collins, B. S. L.; Feringa, B. L. Chem.-Eur. J.2016, 22, 17080. (d) van Dijk, L.; Tilby, M. J.; Szpera, R.; Smith, O. A.; Bunce, H. A. P.; Fletcher, S. P. Nat. Rev. Chem.2018, 2, 0117.
Sud, D.; Norsten, T. B.; Branda, N. R. Angew. Chem., Int. Ed.2005, 44, 2019. doi: 10.1002/anie.200462538
[13]
(a) Koumura, N.; Zijlstra, R. W. J.; van Delden, R. A.; Harada, N.; Feringa, B. L. Nature1999, 401, 152. (b) Eelkema, R.; Pollard, M. M.; Vicario, J.; Katsonis, N.; Ramon, B. S.; Bastiaansen, C. W. M.; Broer, D. J.; Feringa, B. L. Nature2006, 440, 163.
[14]
Dorel, R.; Feringa, B. L. Chem. Commun.2019, 55, 6477. doi: 10.1039/C9CC01891C
Vlatković, M.; Bernardi, L.; Otten, E.; Feringa, B. L. Chem. Commun.2014, 50, 7773. doi: 10.1039/c4cc00794h
[17]
Trost, B. M.; Van Vranken, D. L.; Bingel, C. J. Am. Chem. Soc.1992, 114, 9327. doi: 10.1021/ja00050a013
[18]
Zhao, D.; Neubauer, T. M.; Feringa, B. L. Nat. Commun.2015, 6, 6652. doi: 10.1038/ncomms7652
[19]
(a) Juwarker, H.; Lenhardt, J. M.; Pham, D. M.; Craig, S. L. Angew. Chem., Int. Ed.2008, 47, 3740. (b) Juwarker, H.; Jeong, K.-S. Chem. Soc. Rev.2010, 39, 3664.
[20]
Dorel, R.; Feringa, B. L. Angew. Chem., Int. Ed.2020, 59, 785. doi: 10.1002/anie.201913054
[21]
(a) Koumura, N.; Geertsema, E. M.; van Gelder, M. B.; Meetsma, A.; Feringa, B. L. J. Am. Chem. Soc.2002, 124, 5037. (b) Vicario, J.; Walko, M.; Meetsma, A.; Feringa, B. L. J. Am. Chem. Soc.2006, 128, 5127. (c) Klok, M.; Walko, M.; Geertsema, E. M.; Ruangsupapichat, N.; Kistemaker, J. C. M.; Meetsma, A.; Feringa, B. L. Chem.-Eur. J.2008, 14, 11183.
[22]
Pizzolato, S. F.; Collins, B. S. L.; van Leeuwen, T.; Feringa, B. L. Chem.-Eur. J.2017, 23, 6174. doi: 10.1002/chem.201604966
[23]
Pizzolato, S. F.; Štacko, P.; Kistemaker, J. C. M.; van Leeuwen, T.; Otten, E.; Feringa, B. L. J. Am. Chem. Soc.2018, 140, 17278. doi: 10.1021/jacs.8b10816
[24]
Pizzolato, S. F.; Štacko, P.; Kistemaker, J. C. M.; van Leeuwen, T.; Feringa, B. L. Nat. Catal.2020, 3, 488. doi: 10.1038/s41929-020-0452-y
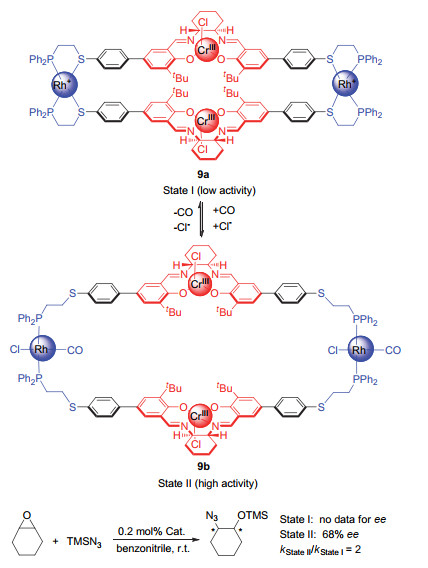
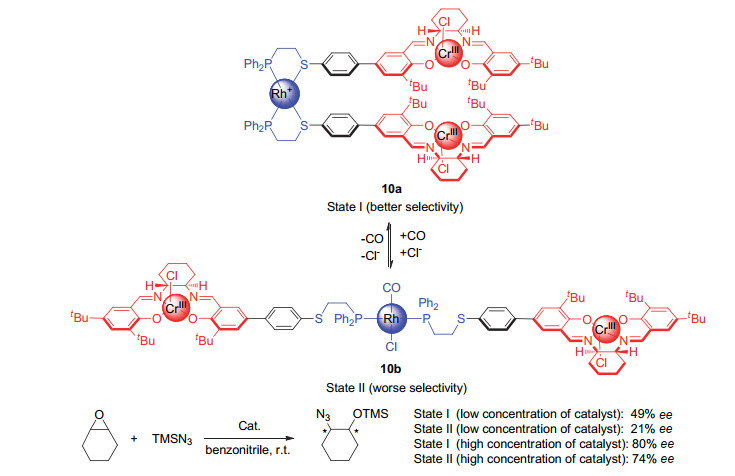
(a) Holliday, B. J.; Mirkin, C. A. Angew. Chem., Int. Ed.2001, 40, 2022. (b) Caulder, D. L.; Raymond, K. N. Acc. Chem. Res.1999, 32, 975.
[28]
Oliveri, C. G.; Ulmann, P. A.; Wiester, M. J.; Mirkin, C. A. Acc. Chem. Res.2008, 41, 1618. doi: 10.1021/ar800025w
[29]
Wiester, M. J.; Ulmann, P. A.; Mirkin, C. A. Angew. Chem., Int. Ed.2011, 50, 114. doi: 10.1002/anie.201000380
[30]
Gianneschi, N. C.; Masar Ⅲ, M. S.; Mirkin, C. A. Acc. Chem. Res.2005, 38, 825. doi: 10.1021/ar980101q
[31]
Yoon, H. J.; Kuwabara, J.; Kim, J.-H.; Mirkin, C. A. Science2010, 330, 66. doi: 10.1126/science.1193928
[32]
Gianneschi, N. C.; Bertin, P. A.; Nguyen, S. T.; Mirkin, C. A.; Zakharov, L. N.; Rheingold, A. L. J. Am. Chem. Soc.2003, 125, 10508. doi: 10.1021/ja035621h
[33]
Hansen, K. B.; Leighton, J. L.; Jacobsen, E. N. J. Am. Chem. Soc.1996, 118, 10924. doi: 10.1021/ja962600x
[34]
Gianneschi, N. C.; Cho, S.-H.; Nguyen, S. T.; Mirkin, C. A. Angew. Chem., Int. Ed.2004, 43, 5503. doi: 10.1002/anie.200460932
(a) Coskun, A.; Banaszak, M.; Astumian, R. D.; Stoddart, J. F.; Grzybowski, B. A. Chem. Soc. Rev.2012, 41, 19. (b) Zhang, L.; Marcos, V.; Leigh, D. A. Proc. Natl. Acad. Sci. U. S. A.2018, 115, 9397.
[37]
Leigh, D. A.; Marcos, V.; Wilson, M. R. ACS Catal.2014, 4, 4490. doi: 10.1021/cs5013415
[38]
Blanco, V.; Carlone, A.; Hänni, K. D.; Leigh, D. A.; Lewandowski, B. Angew. Chem., Int. Ed.2012, 51, 5166. doi: 10.1002/anie.201201364
[39]
Blanco, V.; Leigh, D. A.; Marcos, V.; Morales-Serna, J. A.; Nussbaumer, A. L. J. Am. Chem. Soc.2014, 136, 4905. doi: 10.1021/ja501561c
[40]
(a) Alvarez-Pérez, M.; Goldup, S. M.; Leigh, D. A.; Slawin, A. M. Z. J. Am. Chem. Soc.2008, 130, 1836. (b) Cakmak, Y.; Erbas-Cakmak, S.; Leigh, D. A. J. Am. Chem. Soc.2016, 138, 1749.
[41]
Dommaschk, M.; Echavarren, J.; Leigh, D. A.; Marcos, V.; Singleton, T. A. Angew. Chem., Int. Ed.2019, 58, 14955. doi: 10.1002/anie.201908330
[42]
De Bo, G.; Leigh, D. A.; McTernan, C. T.; Wang, S. Chem. Sci.2017, 8, 7077. doi: 10.1039/C7SC02462B
[43]
(a) Chaur, M. N.; Collado, D.; Lehn, J.-M. Chem.-Eur. J.2011, 17, 248. (b) Su, X.; Aprahamian, I. Chem. Soc. Rev.2014, 43, 1963.
[44]
(a) Allgeier, A. M.; Mirkin, C. A. Angew. Chem., Int. Ed.1998, 37, 894. (b) Praneeth, V. K. K.; Ringenberg, M. R.; Ward, T. R. Angew. Chem., Int. Ed.2012, 51, 10228.
(a) Kassem, S.; Lee, A. T. L.; Leigh, D. A.; Marcos, V.; Palmer, L. I.; Pisano, S. Nature2017, 549, 374. (b) Kelly, T. R.; Snapper, M. L. Nature2017, 549, 336.
[50]
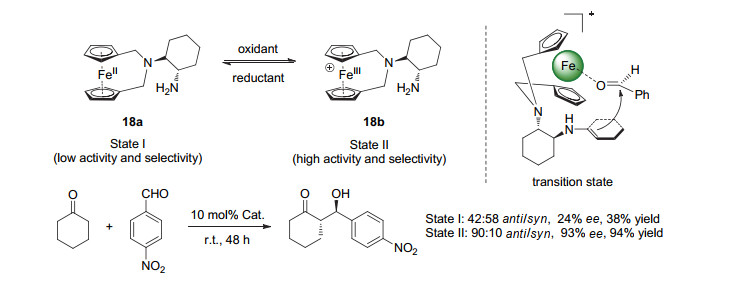
(a) Neel, A. J.; Hilton, M. J.; Sigman, M. S.; Toste, F. D. Nature2017, 543, 637. (b) Huang, G.; Chen, Z.; Wei, X.; Chen, Y.; Li, X.; Zhong, H.; Tan, M. Chin. J. Org. Chem.2020, 40, 614 (in Chinese). (黄国保, 陈志林, 韦贤生, 陈钰, 李秀英, 仲辉, 谭明雄, 有机化学, 2020, 40, 614.) (c) Wang, Y.; Liu, H.; Zhu, X. Acta Chim. Sinica2020, 78, 746 (in Chinese). (王友付, 刘航海, 朱新远, 化学学报, 2020, 78, 746.
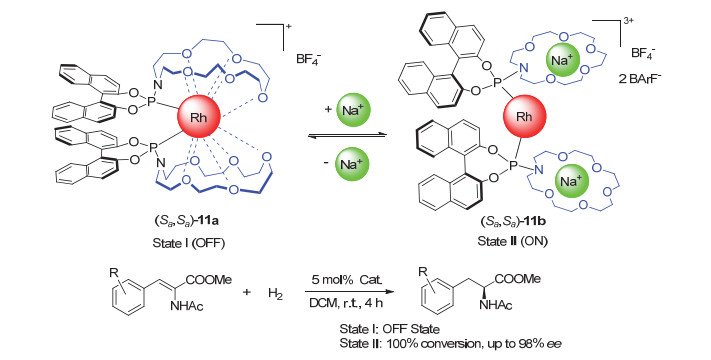
Scheme 1
A photo-driven catalyst based on 1, 2-dithienyl- ethylene and its application in switchable asymmetric cyclopropanation reactions developed by Branda group
Scheme 2
A bifunctional organic catalyst based on the first generation molecular motor and its application in switchable asymmetric Michael addition reaction developed by Feringa group
Scheme 3
Highly active bifunctional organic catalyst based on the first generation molecular motor and its application in switchable asymmetric Henry reactions developed by Feringa group
Scheme 4
A Pd/bisphosphine catalyst based on the first generation molecular motor and its application in switchable asymmetric reactions developed by Feringa group
Scheme 5
An anion-binding catalyst based on the first generation molecular motor and its application in switchable asymmetric reactions developed by Feringa group
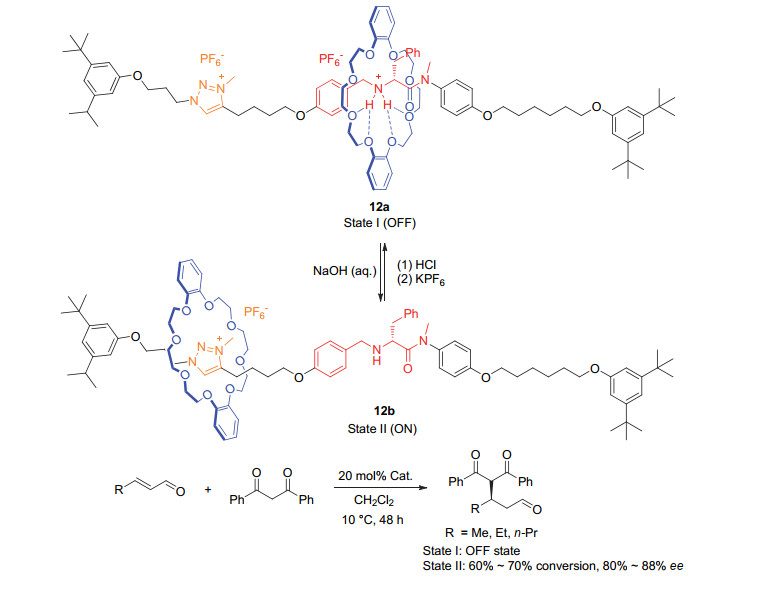
Scheme 6
Chiral diphenol ligand based on the second generation molecular motor and its applicaton in switchable asymmetric reactions developed by Feringa group
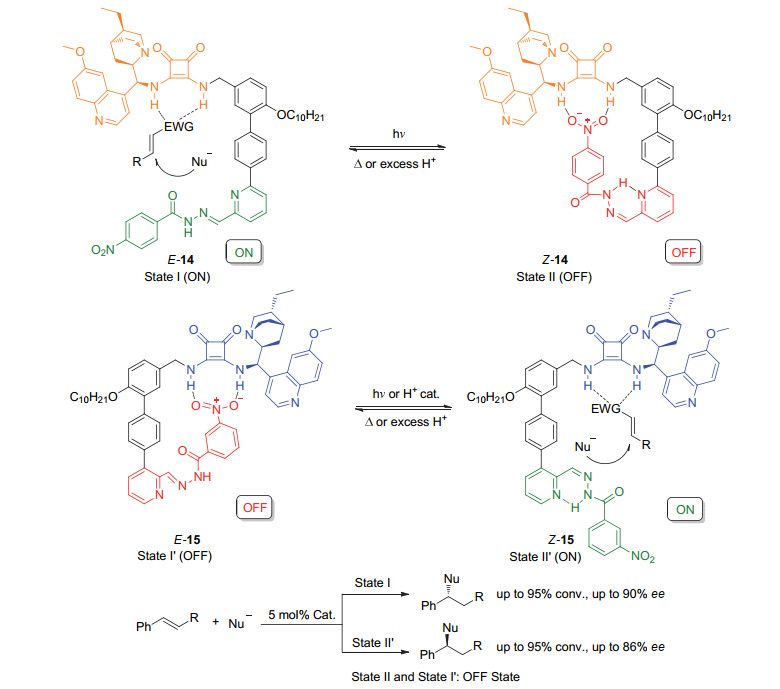
Scheme 7
Chiral phosphoramidite ligands based on the second generation molecular motor and their application in switchable asymmetric reactions developed by Feringa group
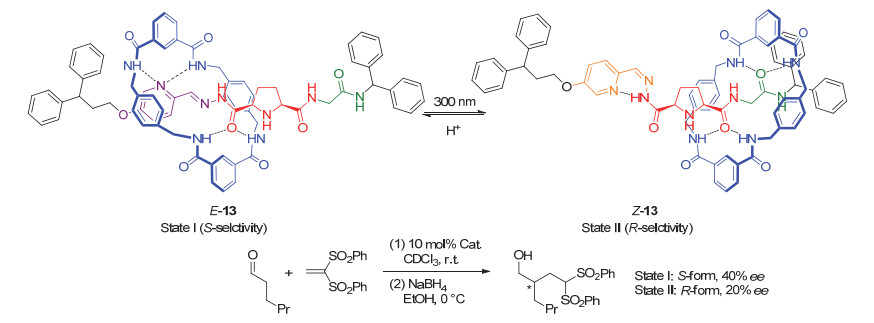
Scheme 8
A switchable chiral helicene catalyst and its application in switchable asymmetric Steglich rearrangement reactions developed by Chen and Yu et al.

 下载:
下载:


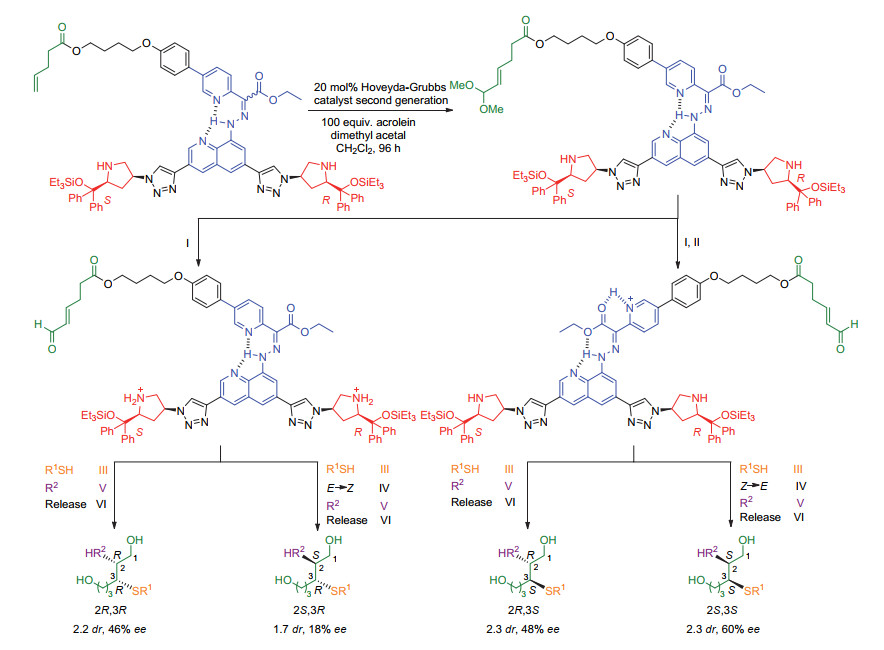

 下载:
下载:


















