Scheme 1.
Methylation of aromatic halides
Palladium-Catalyzed Reductive Coupling of Aromatic Bromides and Trimethylsilyldiazomethane: Its Application to Methylation of Aromatic Compounds
Shuai Wang , Cheng Yang , Shuo Sun , Hanli Sun , Jianbo Wang

Methyl group (Me), as the smallest alkyl group, is ubiquitous in organic compounds, especially in a large number of bioactive molecules.[1] The introduction of methyl groups has been demonstrated to have significant influence on the bioactivity of molecules, which is very important for drug discovery and development.[2] The optimization of many properties by introducing various groups, which is referred as "magic methyl effect", can be attributed to the change of solubility, hydrophilicity, and molecular conformation.[3] For this reason, the development of efficient methylation methods has attracted considerable attentions in recent years.[4]
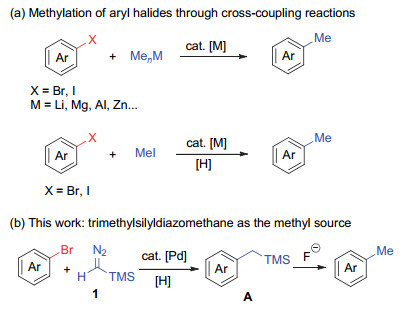
The traditional methylation methods involve nucleophilic substitution of in situ generated aryl lithium reagent and methyl electrophile, and Minisci-type reaction of electron-deficient arenes.[5] These methods commonly suffered from rigorous conditions and limited substrate scope. In the past decades, transition-metal-catalyzed cross-coupling reactions have been widely studied and have emerged as a powerful tool for the construction of C—C bonds. In recent years, synthetic methods for both methylation of C—X bonds and directing-group promoted C—H methylation were developed with transition metal complexes as the catalysts (Scheme 1a).[6] In general, the methylation reagents used in these transformations involve pre-synthesized methyl organometallic reagents and highly reactive, toxic methyl electrophiles.[7] Thus, the employment of easily-handled methylation reagents is crucial for the further development of methylation methods. A few of new methylation reagents were reported in recent years. In 2016, Chatani and co-workers reported that phenyltrimethylamnium salts could be employed as methylation reagents in nickel-catalyzed C—H bond methylation reactions.[8] In 2018, Hartwig and co-workers[9] reported the copper-catalyzed methylation of arylboronic esters with trimethylphosphate as stable methylation reagent. In addition, several radical-based methylation reagents, such as N-hydroxyphthalimide esters and trimethyl orthoformate, have been reported by the groups of Weix[10] and Doyle.[11] While significant advances have been achieved, new and efficient methylation methods with easily-handled methylation reagents are still highly demanded.
On the other hand, transition-metal-catalyzed cross- coupling reactions involving metal carbene species have been actively explored in the past decade, and now emerged as powerful tool for the construction of C—C and C—X bonds.[12] We conceived that trimethylsilyldiazomethane (TMSCHN2, 1), which is used as a stable alternative of diazomethane and widely employed in the synthesis of methyl esters from the corresponding acids, may be functioned as C1 carbene precursor undergoing reductive coupling with aryl bromides through migratory insertion process, giving silylmethylated arenes A as the intermediate products.[13-14] Followed by desilicification step with fluoride, the methylated products are expected to be easily obtained (Scheme 1b).
Moreover, the benzyltrimethylsilane intermediate products A are also valuable compounds in organic synthesis.[15] However, methodologies for the direct introduction of silylmethyl groups are limited. In general, pre-synthesized silylmethyl metal compounds are employed as the silylmethylation reagents, which restrict the functional group tolerance.[15a, 16] In this context, the palladium-catalyzed reductive coupling we report herein can also be regarded as an efficient way for the direct introduction of silylmethyl group.
To test our hypothesis, methyl 4-bromobenzoate (2a) was initially selected as the standard substrate in the Pd-catalyzed reductive coupling reaction. To our disappointment, the reaction of 1 and 2a only gave trace of the desire product 3a with 5 mol% Pd(PPh3)4 as catalyst and 5 equiv. of iPr3SiH as the reductant in toluene (Eq. 1). This result indicates that the benzyl palladium intermediate generated from the migratory insertion process reacts faster with the second molecule of 1 rather than the reductant (vide infra). Thus, the optimization of the reaction conditions by investigating the reaction parameters was continued, with slow addition of 1 using syringe pump.
|
|
(1) |
The study showed that 40% yield of 3a could be obtained with 4.0 equiv. of 1 added slowly in 6 h (Table 1, Entry 1). Then the ratio of palladium catalyst and the ligand with weak-coordinated Pd2(dba)3 and PPh3 instead of Pd(PPh3)4 was examined, and the 1:3 ratio (Pd:L) gave a better yield of 55% (Table 1, Entry 2). By testing several palladium catalysts and ligands, it was found that the combination of palladium acetate and tris(p-chlorophenyl)phos- phane was more effective for this transformation (Table 1, Entries 3~5). Then several reductants were screened. To our delight, by using 3 equiv. of Me(EtO)2SiH as the reductant instead of 6 equiv. of iPr3SiH, both the loading and the addition time of 1 could be reduced (Table 1, Entry 6). Furthermore, solvent effect was then examined. Replacement of toluene with 1, 4-dioxane slightly improved the yield to 73% (Table 1, Entry 7). When the reaction was carried out under a higher or lower temperature, the product 3a was obtained in a lower yield (Table 1, Entries 8, 9).
 下载:
导出CSV
下载:
导出CSV
 |
|||||||||
| Entry | x/equiv. | y/equiv. | z/h | Cat. [Pd] | L | [H] | Solvent | T/℃ | Yieldb/% |
| 1 | 4 | 5 | 6 | Pd(PPh3)4 | — | iPr3SiH | Toluene | 100 | 40 |
| 2 | 4 | 5 | 6 | Pd2(dba)3 | PPh3 | iPr3SiH | Toluene | 100 | 55 |
| 3 | 4 | 5 | 6 | Pd2(dba)3 | ( p-ClC6H4)3P | iPr3SiH | Toluene | 100 | 62 |
| 4 | 4 | 5 | 6 | Pd(OAc)2 | ( p-ClC6H4)3P | iPr3SiH | Toluene | 100 | 68 |
| 5 | 4 | 5 | 6 | Pd(OAc)2 | ( p-FC6H4)3P | iPr3SiH | Toluene | 100 | 54 |
| 6 | 3 | 2 | 3 | Pd(OAc)2 | ( p-ClC6H4)3P | Me(EtO)2SiH | Toluene | 100 | 70 |
| 7 | 3 | 2 | 3 | Pd(OAc)2 | ( p-ClC6H4)3P | Me(EtO)2SiH | Dioxane | 100 | 73 |
| 8 | 3 | 2 | 3 | Pd(OAc)2 | ( p-ClC6H4)3P | Me(EtO)2SiH | Dioxane | 110 | 66 |
| 9 | 3 | 2 | 3 | Pd(OAc)2 | ( p-ClC6H4)3P | Me(EtO)2SiH | Dioxane | 90 | 50 |
| a Reaction conditions: p-bromobenzoate 2a (0.3 mmol), TMSCHN2 1 (2 mol/L in hexane, 1.2 mmol, 4 equiv.), iPr3SiH (1.5 mmol, 5 equiv.), Pd(PPh3)4 (0.015 mmol, 5 mol%) in toluene (2 mL) under 100 ℃ for 8 h, in which 1 was added in 6 h. bAll the yields refer to isolated products after silica gel column chromatography. | |||||||||
With the optimized conditions in hand, the substrate scope of the aryl bromides was further investigated (Scheme 2). A variety of electron-donating and electron-withdrawing functional groups, including alkyl (3b and 3r), phenyl (3c), methoxyl (3d, 3l, 3p and 3r), nitro (3e), cyano (3f and 3n), acetamido (3g), acetyl (3h and 3m), sulfonyl (3j), alkenyl (3k), fluorine (3p), 1, 3-dioxolan-2-yl (3q), and naphthyl (3s), were all well-tolerated and gave the corresponding products in moderate to good yields, and the results showed that electron effect has obvious influence on the yields. In addition, the influence of steric effect was also observed. The ortho-substituted aryl bromide gave relatively lower yield as compared to the meta- and para-sub- stituted substrates with the same functional group (3l compared to 3d and 3o). Notably, the reaction tolerates the functional group bearing relatively active hydrogen (3g).
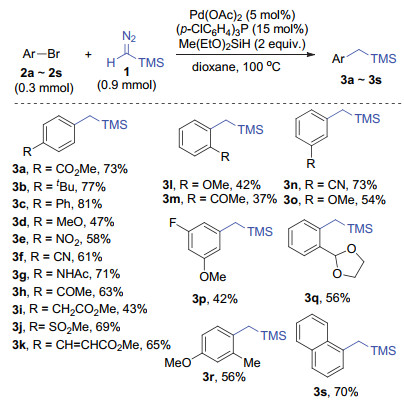
Reaction conditions: aryl bromide (0.3 mmol), TMSCHN2 1 (2 mol/L in hexane, 0.9 mmol, 3 equiv.), Me(EtO)2SiH (0.6 mmol, 2 equiv.), Pd(OAc)2 (0.015 mmol, 5 mol%), (p-ClC6H4)3P (0.045 mmol, 15 mol%) in dioxane (2 mL) under 100 ℃ for 8 h. 1 was added slowly with a syringe pump over 3 h. All the yields refer to the isolated products after silica gel column chromatography
The substrate scope to heteroaromatic bromides under the standard conditions was then expanded, and the results are shown in Scheme 3. Substrates with a series of heteroaromatic rings, including pyrimidine (5a), quinoline (5b), indole (5d~5g), benzofuran (5j), thiophene (5i), benzothiophene (5c) and benzoxazole (5h), could undergo this transformation and give the corresponding products in moderate yields.
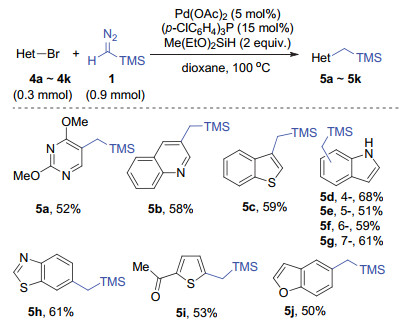
Reaction conditions: heteroaromatic bromide (0.3 mmol), TMS-CHN2 1 (2 mol/L in hexane, 0.9 mmol, 3 equiv.), Me(EtO)2SiH (0.6 mmol, 2 equiv.), Pd(OAc)2 (0.015 mmol, 5 mol%), (p-ClC6H4)3P (0.045 mmol, 15 mol%) in dioxane (2 mL) under 100 ℃ for 8 h. 1 was added slowly with a syringe pump over 3 h. All the yields refer to isolated products after silica gel column chromatography
For a further demonstration of the practical synthetic application of this method, the Pd-catalyzed reductive coupling reactions of 2a and 2b were carried out in 6 mmol scale, and the corresponding products 3a and 3b were isolated in gram-scale in similar yields to those in the small scale reactions (Eq. 2).
|
|
(2) |
Subsequently, the desilicification reactions were carried out to verify the feasibility of methylation process (Scheme 4). The desilicification process was efficient and easy to operate with excess amount of tetrabutylammonium fluoride (TBAF). The methylated products 6a~6j could be isolated in moderate yields from the corresponding benzyltrimethylsilanes.
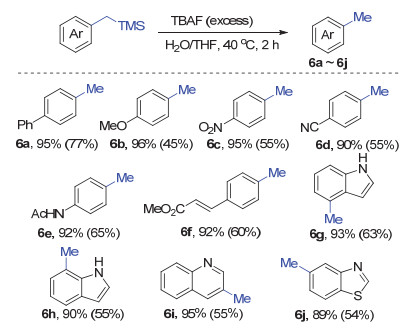
Reaction conditions: the purified benzyltrimethylsilanes (obtained from the 0.3 mmol-scale reductive coupling), TBAF (1 mol/L in THF, 0.4 mmol), H2O (36 mg, 2 mmol) in THF (1 mL), 40 ℃ for 2 h. All the yields refer to isolated products after silica gel column chromatography; the number in the bracket refers to the overall yield of the two steps
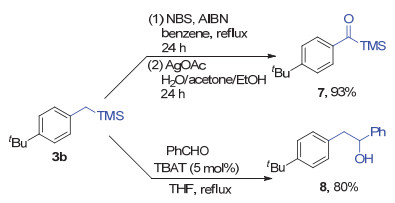
Furthermore, the benzyltrimethylsilane products obtained from the reductive cross-coupling are valuable compounds in organic synthesis. For examples, (p-(tert- butyl)benzyl)trimethylsilane (3b) could undergo oxidation reaction to afford 7, while reaction of 3b with benzaldehyde could afford addition product 8 in a moderate yield (Scheme 5).
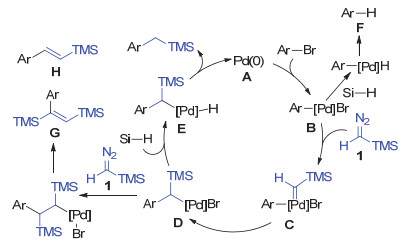
The proposed mechanism is shown in Scheme 6. The catalytically active Pd(0) species is generated from the reduction of Pd(OAc)2. The oxidative addition of the aryl halide to the Pd(0) A gives aryl palladium(Ⅱ) complex B. Reaction of trimethylsilyldiazomethane 1 and B leads to the generation of metal carbene species C. Subsequently, migratory insertion occurs to afford the benzyl palladium intermediate D. Intermediate D undergoes ligand exchange with Me(EtO)2SiH to afford intermediate E, and the product is generated in a following reductive elimination process. Two side reactions may occur in this transformation: (1) the ligand exchange between B and Me(EtO)2SiH, followed by reductive elimination provides arenes F, namely the direct reduction of arylbromide; (2) D may react with the second molecule of 1 to afford alkene by-products G and H. It is noteworthy that the latter process is reported as a synthetic method for silyl-substituted alkenes by Xu, Chen and co-workers.[17] These two kinds of by-products are both observed in our reaction.
Overall, we have developed a new methylation strategy using a reductive coupling/desilicification cascade process with trimethylsilyldiazomethane as methylation reagent. A wide range of functional groups and heteroaromatic rings were well-tolerated in this reaction. The transformation provides a new method for the introduction of methyl group and silylmethyl group into aromatic rings.
All necessary reagents were purchased from commercial suppliers and used without further purification. The solvents were all distilled prior to use. Purification of products were accomplished by flash chromatography on silica gel (200~300 mesh, from Qingdao, China). NMR spectra were measured on a Bruker ARX400 (1H NMR at 400 MHz, 13C NMR at 100 MHz) magnetic resonance spectrometer. IR spectra were recorded on a Nicolet Avatar 330 Fourier transform spectrometer (FT-IR) and are reported in wave numbers (cm-1). For HRMS measurements, the mass analyzer is FT-ICR.
Under nitrogen atmosphere, aryl bromide (0.3 mmol), Pd(OAc)2 (0.015 mmol, 3.4 mg), (p-ClC6H4)3P (0.045 mmol, 16.5 mg), Me(EtO)2SiH (0.3 mmol, 40 mg) and dioxane (1.6 mL) were mixed in a 10 mL reaction flask. The solution was stirred at 100 ℃, and the solution of TMSCHN2 1 (2 mol/L in hexane, 0.45 mL) and Me(EtO)2- SiH (0.3 mmol, 40 mg) in dioxane (0.4 mL) was added to this mixture through a syringe pump over 3 h. The reaction was stirred at 100 ℃ for another 5 h. Then the mixture was cooled to room temperature and filtered through a short column filled with silica gel. Solvent was evaporated under reduced pressure, and the crude residue was purified by thin layer chromatography on silica gel to afford the product.
Methyl 4-((trimethylsilyl)methyl)benzoate (3a): 73% (48.6 mg) yield, colorless oil. The 1H NMR and 13C NMR data were consistent with the literature. [18]
(4-(tert-Butyl)benzyl)trimethylsilane (3b): 77% (50.8 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[16a]
([1, 1'-Biphenyl]-4-ylmethyl)trimethylsilane (3c): 81% (58.3 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[16a]
(4-Methoxybenzyl)trimethylsilane (3d): 47% (27.4 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[16a]
Trimethyl(4-nitrobenzyl)silane (3e): 58% (36.4 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[19]
4-((Trimethylsilyl)methyl)benzonitrile (3f): 61% (34.6 mg) yield, colorless oil. The 1H NMR and 13C NMR are were consistent with the literature.[16a]
N-(4-((Trimethylsilyl)methyl)phenyl)acetamide (3g): 71% (47.1 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature. [20]
1-(4-((Trimethylsilyl)methyl)phenyl)ethan-1-one (3h): 63% (38.9 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[16b]
Methyl 2-(4-((trimethylsilyl)methyl)phenyl)acetate (3i): 43% (30.4 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 6.92 (d, J=8.0 Hz, 2H), 6.75 (d, J=8.0 Hz, 2H), 3.49 (s, 3H), 3.37 (s, 2H), 1.86 (s, 2H), -0.21 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 172.4, 139.3, 129.3, 128.9, 128.2, 51.9, 40.7, 26.6, -2.0; IR (film) ν: 1741, 1436, 1259, 1161, 1013, 807 cm-1; HRMS (ESI) calcd for C13H21O2Si [M+H]+ 237.1305, found 237.1302.
Trimethyl(4-(methylsulfonyl)benzyl)silane (3j): 69% (50.1 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.77 (d, J=8.4 Hz, 2H), 7.16 (d, J=8.3 Hz, 2H), 3.04 (s, 3H), 2.20 (s, 2H), 0.00 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 148.1, 136.0, 128.5, 127.3, 44.6, 28.0, -2.0; IR (film) ν: 2957, 1593, 1304 1146, 1091, 851 cm-1; HRMS (ESI) calcd for C11H19O2SSi [M+H]+ 243.0870, found 243.0862.
Methyl (E)-3-(4-((trimethylsilyl)methyl)phenyl)acrylate (3k): 65% (48.4 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.66 (d, J=16.0 Hz, 1H), 7.38 (d, J=8.1 Hz, 2H), 7.01 (d, J=8.1 Hz, 2H), 6.37 (d, J=16.0 Hz, 1H), 3.79 (s, 3H), 2.12 (s, 2H), 0.00 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 167.7, 145.1, 143.9, 130.2, 128.4, 128.1, 115.8, 51.5, 27.6, -2.0; IR (film) ν: 2954, 1719, 1634, 1249, 1168, 850 cm-1; HRMS (ESI) calcd for C14H21O2Si [M+H]+ 249.1305, found 249.1302.
(2-Methoxybenzyl)trimethylsilane (3l): 42% (24.4 mg) yield, colorless oil. The 1H NMR and 13C NMR data were consistent with the literature.[21]
1-(2-((Trimethylsilyl)methyl)phenyl)ethan-1-one (3m): 37% (22.9 mg) yield, colorless oil. The 1H NMR and 13C NMR data were consistent with the literature.[22]
3-((Trimethylsilyl)methyl)benzonitrile (3n): 73% (41.4 mg) yield, colorless oil. The 1H NMR and 13C NMR data were consistent with the literature.[23]
(3-Methoxybenzyl)trimethylsilane (3o): 54% (31.4 mg) yield, colorless oil. The 1H NMR and 13C NMR data were consistent with the literature.[21]
(3-Fluoro-5-methoxybenzyl)trimethylsilane (3p): 42% (26.7 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 6.36~6.30 (m, 3H), 3.76 (s, 3H), 2.05 (s, 2H), 0.01 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 163.5 (d, J=243.5 Hz), 160.5 (d, J=11.9 Hz), 143.8 (d, J=9.8 Hz), 109.5, 107.1 (d, J=21.5 Hz), 97.1 (d, J=25.4 Hz), 55.3, 27.5, -1.91; IR (film) ν: 2956, 1612, 1249, 1156, 982, 850 cm-1; HRMS (ESI) calcd for C11H18FOSi [M+H]+213.1111, found 213.1109.
(2-(1, 3-Dioxolan-2-yl)benzyl)trimethylsilane (3q): 56% (39.6 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.53 (d, J=7.6 Hz, 1H), 7.22 (td, J=8.4, 1.0 Hz, 1H), 7.12 (t, J=7.4 Hz, 1H), 7.00 (d, J=7.6 Hz, 1H), 5.91 (s, 1H), 4.17~4.11 (m, 2H), 4.07~4.01 (m, 2H), 2.27 (s, 2H), 0.02 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 139.3, 133.7, 129.4, 128.7, 126.1, 124.2, 101.7, 65.2, 22.8, -1.4; IR (film) ν: 2954, 2894, 1249, 1087, 944, 846 cm-1; HRMS (ESI) calcd for C13H21O2Si [M+H]+ 237.1311, found 237.1307.
(4-Methoxy-2-methylbenzyl)trimethylsilane (3r): 56% (34.9 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 6.89 (d, J=8.3 Hz, 1H), 6.71~6.71 (m, 1H), 6.68~6.65 (m, 1H), 3.78 (s, 3H), 2.23 (s, 3H), 2.04 (s, 2H), 0.02 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 156.4, 135.6, 130.8, 129.4, 115.6, 111.0, 55.1, 22.4, 20.6, -1.4; IR (film) ν: 2955, 1609, 1501, 1254, 1203, 854 cm-1; HRMS (ESI) calcd for C12H20OSi [M]+ 208.1283, found 208.1277.
Trimethyl(naphthalen-1-ylmethyl)silane (3s): 70% (44.9 mg) yield, colorless oil. The 1H NMR and 13C NMR data were consistent with the literature. [24]
2, 4-Dimethoxy-5-((trimethylsilyl)methyl)pyrimidine (5a): 52% (35.3 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.83 (s, 1H), 3.93 (s, 6H), 1.84 (s, 2H), -0.04 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 168.5, 162.9, 155.3, 113.9, 54.4, 53.5, 15.6, -1.8; IR (film) ν: 2956, 1570, 1486, 1377, 1074, 844 cm-1; HRMS (ESI) calcd for C10H19N2O2Si [M+H]+ 227.1216, found 227.1217.
3-((Trimethylsilyl)methyl)quinoline (5b): 58% (37.4 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.63 (d, J=2.0 Hz, 1H), 8.05 (d, J=8.4 Hz, 1H), 7.72~7.70 (m, 2H), 7.62~7.58 (m, 1H), 7.50~7.74 (m, 1H), 2.24 (s, 2H), 0.04 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 151.8, 145.7, 133.6, 132.5, 129.0, 128.3, 127.8, 126.9, 126.4, 24.3, -2.0; IR (film) ν: 2953, 1492, 1249, 1174, 904, 847, 748 cm-1; HRMS (ESI) calcd for C13H18NSi [M+H]+ 216.1209, found 216.1209.
(Benzo[b]thiophen-3-ylmethyl)trimethylsilane (5c): 59% (38.9 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.86~7.84 (m, 1H), 7.67 (d, J=7.4 Hz, 1H), 7.40~7.31 (m, 2H), 6.86 (s, 1H), 2.33 (s, 2H), 0.04 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 140.2, 139.3, 134.3, 123.8, 123.4, 122.7, 122.1, 118.3, 18.6, -1.4; IR (film) ν: 2953, 1733, 1422, 1246, 838, 761 cm-1; HRMS (EI) calcd for C12H16OSi [M]+ 204.0970, found 204.0966.
4-((Trimethylsilyl)methyl)-1H-indole (5d): 68% (41.4 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.01 (s, 1H), 7.16~7.16 (m, 3H), 6.80 (d, J=6.4 Hz, 1H), 6.53~6.52 (m, 1H), 2.42 (s, 2H), 0.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 135.6, 133.0, 127.0, 122.9, 122.0, 118.8, 106.9, 101.8, 24.0, -1.3; IR (film) ν: 3415, 2955, 1248, 1078, 851, 749 cm-1; HRMS (ESI) calcd for C12H18NSi [M+H]+ 204.1209, found 204.1202.
5-((Trimethylsilyl)methyl)-1H-indole (5e): 51% (31.1 mg) yield, colorless oil. The 1H NMR and 13C NMR data were consistent with the literature. [16a]
6-((Trimethylsilyl)methyl)-1H-indole (5f): 59% (35.9 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.94 (s, 1H), 7.51 (s, J=8.1 Hz, 1H), 7.10 (t, J=2.9 Hz, 1H), 7.02 (s, 1H), 6.82 (d, J=8.1 Hz, 1H), 6.50 (s, 1H), 6.20 (s, 2H), 0.03 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 136.4, 134.4, 124.6, 122.9, 121.3, 120.1, 109.6, 102.3, 27.1, -1.8; IR (film) ν: 3408, 2955, 1454, 1247, 1089, 848 cm-1; HRMS (ESI) calcd for C12H18NSi [M+H]+ 204.1209, found 204.1199.
7-((Trimethylsilyl)methyl)-1H-indole (5g): 61% (37.1 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.92 (s, 1H), 7.44 (d, J=7.9 Hz, 1H), 7.18 (t, J=2.9 Hz, 1H), 7.05 (t, J=7.4 Hz, 1H), 6.87 (d, J=7.1 Hz, 1H), 6.56 (dd, J=3.2, 2.1 Hz, 1H), 2.27 (s, 2H), 0.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 134.6, 127.5, 123.5, 122.8, 121.4, 120.0, 116.7, 103.0, 21.8, -1.3; IR (film) ν: 3435, 1430, 1332, 1249, 1099, 854 cm-1; HRMS (ESI) calcd for C12H18NSi [M+H]+ 204.1209, found 204.1197.
6-((Trimethylsilyl)methyl)benzo[d]thiazole (5h): 61% (40.4 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.94 (s, 1H), 7.79~7.77 (m, 2H), 7.10 (d, J=8.3 Hz, 1H), 2.25 (s, 1H), 0.02 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 153.9, 153.8, 139.2, 126.6, 122.1, 121.1, 27.0, -2.0; IR (film) ν: 3435, 2953, 1430, 1249, 1067, 854 cm-1; HRMS (ESI) calcd for C11H16NSSi [M+H]+ 222.0773, found 222.0765.
1-(5-((Trimethylsilyl)methyl)thiophen-2-yl)ethan-1-one (5i): 53% (33.7 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.49 (d, J=3.8 Hz, 1H), 6.60 (d, J=3.7 Hz, 1H), 2.48 (s, 3H), 2.34 (s, 2H), 0.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 190.1, 154.2, 140.5, 133.5, 124.6, 26.3, 22.5, -2.0; IR (film) ν: 2956, 1655, 1446, 1281, 1250, 843 cm-1; HRMS (ESI) calcd for C10H17OSSi [M+H]+ 213.0769, found 213.0758.
(Benzofuran-5-ylmethyl)trimethylsilane (5j): 50% (30.6 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.57 (d, J=2.1 Hz, 1H), 7.36 (d, J=8.4 Hz, 1H), 7.21 (d, J=1.0 Hz, 1H), 6.94 (dd, J=8.4, 1.6 Hz, 1H), 6.68 (dd, J=2.0, 0.8 Hz, 1H), 2.17 (s, 2H), 0.01 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 152.7, 144.8, 134.7, 127.5, 124.7, 119.5, 110.7, 106.2, 26.6, -1.9; IR (film) ν: 2955, 1467, 1247, 1198, 847, 733 cm-1; HRMS (EI) calcd for C12H16SSi [M]+ 220.0742, found 220.0737.
Under nitrogen atmosphere, aryl bromides (6 mmol), Pd(OAc)2 (0.3 mmol, 67.3 mg), (p-ClC6H4)3P (0.9 mmol, 329 mg), Me(EtO)2SiH (6 mmol, 804 mg) and dioxane (32 mL) were mixed in a 100 mL reaction flask. The mixture was stirred at 100 ℃ and the solution of TMSCHN2 1 (2 mol/L in hexane, 18 mmol, 9 mL), Me(EtO)2SiH (6 mmol, 804 mg) and dioxane (8 mL) was added into the reaction system through syringe pump over 3 h. Then the reaction was stirred at 100 ℃ for another 5 h. Then the mixture was cooled to room temperature and filtered through a short column filled with silica gel. Solvent was evaporated under reduced pressure, and the crude residue was purified by distillation under reduced pressure to give the product.
The purified benzyltrimethylsilanes, obtained by the 0.3 mmol-scale reductive coupling, was transferred into a 10 mL round bottom flask. Then THF (2 mL), TBAF (1 mol/L in THF, 0.3 mmol, 0.3 mL), H2O (2 mmol, 36 mg) was added. The mixture was stirred at 40 ℃ for 2 h. Solvent was evaporated under reduced pressure, and the crude residue was purified by flash chromatography on silica gel to give the product.
4-Methyl-1, 1'-biphenyl (6a): 77% (38.8 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature. [7a]
1-Methoxy-4-methylbenzene (6b): 45% (16.5 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature. [11]
1-Methyl-4-nitrobenzene (6c): 55% (22.6 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[25]
4-Methylbenzonitrile (6d): 55% (19.3 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature. [11]
N-(p-tolyl)acetamide (6e): 65% (29.1 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[26]
Methyl (E)-3-(p-tolyl)acrylate (6f): 60% (31.7 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[27]
4-Methyl-1H-indole (6g): 63% (24.8 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[28]
7-Methyl-1H-indole (6h): 55% (21.6 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature. [28]
3-Methylquinoline (6i): 55% (23.6 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature.[28]
5-Methylbenzo[d]thiazole (6j): 54% (24.1 mg) yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ: 8.95 (s, 1H), 7.94 (s, 1H), 7.83 (d, J=8.2 Hz, 1H), 7.27 (d, J=8.6 Hz, 1H), 2.52 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 153.9, 153.7, 136.3, 130.6, 127.2, 123.5, 121.3, 21.4; IR (film) ν: 3055, 2918, 1435, 1318, 849, 803 cm-1; HRMS (ESI) calcd for C8H8NS [M+H]+ 150.0377, found 150.0371.
Under nitrogen atmosphere, 3b (0.5 mmol, 110 mg), N-bromosuccinimide (NBS) (1.1 mmol, 196 mg), 2, 2'-azobis(2-methylpropionitrile) (AIBN) (0.025 mmol, 4.1 mg) and benzene (2.5 mL) were mixed in a 10 mL reaction flask and refluxed for 24 h. Then the mixture was cooled to room temperature and filtered. Solvent was evaporated under reduced pressure, and the crude residue was dissolved in dichloromethane (DCM) 10 mL and washed with saturated Na2S2O3 aqueous solution 10 mL. The organic layer was washed with 10 mL brine for 3 times and then dried over Na2SO4. The organic layer was separated and solvent was evaporated under reduced pressure to afford the α, α-dibromo-substituted intermediate product. This intermediate product was then transferred into a 10 mL reaction flask, to which the solvent (water/acetone/ethanol, V:V:V=1:2:3, 5 mL) and AgOAc (1 mmol, 167 mg) were added. The mixture was stirred for 24 h in dark. After the reaction was complete, the mixture was filtered and the solvent was evaporated under reduced pressure. The crude residue was dissolved in 20 mL petroleum ether and washed with 10 mL brine for 3 times. The combined aqueous layer was extracted with 10 mL petroleum ether for three times. The combined organic layer was dried over Na2SO4. The organic layer was then filtered and solvent was evaporated under reduced pressure. The crude residue was purified by flash chromatography on silica gel, eluted with petroleum ether to give the product (4-(tert-butyl)phenyl)(trime- thylsilyl)methanone (7):[29] 93% (108.8 mg) yield, colorless oil. The 1H NMR and 13C NMR data were consistent with the literature. [30]
Under nitrogen atmosphere, 3b (0.5 mmol, 110 mg), tetrabutylammonium difluorotriphenylsilicate (TBAT) (0.025 mmol, 13.5 mg), benzaldehyde (0. 5 mmol, 53 mg) and THF (2 mL) were mixed in a 10 mL reaction flask and refluxed for 3 h. The mixture was cooled to room temperature. Then 10 mL ethyl acetate and 10 mL hydrochloric acid (2 mol/L) was added to the mixture. The water layer was extracted with 10 mL ethyl acetate for three times. The organic layer was combined and dried over Na2SO4. Solvent was evaporated under reduced pressure, and the crude residue was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate, V:V=100:1) to give the product 2-(4-(tert-butyl)phenyl)-1-phenylethan-1- ol (8):[21] 80% (101.6 mg) yield, colorless oil. The 1H NMR and 13C NMR data are consistent with the literature. [31]
Supporting Information 1H NMR and 13C NMR spectra of all the products. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(a) McGrath, N. A.; Brichacek, M.; Njardarson, J. T. J. Chem. Educ. 2010, 87, 1348.
(b) Barreiro, E. J.; Kummerle, A. E.; Fraga, C. A. Chem. Rev. 2011, 111, 5215.
For an example, see: Angell, R.; Aston, N. M.; Bamborough, P.; Buckton, J. B.; Cockerill, S.; deBoeck, S. J.; Edwards, C. D.; Holmes, D. S.; Jones, K. L.; Laine, D. I.; Patel, S.; Smee, P. A.; Smith, K. J.; Somers, D. O.; Walker, A. L. Bioorg. Med. Chem. Lett. 2008, 18, 4428.
(a) Schönherr, H.; Cernak, T. Angew. Chem., Int. Ed. 2013, 52, 12256.
(b) Leung, C. S.; Leung, S. S. F.; Tirado-Rives, J.; Jorgensen, W. L. J. Med. Chem. 2012, 55, 4489.
For selected recent examples, see: (a) Feng, K.; Quevedo, R. E.; Kohrt, J. T.; Oderinde, M. S.; Reilly, U.; White, M. C. Nature 2020, 580, 621.
(b) Serpier, F.; Pan, F.; Ham, W. S.; Jacq, J.; Genicot, C.; Ritter, T. Angew. Chem., Int. Ed. 2018, 57, 10697.
(c) Haydl, A. M.; Hartwig, J. F. Org. Lett. 2019, 21, 1337.
(d) Ye, W.; Yan, Z.; Wan, C.; Hou, H.; Wang, Z. Acta Chim. Sinica 2018, 76, 99(in Chinees).
(叶文波, 晏子聪, 万常峰, 侯豪情, 汪志勇, 化学学报, 2018, 76, 99.)
(a) Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinummo, M. Tetrahedron 1971, 27, 3575.
(b) Ochiai, M.; Morita, K. Tetrahedron Lett. 1967, 8, 2349.
(c) Minisci, F.; Galli, R.; Cecere, M.; Malatesta, V.; Caronna, T. Tetrahedron Lett. 1968, 9, 5609.
(d) Sugimori, A.; Yamada, T.; Ishida, H.; Nose, M.; Terashima, K.; Oohata, N. Bull. Chem. Soc. Jpn. 1986, 59, 3905.
For reviews of methylation methods, see: (a) Yan, G.; Borah, A. J.; Wang, L.; Yang, M. Adv. Synth. Catal. 2015, 357, 1333.
(b) Kim, J.; Cho, S. H. Synlett 2016, 27, 2525.
(c) Hu, L.; Liu, Y. A.; Liao, X. Synlett 2018, 29, 375.
For selected examples, see: (a) Hu, L.; Liu, X.; Liao, X. Angew. Chem., Int. Ed. 2016, 55, 9743.
(b) Yang, C. T.; Zhang, Z. Q.; Liu, Y. C.; Liu, L. Angew. Chem., Int. Ed. 2011, 50, 3904.
(c) Agrawal, T.; Cook, S. P. Org. Lett. 2014, 16, 5080.
(d) Shang, R.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2015, 137, 7660.
(e) Wang, J.; Zhao, J.; Gong, H. Chem. Commun. 2017, 53, 10180.
(f) Liang, Z.; Xue, W.; Lin, K.; Gong, H. Org. Lett. 2014, 16, 5620.
(g) Shi, W.-J.; Shi, Z.-J. Chin. J. Chem. 2018, 36, 183.
Uemura, T.; Yamaguchi, M.; Chatani, N. Angew. Chem., Int. Ed. 2016, 55, 3162. doi: 10.1002/anie.201511197
He, Z.-T.; Li, H.; Jaudl, A. M.; Whiteker, G. T.; Hartwig, J. F. J. Am. Chem. Soc. 2018, 140, 17197. doi: 10.1021/jacs.8b10076
Huihui, K. M. M.; Caputo, J. A.; Melchor, Z.; Olivares, A. M.; Spiewak, A. M.; Johnson, K. A.; DiBenedetto, T. A.; Kim, S.; Ackerman, L. K. G.; Weix, D. J. J. Am. Chem. Soc. 2016, 138, 5016. doi: 10.1021/jacs.6b01533
Kariofillis, S. K.; Shields, B. J.; Tekle-Smith, M. A.; Zacuto, M. J.; Doyle, A. G. J. Am. Chem. Soc. 2020, 142, 7683. doi: 10.1021/jacs.0c02805
(a) Xiao, Q.; Zhang, Y.; Wang, J. Acc. Chem. Res. 2013, 46, 236.
(b) Xia, Y.; Qiu, D.; Wang, J. Chem. Rev. 2017, 117, 13810.
(c) Xia, Y.; Wang, J. J. Am. Chem. Soc. 2020, 142, 10592.
For selected examples for reductive coupling reactions involving metal carbene species, see: (a) Xia, Y.; Hu, F.; Liu, Z.; Qu, P.; Ge, R.; Ma, C.; Zhang, Y.; Wang, Org. Lett. 2013, 15, 1784.
(b) Xia, Y.; Hu, F.; Xia, Y.; Liu, Z.; Ye, F.; Zhang, Y.; Wang, J. Synthesis 2017, 49, 1073.
For the examples of using TMSCHN2 in cross-coupling reactions, see: (a) Kudirka, R.; Van Vranken, D. L. J. Org. Chem. 2008, 73, 3585.
(b) Xu, S.; Chen, R.; Fu, Z.; Zhou, Q.; Zhang, Y.; Wang, J. ACS Catal. 2017, 7, 1993.
(a) Leiendecker, M.; Hsiao, C.-C.; Guo, L.; Alandini, N.; Rueping, M. Angew. Chem., Int. Ed. 2014, 53, 12912.
(b) Zhang, W.-X.; Ding, C.-H.; Luo, Z.-B.; Hou, X.-L.; Dai, L.-X. Tetrahedron Lett. 2006, 47, 8391.
(c) Baciocchi, E.; Rol, C.; Rosato, G. C.; Sebastiani, G. V. J. Chem. Soc., Chem. Commun. 1992, 59.
(a) Cai, G.; Huang, Y.; Du, T.; Zhang, S.; Yao, B.; Li, X. Chem. Commun. 2016, 52, 5425.
(b) Perez, I.; Sestelo, J. P.; Sarandeses, L. A. J. Am. Chem. Soc. 2001, 123, 4155.
Mu, Q.-C.; Wang, X.-B.; Ye, F.; Sun, Y.-Li.; Bai, X.-F.; Chen, J.; Xia, C.-G.; Xu, Li.-W. Chem. Commun. 2018, 54, 12994.
Tobisu, M.; Kita, Y.; Ano, Y.; Chatani, N. J. Am. Chem. Soc. 2008, 130, 15982. doi: 10.1021/ja804992n
Suzuki, H.; Murashima, T.; Kozai, I.; Mori, T. J. Chem. Soc., Perkin Trans. 11993, 1591.
Molander, G. A.; Yun, C.-S.; Ribagorda, M.; Biolatto, B. J. Org. Chem. 2003, 68, 5534. doi: 10.1021/jo0343331
Das, M.; O'Shea, D. F. Tetrahedron 2013, 69, 6448. doi: 10.1016/j.tet.2013.05.078
Kalvet, I.; Sperger, T.; Scattolin, T.; Magnin, G.; Schoenebeck, F. Angew. Chem., Int. Ed. 2017, 56, 7078. doi: 10.1002/anie.201701691
Wu, Y.; Bouvet, S.; Izquierdo, S.; Shafir, A. Angew. Chem., Int. Ed. 2019, 58, 2617. doi: 10.1002/anie.201809657
Heijnen, D.; Hornillos, V.; Corbet, B. P.; Giannerini, M.; Feringa, B. L. Org. Lett. 2015, 17, 2262. doi: 10.1021/acs.orglett.5b00905
Al-Masum, M.; Welch, R. L. Tetrahedron Lett. 2014, 55, 1726. doi: 10.1016/j.tetlet.2014.01.102
Patel, P.; Borah, G. Chem. Commun. 2017, 53, 443. doi: 10.1039/C6CC08788D
Ruan, J.; Li, X.; Saidi, O.; Xiao, J. J. Am. Chem. Soc. 2008, 130, 2424. doi: 10.1021/ja0782955
He, K.-H.; Tan, F.-F.; Zhou, C.-Z.; Zhou, G.-J.; Yang, X.-L.; Li, Y. Angew. Chem., Int. Ed. 2017, 56, 3080. doi: 10.1002/anie.201612486
Huckins, J. R.; Rychnovsky, S. D. J. Org. Chem. 2003, 68, 10135. doi: 10.1021/jo035260o
Scala, A. D.; Garbacia, S.; Hélion, F.; Lannou, M.-I.; Namy, J.-L. Eur. J. Org. Chem. 2002, 2989.
Wu, X.-F.; Neumann, H.; Beller, M. Tetrahedron Lett. 2012, 53, 582. doi: 10.1016/j.tetlet.2011.11.104

Scheme 2 Substrate scope of aryl bromides
Reaction conditions: aryl bromide (0.3 mmol), TMSCHN2 1 (2 mol/L in hexane, 0.9 mmol, 3 equiv.), Me(EtO)2SiH (0.6 mmol, 2 equiv.), Pd(OAc)2 (0.015 mmol, 5 mol%), (p-ClC6H4)3P (0.045 mmol, 15 mol%) in dioxane (2 mL) under 100 ℃ for 8 h. 1 was added slowly with a syringe pump over 3 h. All the yields refer to the isolated products after silica gel column chromatography

Scheme 3 Substrate scope of heteroaromatic bromides
Reaction conditions: heteroaromatic bromide (0.3 mmol), TMS-CHN2 1 (2 mol/L in hexane, 0.9 mmol, 3 equiv.), Me(EtO)2SiH (0.6 mmol, 2 equiv.), Pd(OAc)2 (0.015 mmol, 5 mol%), (p-ClC6H4)3P (0.045 mmol, 15 mol%) in dioxane (2 mL) under 100 ℃ for 8 h. 1 was added slowly with a syringe pump over 3 h. All the yields refer to isolated products after silica gel column chromatography

Scheme 4 Substrate scope of the methylation reaction
Reaction conditions: the purified benzyltrimethylsilanes (obtained from the 0.3 mmol-scale reductive coupling), TBAF (1 mol/L in THF, 0.4 mmol), H2O (36 mg, 2 mmol) in THF (1 mL), 40 ℃ for 2 h. All the yields refer to isolated products after silica gel column chromatography; the number in the bracket refers to the overall yield of the two steps
Table 1. Optimization of the reaction conditionsa
|
|||||||||
| Entry | x/equiv. | y/equiv. | z/h | Cat. [Pd] | L | [H] | Solvent | T/℃ | Yieldb/% |
| 1 | 4 | 5 | 6 | Pd(PPh3)4 | — | iPr3SiH | Toluene | 100 | 40 |
| 2 | 4 | 5 | 6 | Pd2(dba)3 | PPh3 | iPr3SiH | Toluene | 100 | 55 |
| 3 | 4 | 5 | 6 | Pd2(dba)3 | ( p-ClC6H4)3P | iPr3SiH | Toluene | 100 | 62 |
| 4 | 4 | 5 | 6 | Pd(OAc)2 | ( p-ClC6H4)3P | iPr3SiH | Toluene | 100 | 68 |
| 5 | 4 | 5 | 6 | Pd(OAc)2 | ( p-FC6H4)3P | iPr3SiH | Toluene | 100 | 54 |
| 6 | 3 | 2 | 3 | Pd(OAc)2 | ( p-ClC6H4)3P | Me(EtO)2SiH | Toluene | 100 | 70 |
| 7 | 3 | 2 | 3 | Pd(OAc)2 | ( p-ClC6H4)3P | Me(EtO)2SiH | Dioxane | 100 | 73 |
| 8 | 3 | 2 | 3 | Pd(OAc)2 | ( p-ClC6H4)3P | Me(EtO)2SiH | Dioxane | 110 | 66 |
| 9 | 3 | 2 | 3 | Pd(OAc)2 | ( p-ClC6H4)3P | Me(EtO)2SiH | Dioxane | 90 | 50 |
| a Reaction conditions: p-bromobenzoate 2a (0.3 mmol), TMSCHN2 1 (2 mol/L in hexane, 1.2 mmol, 4 equiv.), iPr3SiH (1.5 mmol, 5 equiv.), Pd(PPh3)4 (0.015 mmol, 5 mol%) in toluene (2 mL) under 100 ℃ for 8 h, in which 1 was added in 6 h. bAll the yields refer to isolated products after silica gel column chromatography. | |||||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们