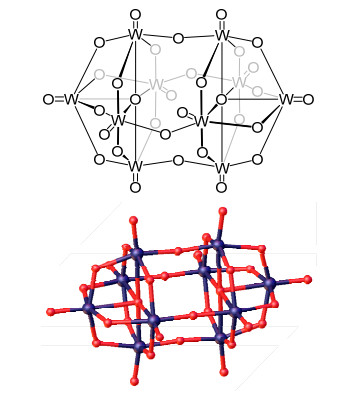
图式 1.
十聚钨酸阴离子[W10O32]4-的结构式和单晶结构
Scheme 1.
Structure of decatungstate [W10O32]4-

多金属氧酸盐(Polyoxometalate, POM)是一类由高价态的前过渡金属通过氧原子连接而成的金属氧簇化合物.由于其独特的光致电荷转移性质、氧化还原性质、酸碱性质和化学反应性, 多金属氧酸盐在合成化学、催化化学、能源化学、材料化学等领域得到了广泛应用[1].在光催化方面, Yamase[2], Papaconstantinou[3], Hill[4]等课题组在20世纪80年代系统研究了多金属氧酸盐催化烷烃、烯烃、醇、硫醚等有机物的氧化反应.此外, 多金属氧酸盐在光解水产氢/产氧、二氧化碳光还原、光催化降解污染物等方面也有着重要应用[5].
特别值得注意的是, 十聚钨酸盐([W10O32]4-)由于其特殊的光化学性能, 近年来在光催化有机合成方面备受关注.这类钨氧簇化合物的最高占据分子轨道(HOMO)分布在氧原子上, 最低未占据分子轨道(LUMO)分布在钨原子上.在紫外光或紫光照射下, 氧的2p轨道电子可以向钨的5d轨道转移, 通过这种“配体-金属电荷转移(Ligand-to-Metal Charge Transfer, LMCT)”作用, 实现分子内电荷转移, 产生高活性的激发态.此外, 十聚钨酸阴离子[W10O32]4-的结构中存在四条近乎于线型的W-O-W桥(Scheme 1), 有利于分子内的O→W电子转移, 因此[W10O32]4-在紫外光照射下很容易通过LMCT作用形成活性的激发态[6].
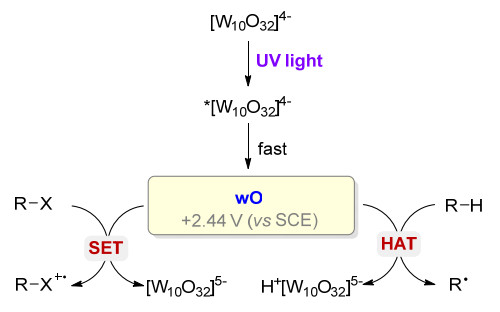
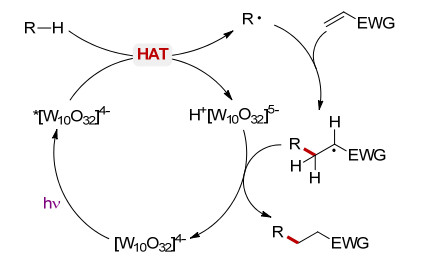
在紫外光或紫光照射下, 十聚钨酸盐被激发产生S1激发态(*[W10O32]4-), 随后很快(小于30 ps)转化为活性物种wO(量子产率约0.5~0.6). wO是一种驰豫激发态, 其形成可能经过氧原子中未成对电子占据轨道的重组, 产生亲电性较强的氧原子中心, 其氧化还原电位E(wO/[W10O32]5-)为+2.44 V (vs. SCE).因此, 当反应物R—X的氧化电位小于+2.44 V (vs. SCE)时, R—X与wO发生单电子转移(single electron transfer, SET)产生相应的自由基正离子R—X+•; 反之, 当反应物R—H键的氧化电位大于+2.44 V (vs. SCE)时, 发生氢原子转移(hydrogen atom transfer, HAT)产生相应的自由基R• (Scheme 2)[7].因此, 在光激发下十聚钨酸盐通过HAT或SET途径活化有机反应物, 为发展新型的光催化有机合成方法奠定了基础.
C—H键是有机化合物中普遍存在的化学键, C—H键的直接官能团化可以避免传统合成方法中的原料预合成、保护/脱保护等步骤, 使反应更具原子经济性和步骤经济性.正是由于这些优点, C—H键官能团化反应在过去几十年里得到了极大的关注和发展, 为有机功能分子的构筑提供了高效简便的新方法.通常情况下C—H键较为惰性, 难以直接发生化学转化, 往往需要贵金属催化剂、氧化剂、高温等反应条件才能转化[8].因此, 开发温和条件下的C—H键的官能团化反应具有重大意义.近年来, 光催化的有机合成反应得到了极大的复兴和发展, 为开发温和高效的新型合成方法提供了强有力的手段[9].十聚钨酸盐具有良好的光催化性能, 在紫外光激发下能直接攫取惰性C—H键的氢原子产生相应的碳中心自由基, 为温和条件下的C—H键官能团化反应提供了新策略[10].由于多金属氧酸盐催化氧化C—H键的相关研究开始较早, 本文综述了近年来光促进下十聚钨酸盐催化的C—H键官能团化反应.根据所构建化学键的类型(如C—C键、C—N键和C—F键)以及反应物的种类(饱和烷烃/醇/醚、醛、酮等), 对光促进下十聚钨酸盐催化的C—H键官能团化反应的研究进展进行了总结.
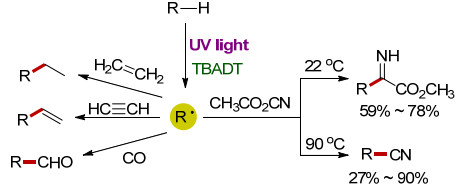
1993年, Hill课题组[11]采用十聚钨酸四丁基铵盐([nBu4N]4[W10O32], TBADT)为催化剂, 在紫外光照射下分别实现了饱和烷烃(如环己烷、环辛烷、正己烷等)与乙烯、乙炔、一氧化碳的氢化加成反应, 得到了相应的氢化烷基化产物.此外, 该催化体系也适用于饱和烷烃与氰基甲酸甲酯的反应[12].在22 ℃条件下, 主产物是α-亚氨基酯, 最高产率达到78%;在90 ℃条件下, 主产物是烷基腈化合物, 产率最高达90% (Scheme 3).值得一提的是, 在该反应中TBADT的浓度越低, 选择性越高; 当TBADT的浓度小于0.05 mmol/L时, 低温反应体系的选择性最高.
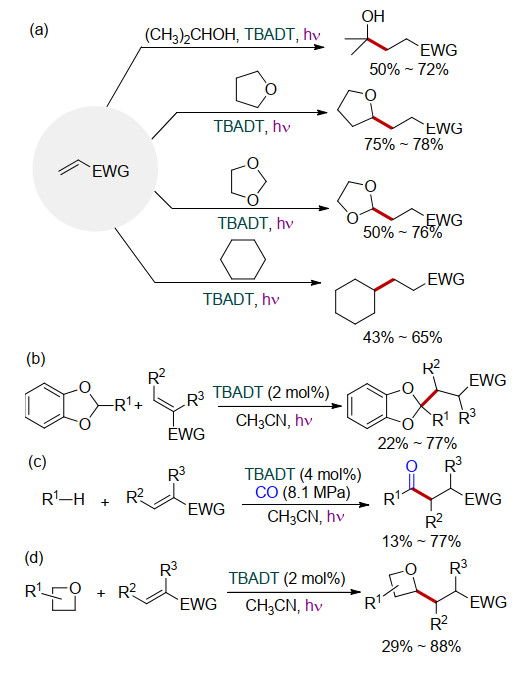
Albini课题组[13]采用TBADT为催化剂, 在光(310 nm)照射下实现了缺电子烯烃的氢化烷基化反应.环己烷、四氢呋喃(THF)、1, 3-二氧戊环、醇类化合物等作为自由基前体, 可以顺利地与缺电子烯烃发生反应, 以中等至良好的收率得到相应的氢化烷基化产物(Scheme 4a). 2011年, Fagnoni课题组[14]利用1, 3-苯并间二氧杂环戊烯作为自由基前体, 在TBADT催化下与缺电子烯烃反应, 以46%~77%的产率得到了一系列2-取代的1, 3-苯并间二氧杂环戊烯衍生物(Scheme 4b).随后, Ryu课题组[15]实现了TBADT催化三组分反应(Scheme 4c), 在环烷烃与缺电子烯烃的反应体系中加入8.1 MPa的CO, 得到了中等至良好收率的酰基化产物, 该反应体系烷烃与缺电子烯烃的化学计量比为20:1. 2014年, Ravelli课题组[16]利用TBADT催化氧杂环丁烷与缺电子烯烃的反应, 实现了氧杂环丁烷的2-烷基化反应(Scheme 4d).
反应机理如Scheme 5所示: TBADT首先在光激发下产生激发态*[W10O32]4-, 激发态*[W10O32]4-与饱和烷烃发生HAT过程, 直接攫取C(sp3)—H键的氢原子产生烷基自由基R•并生成H+[W10O32]5-.自由基R•进而与缺电子烯烃的C=C双键加成得到自由基加合物, 最后自由基加合物与H+[W10O32]5-发生氢原子转移, 生成产物并再生TBADT催化剂.传统的自由基链式反应通常需要加入自由基引发剂, 且反应进度不可控, 该过程为非链式反应过程, 不仅反应过程可控, 而且实现了100%的原子利用率.
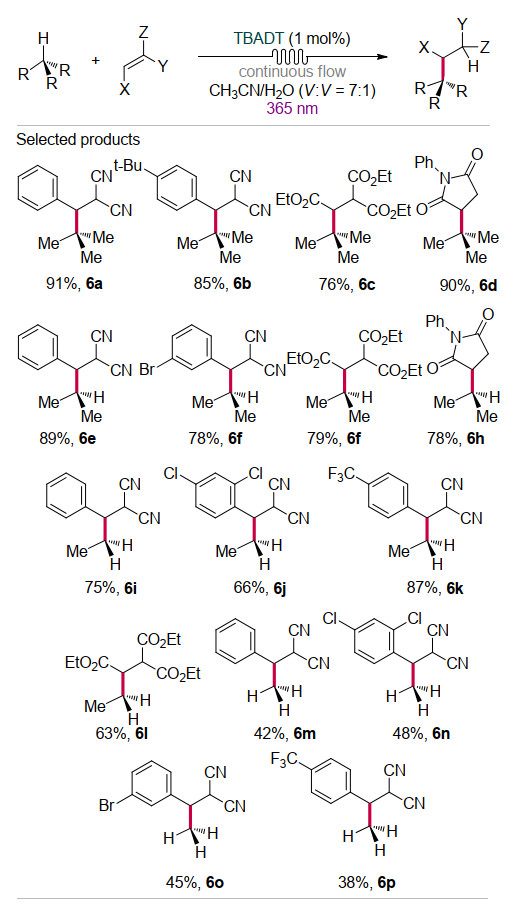
最近, Noël课题组[17]将TBADT催化和流式反应器相结合, 在温和条件下实现了惰性的气态低碳烷烃(甲烷、乙烷、丙烷和异丁烷)与缺电子烯烃的加成反应(Scheme 6).该方法采用的流式反应技术有利于低碳烷烃在反应溶剂中的溶解, 可以提升催化剂和反应物的传质效率, 避免过量气体反应物的使用.该反应在流式反应器中, 以乙腈/水(溶剂体积比为7:1)为溶剂通过波长为365 nm的LED紫外灯照射, 通过TBADT的HAT作用成功实现了在室温条件下具有不同键解离能(BDE)惰性C(sp3)—H键的活化(BDE:异丁烷403.9 kJ/mol、丙烷414.4 kJ/mol、乙烷422.8 kJ/mol以及甲烷439.5 kJ/mol).该方法具有较广的底物适用范围, 高选择性、高收率地实现了38例氢化烷基化产物的有效合成.与经典的共轭加成策略相比, 该方法直接活化惰性C—H键, 避免传统策略中冗长的预官能化过程.
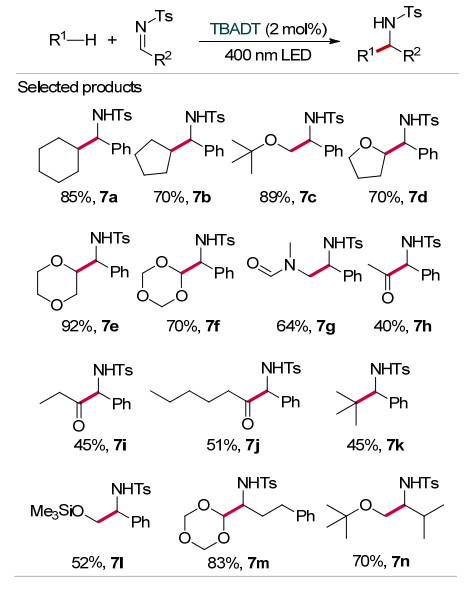
2019年, Dilman等[18]在400 nm光促进下以TBADT为催化剂实现了N-对甲苯磺酰亚胺类化合物的烷基化反应(Scheme 7).以饱和烷烃、醚或酰胺为自由基前体, 对芳香族N-对甲苯磺酰亚胺实现加成烷基化, 以中等至良好的收率得到相应的磺酰胺(7a~7g, 7l), 产率最高可达92%;脂肪族N-对甲苯磺酰亚胺也同样具有很好的反应性(7m~7n).此外, 采用脂肪醛(乙醛、丙醛、己醛等)为自由基前体, 可得到中等产率的羰基化产物(7h~7j).有趣的是, 当新戊醛为自由基前体参与反应, 没有生成预期的羰基化产物, 而是得到脱羰烷基化产物7k.这种结果与Fagnoni等[19]在2007年的报道一致.值得注意的是, 由于多金属氧酸盐的特殊性质, TBADT受光照激发之后具有较强的氧化性.当反应中采用未保护的亚胺为反应物时, 很容易发生氧化还原反应而淬灭催化剂.因此, 该方法只适用于氮原子上带保护基的亚胺类化合物, 且底物分子中不含易被氧化的基团.
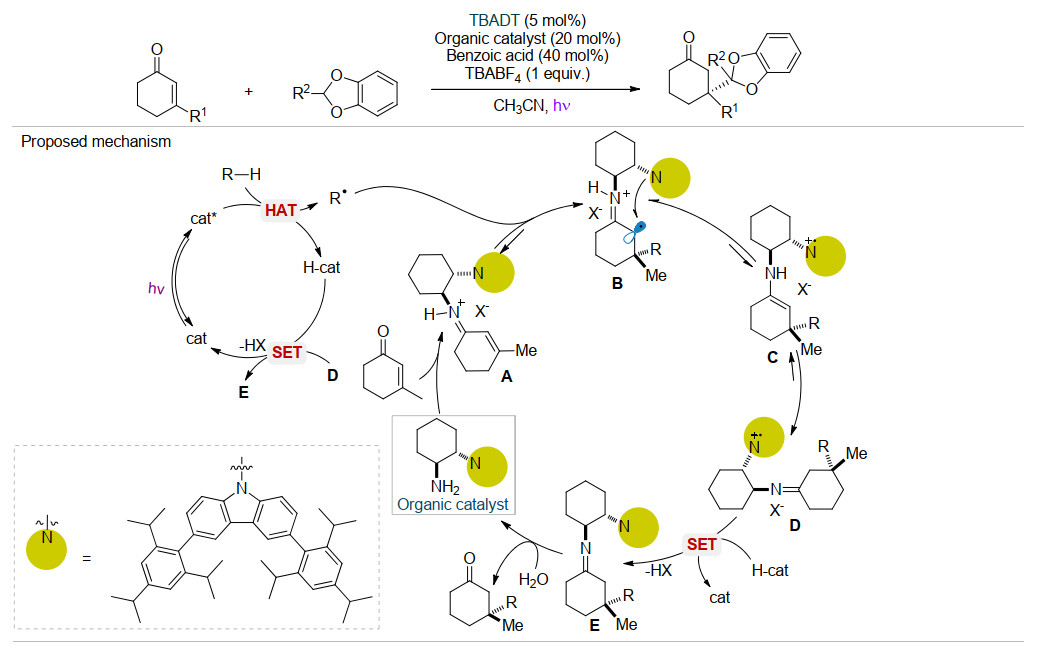
近年来, 光催化剂与手性催化剂的协同催化引起了化学工作者的广泛关注[20].例如, 2016年Melchiorre等[21]在光催化条件下采用TBADT与手性胺催化剂结合的策略, 通过自由基反应首次实现了季碳立体中心构筑(Scheme 8).该反应中大位阻手性胺催化剂首先与反应底物α, β-不饱和酮结合产生手性亚胺阳离子A, 随后在TBADT催化下HAT作用产生的自由基R•对中间体A的碳碳双键加成(由于中间体的位阻效应, 使自由基R•选择性地进攻烯亚胺中间体的Re面, 从而得到ee值较高的烷基化产物), 产生不稳定中间体B.随后中间体B发生分子内的电子转移生成C, 再经过互变异构产生中间体D.随后D经过单电子转移得到E, 进一步水解产生季碳产物, 释放有机催化剂.该反应通过对映选择性自由基共轭加成反应, 成功实现了季碳立体中心的构筑.
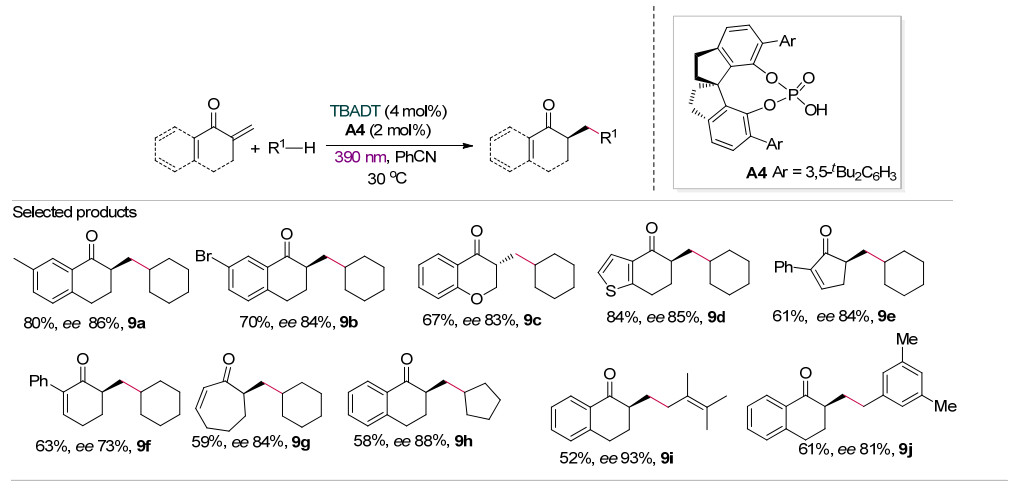
2020年, 汪普生等[22]在390 nm近紫外光照射下, 利用TBADT与手性磷酸的协同催化作用, 实现了环外α, β-不饱和酮与非活化烷烃C—H键的直接氢化烷基化反应.在该反应中光催化剂TBADT作为氢原子转移催化剂, 而手性螺环磷酸A4在对映选择性质子化过程中起到手性质子梭(chiral proton-transfer shuttle, CPTS)的作用(Scheme 9).该方法不仅底物适用范围广, 环状烷烃的C—H键(9a~9h)、苄位C—H键(9j)、烯丙位C—H键(9i)均能在该反应中实现官能团化, 具有较高的对映选择性.
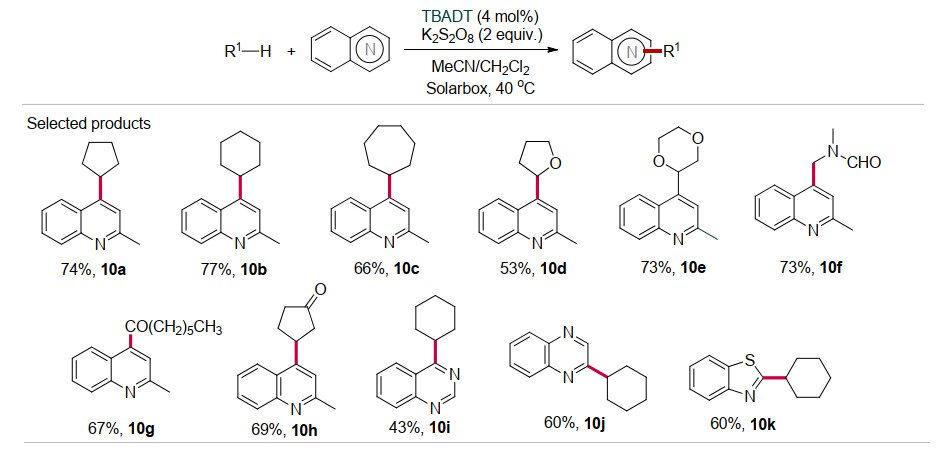
除了上述的氢化烷基化反应, 在TBADT催化下饱和烷烃(或醚等)也可以和烯烃(或杂环)的双键发生脱氢烷基化反应, 反应通常需要氧化剂、碱性添加剂或者直接放出氢气.例如, 2017年Ravelli和Ryuzu等[23]利用TBADT为光催化剂, 过硫酸钾K2S2O8为氧化剂, 实现了多种杂环与环状烷烃、醚、醛等的交叉脱氢偶联反应(Scheme 10).该催化体系的反应条件温和、底物适用面广, 其中自由基前体, 包括环状烷烃(环戊烷、环己烷、环庚烷)(10a~10c)、环醚(THF、1, 4-二氧六环)(10d~10e)、酰胺(10f)、醛(10g)、酮(10h)等, 杂环化合物的类型也非常丰富, 包括喹啉、喹唑啉、喹喔啉、酞嗪、苯并噻唑等.
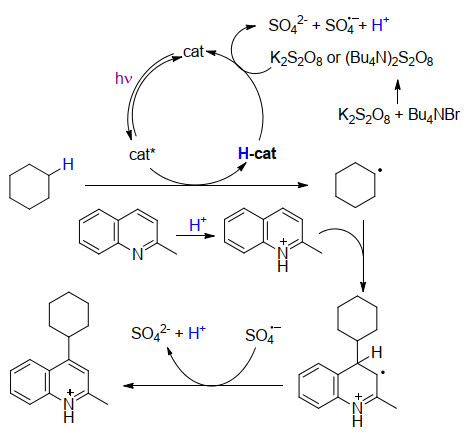
该反应中TBADT在光激发下产生高活性激发态, 直接攫取环状烷烃的C(sp3)—H键的氢原子, 生成相应的碳中心烷基自由基.同时, 催化剂攫取氢原子之后生成中间体H-cat, 在氧化剂K2S2O8作用下再生催化剂TBADT.另一方面, 烷基自由基与质子化的喹啉4位发生加成, 生成自由基加合物, 进一步在硫酸根自由基负离子作用下氧化脱氢得到4-烷基化喹啉(Scheme 11).
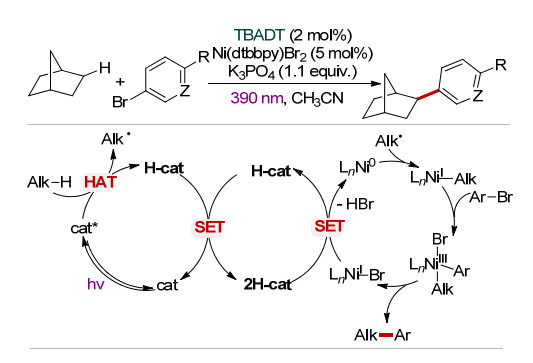
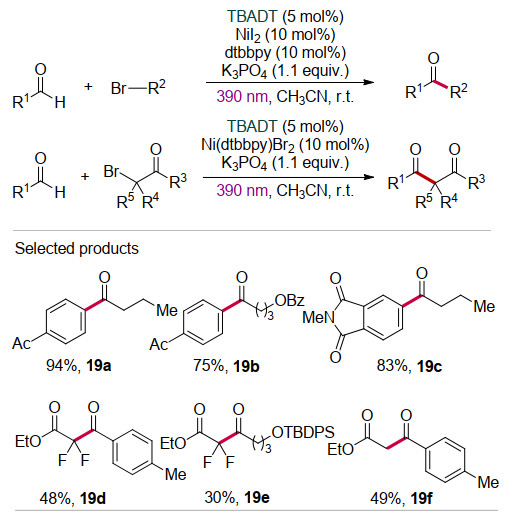
2018年, MacMillan课题组[24]利用TBADT光催化与镍配合物Ni(dtbbpy)Br2 [dtbbpy=4, 4'-二叔丁基-2, 2'-啶]的协同催化, 实现了芳基卤化物与饱和烷烃的交叉脱氢偶联反应.在该反应中, 光催化剂TBADT在390 nm近紫外光激发下通过HAT作用产生烷基自由基Alk•和还原态光敏剂H-cat. H-cat经过歧化反应再生催化剂TBADT和2H-cat.另一方面, 催化剂Ni(dtbbpy)Br2与2H-cat作用, 经历两次单电子还原生成活性的Ni(0)物种.随后, 烷基自由基Alk•对Ni(0)加成产生Ni(Ⅰ)-Alk中间体.进而, Ni(Ⅰ)-Alk中间体与芳基溴化物ArBr发生氧化加成产生Ni(Ⅲ)物种.最后, Ni(Ⅲ)物种发生还原消除产生相应的偶联产物Alk-Ar, 并释放出Ni(Ⅰ)—Br中间体. Ni(Ⅰ)—Br中间体与2H-cat发生单电子还原重生Ni(0)活性物种(Scheme 12).该方法底物适用范围广, 其中烷基化试剂前体包括饱和环状烷烃、桥环、螺环烷烃、环状脂肪醚、酮、苄位C—H键等, 溴化物包括芳基溴化物、杂环溴化物(吲哚、吡啶、嘧啶和噻唑)等.此外, 醇、磺胺等较敏感的官能团在该催化体系中也能很好地兼容.
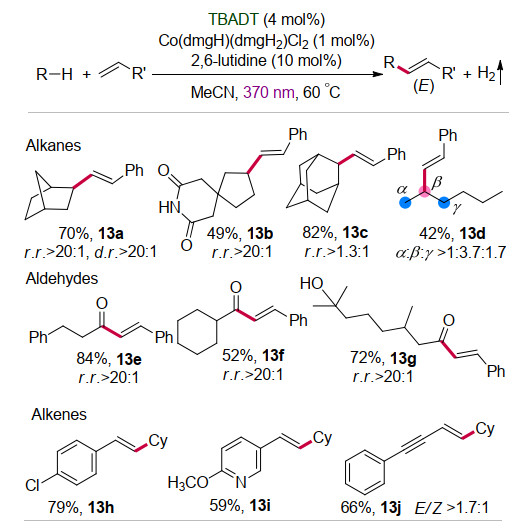
经典的交叉脱氢偶联反应往往需要添加化学计量的氧化剂或者碱性添加剂来促进反应的进行[25], 反应后产生相应的废盐为反应的后处理带来诸多不便.近年来在光/电催化等新型反应手段下, 交叉脱氢偶联反应可以直接放出氢气[26], 有效地避免了化学计量氧化剂或者添加剂的使用, 是一种环境友好的反应策略.基于钴配合物在放氢偶联反应中的关键作用, 吴杰课题组[27]采用TBADT光催化与钴肟配合物Co(dmgH)- (dmgH2)Cl2协同催化的策略, 在370 nm光照射的条件下实现了饱和烷烃(或醛)与烯烃的交叉偶联反应, 高选择性地合成了系列E-式取代烯烃(Scheme 13).反应中产生的Co(Ⅲ)—H中间体与质子发生反应直接放出氢气.该反应不仅立体选择性高, 而且具有较广的底物适用范围:饱和烷烃和醛的C—H键均可以直接实现官能团化(13a~13g), 富电子烯烃也很好地发生反应(13h~13j).此外, 该方法还适用于各种天然产物及其衍生物中饱和C—H键的选择性烯基化反应, 为复杂分子的后修饰提供了新方法.
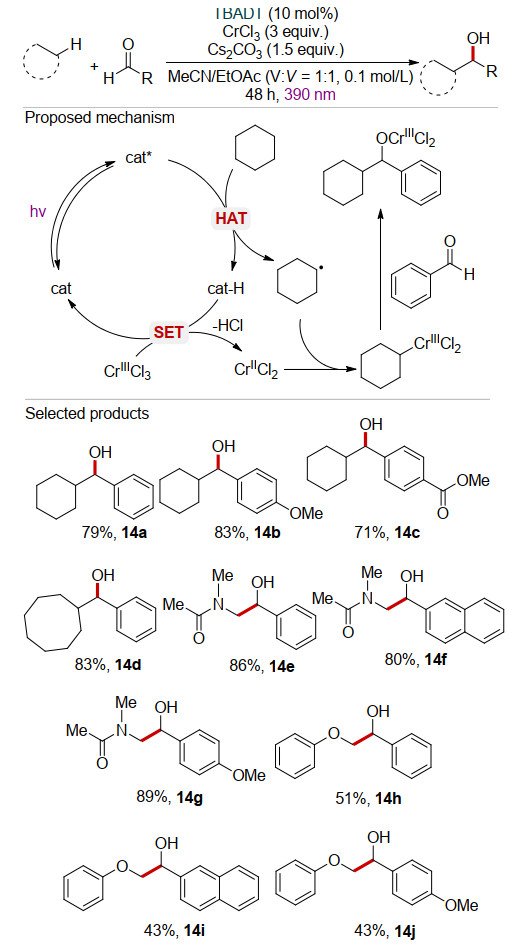
近期, Yahata课题组[28]利用TBADT与过量三氯化铬(CrCl3)的协同作用, 以乙腈/乙酸乙酯(体积比为1:1)为溶剂, 在碱的作用下实现了饱和烷烃与醛类化合物的加成反应.反应机理如Scheme 14所示, 烷烃在激发态cat*作用下发生HAT过程, 生成烷基自由基和还原性物种cat-H.随后cat-H还原CrⅢCl3产生CrⅡCl2, 并再生光催化剂TBADT. CrⅡCl2捕获烷基自由基, 生成具有亲核性质的烷基铬化物, 进一步与醛羰基发生亲核加成实现C—C键的构建.该反应具有较广的底物适用范围, 不仅对环烷烃的C(sp3)—H键有较好的作用(14a~14d), 对N, N-二甲基乙酰胺的甲基C—H键也可适用(14e~14g); 此外, 该反应也适用于苯甲醚的甲基C—H键, 以中等至较优的产率得到相应的仲醇类化合物(14h~14j).
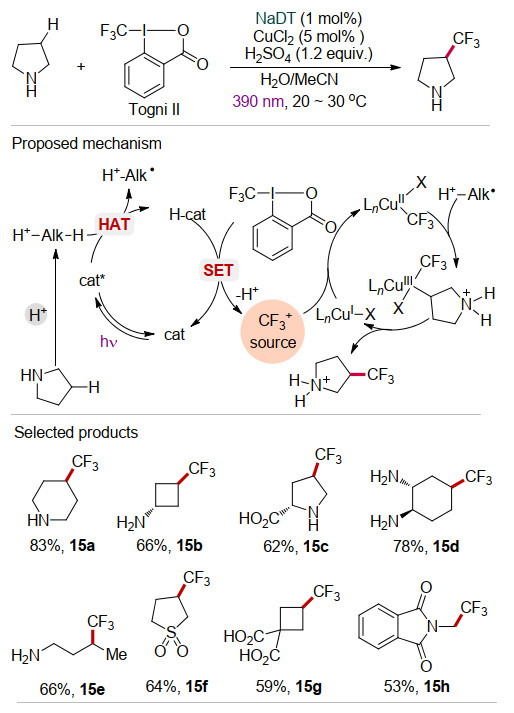
由于三氟甲基在药物化学和生物化学方面的重要性, 开发新型高效的三氟甲基化反应具有重要应用价值[29].最近MacMillan等[30]采用十聚钨酸钠盐(Na4- [W10O32], NaDT)光催化与铜协同催化的策略实现了C(sp3)—H键的直接三氟甲基化反应(Scheme 15).以商品化的Togni-Ⅱ试剂为三氟甲基源, 以简单的CuCl2为共催化剂, 无需添加配体, 在390 nm近紫外光照射下, 较为惰性的脂肪族C(sp3)—H键和苄位C—H键均能发生三氟甲基化反应.通过19F NMR、EPR等手段研究表明, 在该反应中铜催化剂不仅在Togni-Ⅱ试剂的单电子还原过程中发挥重要作用, 还可能生成关键中间体CuⅡ(CF3)x, 实现C(sp3)—CF3键的形成.值得注意的是, 该反应仅需要化学计量的反应物(反应物物质的量比为1/1.25).该方法对于未保护的胺远端的位置具有优异的选择性(15a~15e), 并且可以用于药物和天然产物中惰性C—H键的直接三氟甲基化(15f~15h), 为合成有潜在应用价值的三氟甲基化衍生物提供了新方法.
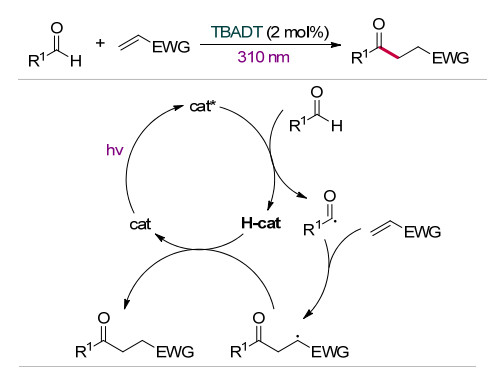
相较于酰氯、酰溴等酰基自由基前体来说, 醛的C—H键均裂具有较高的解离能(BDE), 因此醛C—H键的直接切断产生酰基自由基具有一定的挑战性.除了前述的饱和烷烃、醇、醚中的C(sp3)—H键可以经过HAT作用产生相应的自由基, 激发态的十聚钨酸盐通过HAT作用也可以将醛转化为相应的酰基自由基. 2007年, Fagnoni课题组[19]发现TBADT在310 nm紫外光照射下可以与醛的C—H键发生HAT作用, 产生相应的酰基自由基, 实现缺电子烯烃的氢化酰基化反应, 合成了系列酮类化合物.此前, 通过烯烃的自由基酰基化反应合成酮类化合物往往需要使用硫化物、硒化物等具有恶臭的试剂作为自由基引发剂, 或者采用有毒的一氧化碳气体作为羰基源[31].该反应仅需要2 mol%的TBADT为催化剂, 避免使用化学计量的自由基引发剂或有毒的一氧化碳, 以100%的原子利用率为酮类化合物的合成提供了绿色的新方法.其反应机理如Scheme 16所示, 激发态的TBADT直接从醛中攫取醛基C—H的氢原子生成酰基自由基, 酰基自由基与缺电子烯烃加成生成自由基加合物.随后自由基加合物与H-TBADT发生氢原子转移生成目标产物.
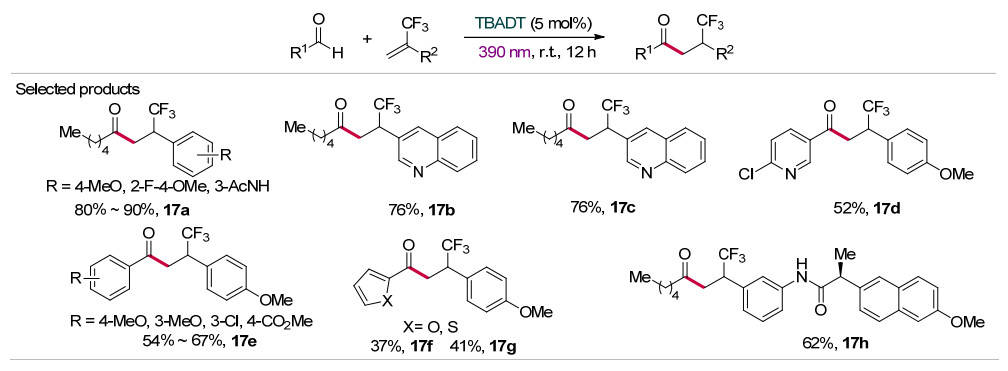
最近, 王川课题组[32]采用TBADT光催化的策略, 在温和条件下通过醛与三氟甲基取代烯烃的自由基加成反应, 实现了β-三氟甲基酮类化合物的高效合成(Scheme 17).值得注意的是, 通常情况下三氟甲基烯烃在加氢官能团化反应过程中往往伴随β-F消除反应, 但TBADT光催化体系经历HAT历程, 可成功避免β-F消除反应的发生.该催化体系表现出良好的底物适用范围, 各种脂肪醛(17a~17c)、芳香醛(17d~17e)、杂环芳香醛(17f~17g)都能取得良好的产率.对于含有雌素酮、萘普生等结构片段的复杂三氟甲基烯烃也能顺利发生氢化羰基化反应, 且保持反应物的构型不变(如含萘普生结构的17h).
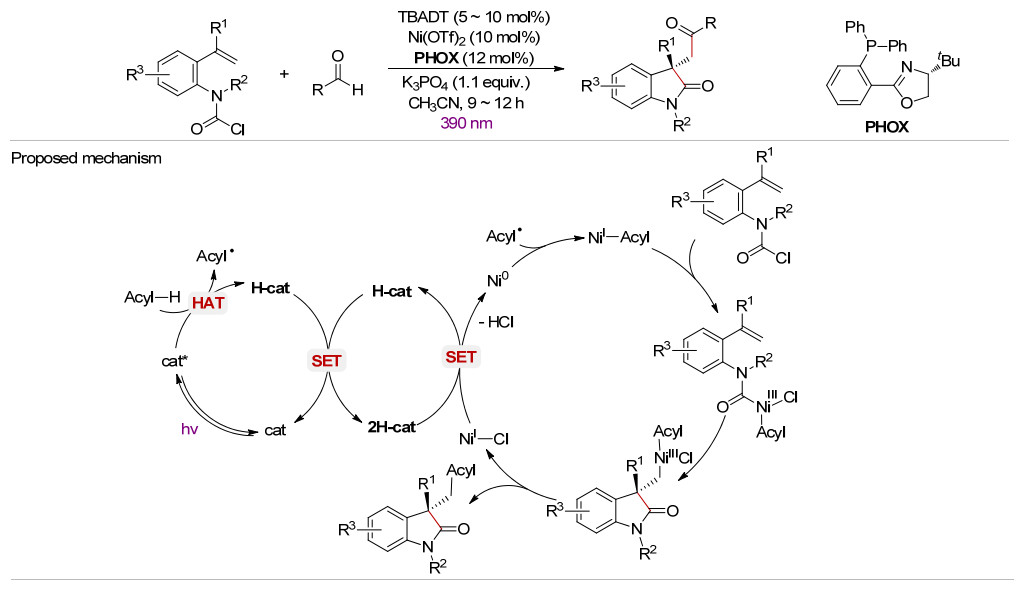
随后, 该课题组[33]采用TBADT与手性Ni-PHOX协同催化, 在390 nm紫外光照射下以醛为酰基源实现了烯烃的不对称酰基氨甲酰基化反应(Scheme 18).该反应是“氢原子转移光化学”与“不对称过渡金属催化”相结合实现烯烃双官能化的首次报道.该方法具有良好的底物适用范围, 可以在温和条件下以优异的对映选择性(ee值高达96%)合成各种具有手性季碳中心的氧化吲哚类产物.所推测的反应机理与Scheme 12所述的协同催化相似.
如前文所述, 2018年MacMillan课题组[24]利用TBADT光催化与镍配合物Ni(dtbbpy)Br2的协同催化, 实现了芳基卤化物与饱和烷烃的交叉脱氢偶联反应(Scheme 12).随后, 王川等[34]采用类似的协同催化策略实现了醛C—H键与芳基卤化物(或α-溴代乙酸乙酯)的偶联反应(Scheme 19).值得注意的是, 此前König课题组[35]用Ni(dmbpy)Br2 (dmbpy=4, 4'-二甲基-2, 2'-联吡啶)为催化剂在395 nm光照条件下实现了醛与芳基卤化物的交叉偶联, 但该方法仅适用于芳香醛.与之相比, TBADT/Ni(dtbbpy)X2的协同催化体系的底物适用范围非常广, 无论是芳香醛还是脂肪醛都能与芳基溴代物(或α-溴代乙酸乙酯)发生芳基化(或烷基化)反应(19a~19f), 以良好至较优的收率转化为相应的产物酮.特别是对脂肪醛的反应, 产率几乎都达到了90%, 且表现出良好的官能团兼容性.
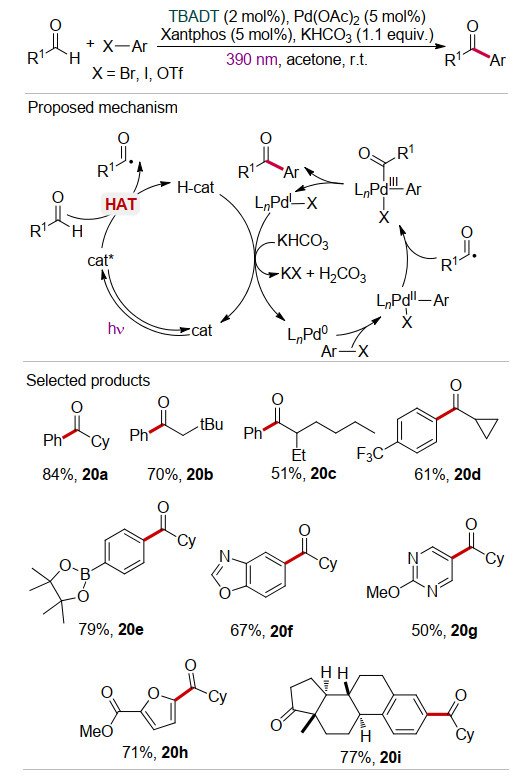
最近, 郑超等[36]采用TBADT/Pd(OAc)2协同催化的策略, 在光催化下实现了醛与芳基溴/碘化物的交叉偶联反应, 合成了一系列酮类产物(Scheme 20).该方法的底物适用性较好, 不仅适用于各种取代芳基卤代烃(20a~20e), 而且也适用于杂环芳香卤代烃(20f~20h), 一些天然产物骨架的卤代烃也能顺利地发生反应(20j).密度泛函理论(DFT)计算表明该反应可能经历“Pd0-PdⅡ- PdⅢ-PdⅠ-Pd0”的钯催化循环.类似地, 光激发的TBADT通过HAT作用将醛转化为相应的酰基自由基.另一方面, 芳基卤化物对Pd(0)催化剂氧化加成, 产生Pd(Ⅱ)中间体.随后酰基自由基插入Pd(Ⅱ)中间体, 产生Pd(Ⅲ)中间体并经过还原消除生成目标产物.在KHCO3作用下Pd(0)催化剂和光催化剂TBADT同时实现再生.
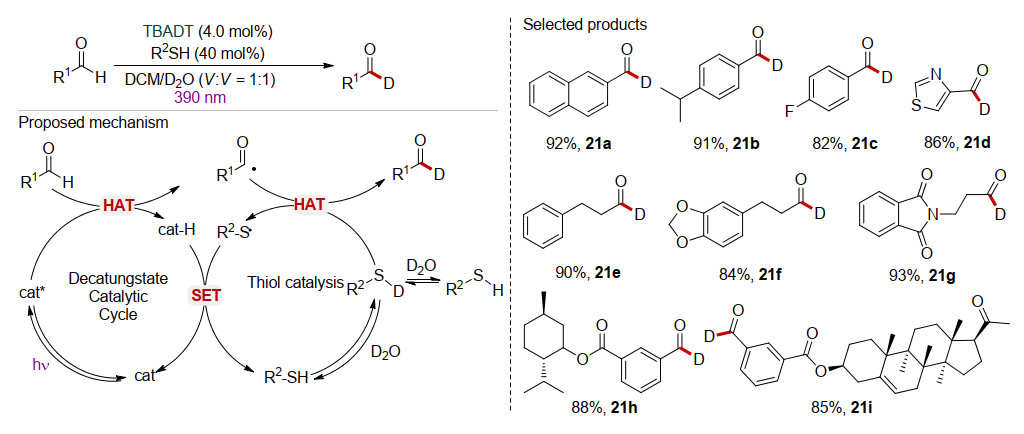
醛的C—H键选择性氘代对于标记和优化药物性质具有重要意义.最近, 汪清民课题组[37]利用TBADT和硫醇R2SH共催化实现了醛C—H键的直接氢同位素交换(hydrogen isotope exchange, HIE).如Scheme 21所示, 硫醇与D2O作用生成氘代硫酚R2SD.另一方面, 醛在TBADT催化下发生HAT作用生成相应的酰基自由基和cat-H.酰基自由基与氘代硫酚R2SD发生HAT过程, 生成产物氘代醛和硫自由基R2S•.更进一步, 硫自由基R2S•与cat-H发生HAT过程再生光催化剂TBADT和硫醇R2SH.该反应利用TBADT和硫酚作为HAT催化剂, 通过两步HAT过程, 实现了醛的C—H键的直接H-D交换生成氘代醛.该方法利用相对廉价的氘水作为氘源, 在温和条件下实现了醛的氘代, 不仅底物范围广, 且氘化的产率在76%以上.此外, 相关药物分子也能以较优的产率实现氘代(21h~21i).
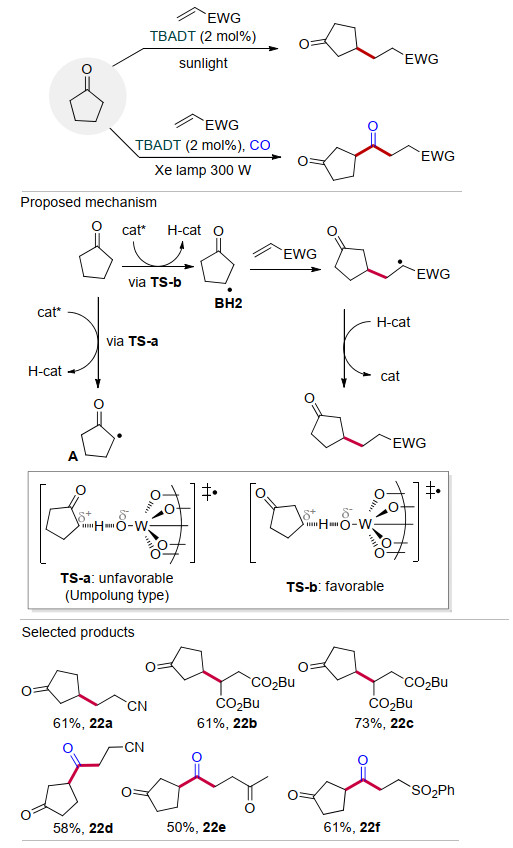
2014年, Fagnoni课题组[38]以TBADT为催化剂, 研究了环戊酮与缺电子烯烃的加成反应(Scheme 22), 以中等至较好的产率合成了系列β-烷基化环戊酮, 该方法有较广的底物适用范围(22a~22c).当反应在CO (20.2 MPa)氛围下进行时, 得到环戊酮、CO和缺电子烯烃的三组分反应(22d~22f), 以较好的产率合成了系列β-酰基环戊酮.虽然环戊酮中α位的C—H键比β位的C—H键更弱, 但是反应并没有发生在α位.主要由于该反应中催化剂TBADT与C—H键的HAT作用经历极性的双分子均裂取代(bimolecular homolytic substitution) SH2过渡态.在如Scheme 22所示的反应路径中, 激发态催化剂分别攫取α位或β位氢原子的时候, 经历过渡态TS-a和TS-b, 相应的碳原子上会产生部分正电荷.其中, α位C—H键参与反应形成的过渡态TS-a中α位产生不稳定的缺电子碳中心(Umpolung type), 而β位C—H键参与反应形成的过渡态TS-b具有较好的稳定性.因此, 该反应主要发生在环戊酮的β位.
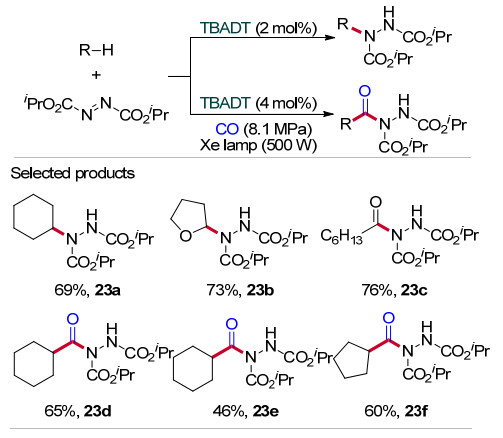
2013年, Ryu和Fagnoni等[39]将TBADT光催化的策略用于构成C—N键, 在500 W氙灯照射下实现了饱和烷烃、环状醚、醛中惰性C—H键与偶氮二甲酸二异丙酯中N=N双键的加成反应(Scheme 23).例如, 以环己烷作为自由基前体, 与偶氮二甲酸二异丙酯反应2 h能以69%的产率得到加成产物(23a); 同样的条件下, 用四氢呋喃或庚醛代替环己烷, 得到73%的烷基化产物(23b)和76%的酰基化产物(23c).此外, 该反应在CO (8.1 MPa)氛围下发生环烷烃、CO与偶氮二甲酸二异丙酯的三组分反应, 得到较高产率的酰肼化合物(23d~23f).
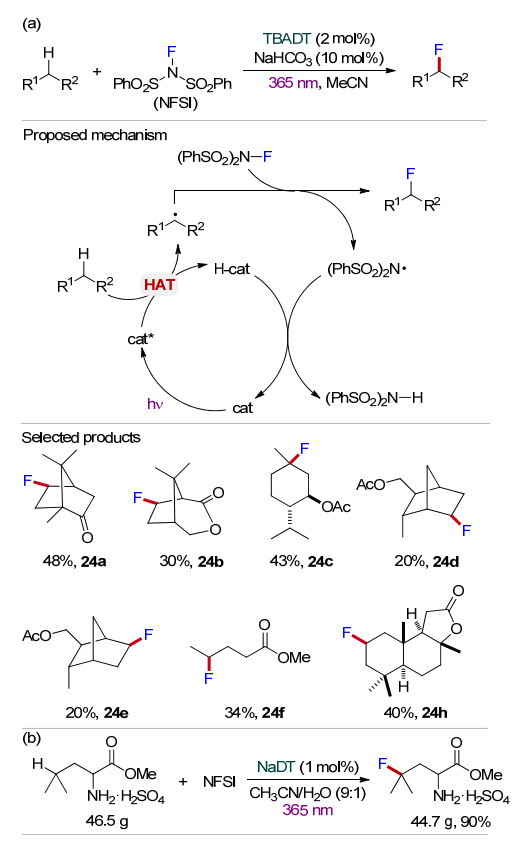
发展温和条件下简单高效的氟化反应, 将氟原子引入有机分子一直是有机化学和药物化学的重要研究课题. 2014年, Britton课题组[40]利用TBADT为光催化剂在365 nm紫外光照射下实现了饱和烷烃中未活化C—H键的氟化反应(Scheme 24a).该方法使用N-氟代双苯磺酰胺((PhSO2)2NF, NFSI)为氟化试剂, NaHCO3为添加剂, 在温和条件下实现了多种有机分子(包括复杂天然产物)C(sp3)—H键的氟化, 并且保持反应物的立体构型.可能催化机理如Scheme 24所示, C—H键在光激发TBADT的HAT作用下产生相应的烷基自由基, 与NFSI产生相应的氟化产物, 同时生成中间体(PhSO2)2N•.该中间体(PhSO2)2N•与H-cat发生氢原子转移再生催化剂TBADT.该方法的底物适用范围广, 官能团容忍性较好, 以中等收率得到相应的氟化产物(24a~24g).随后, 该课题组[41]又采用十聚钨酸钠(NaDT)为光催化剂, 实现了催化亮氨酸甲酯的γ位C—H键的直接氟化反应. γ-氟代亮氨酸甲酯是合成odanacatib的重要中间体, 传统的亮氨酸甲酯氟化反应的选择性较差, 通常得到的混合物较难分离, 而NaDT光催化的氟化反应可以高选择性地生成γ-氟代亮氨酸甲酯, 且放大反应产率高达90% (Scheme 24b).
由于十聚钨酸盐独特的光化学性能, 在光激发下生成高活性激发态的其氧化还原电位为+2.44 V (vs. SCE).当反应物中C—H键的氧化电位大于+2.44 V时, 十聚钨酸盐的激发态可直接通过氢原子转移(HAT)作用, 攫取C—H键中的氢原子从而产生相应的自由基, 实现进一步的官能团化.从上述研究进展不难看出, 十聚钨酸盐作为一种“HAT型光催化剂”, 在C—H键直接官能团化反应中具有巨大的潜力和应用价值.目前这方面的主要研究工作集中在C—C键的构建, 构筑其他C—X键方面的研究还比较少, 相信在不久的将来该领域会进一步引起合成化学工作者的兴趣, 进一步发展更多高效的光催化合成方法.虽然十聚钨酸盐在光催化有机合成方面有巨大潜力, 但是十聚钨酸盐的光催化反应还存在一些局限, 主要包括: (1)十聚钨酸盐的结构特征决定了其HOMO和LUMO之间的能级差较大, 多数时候需要用能量较高的紫外光才能将其激发, 因此多数反应需要在紫外光源下进行; (2)多数情况下需要使用大大过量的C—H键反应前体来提升反应效率; (3)十聚钨酸盐的催化剂类型相对较少.针对上述局限, 还需要进一步开发功能化的十聚钨酸盐类催化剂, 使其在可见光照射下具有较高的催化效率, 高效实现温和条件下的C—H键官能团化.
(a) Su, F.; Guo, Y. Green Chem. 2014, 16, 2934.
(b) Omwoma, S.; Gore, C. T.; Ji, Y.; Hu, C.; Song, Y.-F. Coord. Chem. Rev. 2015, 286, 17.
(c) Yu, B.; Zou, B.; Hu, C.-W. J. CO2 Util. 2018, 26, 314.
(d) Wang, S.-S.; Yang, G.-Y. Chem. Rev. 2015, 115, 4893.
(e) Yang, G.-P.; Wu, X.; Yu, B.; Hu, C.-W. ACS Sustainable Chem. Eng. 2019, 7, 3727.
(f) Yang, G.-P.; Zhang, N.; Ma, N.-N.; Yu, B.; Hu, C.-W. Adv. Synth. Catal. 2017, 359, 926.
(g) Ma, P.; Hu, F.; Wang, J.; Niu, J. Coord. Chem. Rev. 2019, 378, 281.
(h) Du, D.-Y.; Qin, J.-S.; Li, S.-L.; Su, Z.-M.; Lan, Y.-Q. Chem. Soc. Rev. 2014, 43, 4615.
(i) Chen, L.; Chen, W.-L.; Wang, X.-L.; Li, Y.-G.; Su, Z.-M.; Wang, E.-B. Chem. Soc. Rev. 2019, 48, 260.
(j) Zhang, J.; Huang, Y.; Li, G.; Wei, Y. Coord. Chem. Rev. 2019, 378, 395.
(k) Zhou, Y.; Guo, Z.; Hou, W.; Wang, Q.; Wang, J. Catal. Sci. Technol. 2015, 5, 4324.
(l) Huang, B.; Yang, D.-H.; Han, B.-H. J. Mater. Chem. A 2020, 8, 4593.
Yamase, T.; Sasaki, R.; Ikawa, T. J. Chem. Soc., Dalton Trans. 1981, 628. https://pubs.rsc.org/en/content/articlelanding/1981/dt/dt9810000628
Papaconstantinou, E.; Dimotikali, D.; Politou, A. Inorg. Chim. Acta 1980, 43, 155. doi: 10.1016/S0020-1693(00)90521-8
Hill, C. L.; Bouchard, D. A. J. Am. Chem. Soc. 1985, 107, 5148. doi: 10.1021/ja00304a019
(a) Wang, M.-Y.; Ma, R.; He, L.-N. Sci. China Chem. 2016, 59, 507.
(b) Lin, J.; Han, Q.; Ding, Y. Chem. Rec. 2018, 18, 1531.
Duncan, D. C.; Netzel, T. L.; Hill, C. L. Inorg. Chem. 1995, 34, 4640. doi: 10.1021/ic00122a021
Waele, V. D.; Poizat, O.; Fagnoni, M.; Bagno, A.; Ravelli, D. ACS Catal. 2016, 6, 7174. doi: 10.1021/acscatal.6b01984
(a) Hartwig, J. F.; Larsen, M. A. ACS Central Sci. 2016, 2, 281.
(b) Fabry, D. C.; Zoller, J.; Rueping, M. Org. Chem. Front. 2019, 6, 2635.
(a) Lü, Q.; Yu, B. J. Liaocheng Univ. (Nat. Sci. Ed.) 2019, 32, 28(in Chinese).
(吕琪妍, 於兵, 聊城大学学报(自然科学版), 2019, 32, 28.)
(b) Jiang, Y.-Q.; Li, J.; Feng, Z.-W.; Xu, G.-Q.; Shi, X.; Ding, Q.-J.; Li, W.; Ma, C.-H.; Yu, B. Adv. Synth. Catal. 2020, 362, 2609.
(c) Mao, P.; Zhu, J.; Yuan, J.; Yang, L.; Xiao, Y.; Zhang, C. Chin. J. Org. Chem. 2019, 39, 1529.
(d) Gao, F.; Lü, Q.; Yu, B. J. Liaocheng Univ. (Nat. Sci. Ed.) 2020, 33, 66(in Chinese).
(高凡, 吕琪妍, 於兵, 聊城大学学报(自然科学版), 2020, 33, 66.)
(a) Ravelli, D.; Fagnoni, M.; Fukuyama, T.; Nishikawa, T.; Ryu, I. ACS Catal. 2018, 8, 701.
(b) Ravelli, D.; Protti, S.; Fagnoni, M. Acc. Chem. Res. 2016, 49, 2232.
(c) Tzirakis, M. D.; Lykakis, I. N.; Orfanopoulos, M. Chem. Soc. Rev. 2009, 38, 2609.
(d) Suzuki, K.; Mizuno, N.; Yamaguchi, K. ACS Catal. 2018, 8, 10809.
(e) Capaldo, L.; Ravelli, D. Eur. J. Org. Chem. 2017, 2017, 2056.
(a) Jaynes, B. S.; Hill, C. L. J. Am. Chem. Soc. 1993, 115, 12212.
(b) Hill, C. L. Synlett 1995, 127.
(c) Jaynes, B. S.; Hill, C. L. J. Am. Chem. Soc. 1995, 117, 4704.
Zheng, Z.; L. Hill, C. Chem. Commun. 1998, 2467.
(a) Dondi, D.; Fagnoni, M.; Albini, A. Chem. Eur. J. 2006, 12, 4153.
(b) Dondi, D.; Fagnoni, M.; Molinari, A.; Maldotti, A.; Albini, A. Chem. Eur. J. 2004, 10, 142.
(c) Protti, S.; Ravelli, D.; Fagnoni, M.; Albini, A. Chem. Commun. 2009, 7351.
Ravelli, D.; Albini, A.; Fagnoni, M. Chem. Eur. J. 2011, 17, 572. doi: 10.1002/chem.201002546
Ryu, I.; Tani, A.; Fukuyama, T.; Ravelli, D.; Fagnoni, M.; Albini, A. Angew. Chem. Int. Ed. 2011, 50, 1869. doi: 10.1002/anie.201004854
Ravelli, D.; Zoccolillo, M.; Mella, M.; Fagnoni, M. Adv. Synth. Catal. 2014, 356, 2781. doi: 10.1002/adsc.201400027
Laudadio, G.; Deng, Y.; van der Wal, K.; Ravelli, D.; Nuño, M.; Fagnoni, M.; Guthrie, D.; Sun, Y.; Noël, T. Science 2020, 369, 92. doi: 10.1126/science.abb4688
Supranovich, V. I.; Levin, V. V.; Dilman, A. D. Org. Lett. 2019, 21, 4271. doi: 10.1021/acs.orglett.9b01450
Esposti, S.; Dondi, D.; Fagnoni, M.; Albini, A. Angew. Chem., Int. Ed. 2007, 46, 2531. doi: 10.1002/anie.200604820
(a) Kuang, Y.; Wang, K.; Shi, X.; Huang, X.; Meggers, E.; Wu, J. Angew. Chem., Int. Ed. 2019, 58, 16859.
(b) Li, F.; Tian, D.; Fan, Y.; Lee, R.; Lu, G.; Yin, Y.; Qiao, B.; Zhao, X.; Xiao, Z.; Jiang, Z. Nat. Commun. 2019, 10, 1774.
Murphy, J. J.; Bastida, D.; Paria, S.; Fagnoni, M.; Melchiorre, P. Nature 2016, 532, 218. doi: 10.1038/nature17438
Dai, Z.-Y.; Nong, Z.-S.; Wang, P.-S. ACS Catal. 2020, 10, 4786. doi: 10.1021/acscatal.0c00610
Quattrini, M. C.; Fujii, S.; Yamada, K.; Fukuyama, T.; Ravelli, D.; Fagnoni, M.; Ryu, I. Chem. Commun. 2017, 53, 2335. doi: 10.1039/C6CC09725A
Perry, I. B.; Brewer, T. F.; Sarver, P. J.; Schultz, D. M.; DiRocco, D. A.; MacMillan, D. W. C. Nature 2018, 560, 70. doi: 10.1038/s41586-018-0366-x
(a) Kong, Y.; Xu, X.; Ye, F.; Weng, J. Chin. J. Org. Chem. 2019, 39, 3065(in Chinese).
(孔瑶蕾; 徐雯秀; 叶飞霞; 翁建全, 有机化学, 2019, 39, 3065.)
(b) Xiong, L.; Hu, H.; Wei, C.-W.; Yu, B. Eur. J. Org. Chem. 2020, 1588.
(a) Chen, F.; Chen, H.; Wu, Q.; Luo, S. Chin. J. Org. Chem. 2020, 40, 339(in Chinese).
(陈锋, 陈浩, 吴庆安, 罗书平, 有机化学, 2020, 40, 339.)
(b) Wang, H.; Gao, X.; Lv, Z.; Abdelilah, T.; Lei, A. Chem. Rev. 2019, 119, 6769.
(c) Zhong, J.; Meng, Q.; Tong, Z.; Wu, L. Acta Chem. Sinica 2017, 75, 34(in Chinese).
(钟建基; 孟庆元; 陈彬; 佟振合; 吴骊珠, 化学学报, 2017, 75, 34.)
(d) Chen, B.; Wu, L.-Z.; Tung, C.-H. Acc. Chem. Res. 2018, 51, 2512.
Cao, H.; Kuang, Y.; Shi, X.; Wong, K. L.; Tan, B. B.; Kwan, J. M. C.; Liu, X.; Wu, J. Nat. Commun. 2020, 11, 1956. doi: 10.1038/s41467-020-15878-6
Yahata, K.; Sakurai, S.; Hori, S.; Yoshioka, S.; Kaneko, Y.; Hasegawa, K.; Akai, S. Org. Lett. 2020, 22, 1199. doi: 10.1021/acs.orglett.0c00096
(a) Ji, X.; Shi, G.; Zhang, Y. Chin. J. Org. Chem. 2019, 39, 929(in Chinese).
(季小明, 史广法, 张扬会, 有机化学, 2019, 39, 929.)
(b) Liu, T.; Qu, C.; Xie, J.; Zhu, C. Chin. J. Org. Chem. 2019, 39, 1613(in Chinese).
(刘涛, 屈川华, 谢劲, 朱成建, 有机化学, 2019, 39, 1613.)
(c) Zeng, F.-L.; Sun, K.; Chen, X.-L.; Yuan, X.-Y.; He, S.-Q.; Liu, Y.; Peng, Y.-Y.; Qu, L.-B.; Lv, Q.-Y.; Yu, B. Adv. Synth. Catal. 2019, 361, 5176.
(d) Wang, J.; Sun, K.; Chen, X.; Chen, T.; Liu, Y.; Qu, L.; Zhao, Y.; Yu, B. Org. Lett. 2019, 21, 1863.
Sarver, P. J.; Bacauanu, V.; Schultz, D. M.; DiRocco, D. A.; Lam, Y.-H.; Sherer, E. C.; MacMillan, D. W. C. Nat. Chem. 2020, 12, 459. doi: 10.1038/s41557-020-0436-1
(a) Chatgilialoglu, C.; Crich, D.; Komatsu, M.; Ryu, I. Chem. Rev. 1999, 99, 1991.
(b) Bennasar, M. L.; Roca, T.; Griera, R.; Bosch, J. Org. Lett. 2001, 3, 1697.
(c) Bath, S.; Laso, N. M.; Lopez-Ruiz, H.; Quiclet-Sire, B.; Zard, S. Z. Chem. Commun. 2003, 204.
(d) Benati, L.; Calestani, G.; Leardini, R.; Minozzi, M.; Nanni, D.; Spagnolo, P.; Strazzari, S. Org. Lett. 2003, 5, 1313.
Fan, P.; Zhang, C.; Lan, Y.; Lin, Z.; Zhang, L.; Wang, C. Chem. Commun. 2019, 55, 12691. doi: 10.1039/C9CC07285C
Fan, P.; Lan, Y.; Zhang, C.; Wang, C. J. Am. Chem. Soc. 2020, 142, 2180. doi: 10.1021/jacs.9b12554
Fan, P.; Zhang, C.; Zhang, L.; Wang, C. Org. Lett. 2020, 22, 3875. doi: 10.1021/acs.orglett.0c01121
Schirmer, T. E.; Wimmer, A.; Weinzierl, F. W. C.; König, B. Chem. Commun. 2019, 55, 10796. doi: 10.1039/C9CC04726C
Wang, L.; Wang, T.; Cheng, G.-J.; Li, X.; Wei, J.-J.; Guo, B.; Zheng, C.; Chen, G.; Ran, C.; Zheng, C. ACS Catal. 2020, 10, 7543. doi: 10.1021/acscatal.0c02105
Dong, J.; Wang, X.; Wang, Z.; Song, H.; Liu, Y.; Wang, Q. Chem. Sci. 2020, 11, 1026. doi: 10.1039/C9SC05132E
Okada, M.; Fukuyama, T.; Yamada, K.; Ryu, I.; Ravelli, D.; Fagnoni, M. Chem. Sci. 2014, 5, 2893. doi: 10.1039/C4SC01072H
Ryu, I.; Tani, A.; Fukuyama, T.; Ravelli, D.; Montanaro, S.; Fagnoni, M. Org. Lett. 2013, 15, 2554. doi: 10.1021/ol401061v
Halperin, S. D.; Fan, H.; Chang, S.; Martin, R. E.; Britton, R. Angew. Chem., Int. Ed. 2014, 53, 4690. doi: 10.1002/anie.201400420
Halperin, S. D.; Kwon, D.; Holmes, M.; Regalado, E. L.; Campeau, L.-C.; DiRocco, D. A.; Britton, R. Org. Lett. 2015, 17, 5200. doi: 10.1021/acs.orglett.5b02532

图式 2 [W10O32]4-在光催化反应中的作用机理
Scheme 2 Mechanism of decatungstate in photocatalyzed reactions

图式 3 TBADT催化饱和烷烃C—H键官能团化反应
Scheme 3 TBADT-catalyzed C—H functionalization of saturated alkanes

图式 4 TBADT催化缺电子烯烃的反应(EWG=Electron withdrawing group)
Scheme 4 TBADT-catalyzed reaction of electron-deficient olefins

图式 6 TBADT催化气态烷烃与缺电子烯烃反应
Scheme 6 TBADT-catalyzed the reaction of gaseous alkanes with electron-deficient olefins

图式 9 TBADT与手性磷酸的协同催化
Scheme 9 Synergistic catalysis of TBADT and chiral spiro phosphoric acid

图式 10 TBADT催化芳香氮杂环化合物C—H官能团化
Scheme 10 TBADT-catalyzed C—H functionalization of N-heterocycles

图式 11 TBADT催化喹啉4位的烷基化反应机理
Scheme 11 Mechanism of TBADT-catalyzed 4-alkylation of quinoline

图式 16 TBADT催化醛与缺电子烯烃反应
Scheme 16 TBADT-catalyzed the reaction of aldehyde and electron-deficient olefins

图式 18 TBADT与手性Ni-PHOX的协同催化
Scheme 18 Synergistic catalysis of TBADT and chiral Ni-PHOX complex

图式 22 TBADT催化环戊酮β位C—H官能团化
Scheme 22 TBADT-catalyzed functionalization of β-C—H of cyclopentanone

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:












