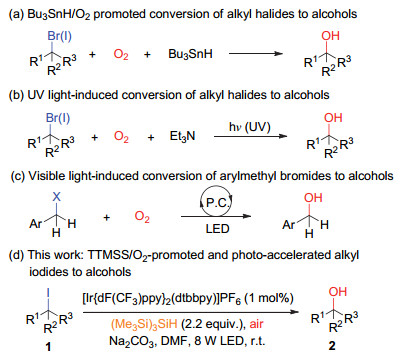
Scheme 1.
Strategies for the conversion of alkyl halides to alcohols via carbon radical processes

The conversion of alkyl halides to alcohols is an important transformation in organic synthesis.[1] The conversion can be achieved by direct hydroxylation of alkyl halides with hydroxyl anion or nucleophilic substitution with oxygenated reagents such as acetate or formate via two-step processes.[1-2] However, these traditional approaches are mainly applied to primary alkyl halides and often suffer from β-elimination side reactions. The C—X bonds undergo heterolytic cleavage during the replacement of the halogen atoms. On the other hand, homolysis of the C—X bonds to generate carbon radicals followed by oxygenated with appropriate O-reagents may provide an alternative method for the transformation from alkyl halides to alcohols.[3-5] For example, Nakamura et al.[3] found that Bu3SnH/O2 could promote the conversion of alkyl bromides and iodides to alcohols (Scheme 1, a). Reductive cleavage of the carbon-halogen bond by the tin hydride generated carbon radical intermediates, which were trapped with O2 and then reduced by Bu3SnH to form alcohol products. Under UV light irradiation conditions, the transformation can be realized without the toxic tin hydride reagent (Scheme 1, b).[4] In the presence of a photoredox catalyst, [6] visible light irradiation also promoted the transformation from benzyl halides to alcohols (Scheme 1, c).[5] Recently, we found that alkyl iodides can be smoothly converted to the corresponding alcohols in moderate to good yields in the presence of (SiMe3)3SiH[7] and air under photoredox catalyzed visible light irradiation conditions (Scheme 1, d). The reaction worked well for primary, secondary as well as tertiary iodides. Herein, we report the preliminary results on this project.
Initial investigation was carried out with alkyl iodide 1a as the standard substrate due to its high fluorescence intensity for thin-layer chromatography (TLC) analysis (Table 1). In the presence of 1 mol% of [Ir{dF(CF3)ppy}2-(dtbbpy)]PF6 as the photoredox catalyst, [8, 9] compound 1a was treated with tris(trimethylsilyl)silane (TTMSS) in N, N-dimethylformamide (DMF) under air atmosphere and blue LED light irradiation to produce alcohol 2a in 94% yield with a trace amount of reduction by-product 3a (Table, Entry 1). Change of the solvent from DMF to dimethylacetamide (DMA) or dimethyl sulfoxide (DMSO) resulted in decreased yields of 2a (Table 1, Entries 2 and 3 vs 1). Silyl reagent TTMSS and O2 both are crucial for the reaction. Without TTMSS or O2, no or a trace amount of alcohol product 2a was detected under otherwise the same conditions (Table 1, Entries 4 and 5). The absence of photoredox catalyst or/and LED light led to much lower yields of 2a (Table 1, Entries 6~8), indicating the photoredox catalyzed irradiation can efficiently accelerate the transformation. In the absence of Na2CO3, the reaction became very messy, leading to an obvious decrease of the yield of 2a (Table 1, Entry 9).
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | Conditions | Yieldb/% | Recovered 1ab/% | |
| 2a | 3a | |||
| 1 | As shown | 94 | Trace | 0 |
| 2 | DMA instead of DMF | 86 | Trace | 7 |
| 3 | DMSO instead of DMF | 24 | 6 | 31 |
| 4 | No TTMSS | 4 | 0 | 95 |
| 5 | Under N2 atmosphere | 0 | 99 | 0 |
| 6 | No photoredox catalyst | 33 | 2 | 63 |
| 7 | No blue LEDs | 39 | 0 | 57 |
| 8 | No photocatalyst and no blue LED | 32 | 0 | 65 |
| 9 | No Na2CO3 | 32 | 6 | 15 |
| a The reaction was carried out with 1a (0.10 mmol), TTMSS (0.22 mmol), Na2CO3 (0.10 mmol), [Ir{dF(CF3)ppy}2(dtbbpy)]PF6 (0.0010 mmol) in DMF (1.0 mL) under 8 W blue LED light irradiation at room temperature for 24 h unless otherwise stated. b The NMR yields of 2a and 3a and the recovery percentage of 1a were determined by 1H NMR analysis of the crude reaction mixture. | ||||
Under the optimal conditions, substrate scope was investigated for the transformation from alkyl halides to alcohols (Table 2). Various primary (for 2a~2k), secondary (for 2l~2p) and tertiary (for 2q and 2r) alkyl iodides were all effective substrates for the reaction, to give the corresponding alcohols in 38%~99% yields. Heteroaromatic rings such as thiophene (for 2j) and heteroatomic groups such as ether (for 2h) and piperine cycles (for 2o and 2p) were all well tolerated. Cyclic iodides could be smoothly converted to the alcohol products (for 2m, 2s, and 2t) but with low trans/cis selectivity, likely due to the nature of the radical process. Under the reaction conditions, only a trace amount of benzylic alcohol was obtained from benzylic iodide. Alkyl bromides such as (3-bromobutyl)benzene and alkyl chlorides such as (3-chloropropyl)benzene are both ineffective for the reaction.
下载:
导出CSV

|
Introducing a radical-trapping reagent, 2, 2, 6, 6-tetra-methylpiperidine 1-oxyl free radical (2, 2, 6, 6-Tetramethyl-piperidinooxy, TEMPO) (4), to the reaction system resulted in the formation of a trace amount of alcohol product 2a along with compound 5 in 14% yield (Scheme 2), demonstrating that a carbon radical related to alkyl iodide 1a was involved in the reaction.
In summary, we have developed an interesting method for the transformation from alkyl iodides to alcohols with a wide substrate scope. Various types of alkyl iodides were efficiently converted to alcohols in 38%~99% yields by TTMSS/O2 under mild conditions. The visible light photoredox catalysis condition can accelerate the transformation greatly.
All commercially available reagents were used without further purification unless otherwise stated. Column chromatography was performed on silica gel (200~300 mesh). 1H NMR spectra were recorded on a 400 MHz NMR spectrometer and 13C NMR spectra were recorded on a 100 MHz NMR spectrometer. 1H NMR and 13C NMR shifts were referenced to CDCl3 (δH 7.26, δC 77.16).
The photoredox catalyst [Ir{dF(CF3)ppy}2(dtbbpy)]PF6 was prepared according to literature mothed[9a].
The alkyl iodides 1a~1g, 1i~1p and 1s~1t were synthesized by the reaction of the corresponding alcohols with I2, PPh3 and imidazole according to the literature procedure.[10a] 1q~1r were prepared from the corresponding alcohols via the reaction with KI and methanesulfonic acid in acetonitrile according to the literature procedure.[10b] 1h was prepared from methylparaben via the reaction with diiodopropane according to the literature procedure.[10c]
To a 10 mL vial equipped with a magnetic stirrer were added [Ir{dF(CF3)ppy}2(dtbbpy)]PF6 (2.5 mg, 0.0020 mmol), Na2CO3 (21.2 mg, 0.20 mmol), 4-(3-iodopropyl)- 1, 1'-biphenyl (1a) (64 mg, 0.20 mmol) and DMF (2.0 mL). To the mixture was added tris(trimethylsilyl)silane (TTMSS) (110 mg, 0.44 mmol). The reaction vial was opened to the air, irradiated with 8W blue LEDs from 13 cm away, and cooled with an electronic fan. After stirring at room temperature for 24 h, ethyl acetate (20 mL) and brine (20 mL) were added to the mixture. The organic layer was separated, washed with brine (20 mL×2), dried over Na2SO4, filtered, concentrated, and purified by chromatography on silica gel [V(petroleum ether):V(ethyl acetate)=10:1] to give 3-([1, 1'-biphenyl]-4-yl)propan- 1-ol (2a)[11a] (39.8 mg, 94%) as a white solid. m.p. 72~74 ℃ (Lit.[11b] 60~63 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.59 (d, J=7.2 Hz, 2H), 7.53 (d, J=8.0 Hz, 2H), 7.44 (t, J=7.2 Hz, 2H), 7.34 (t, J=7.2 Hz, 1H), 7.29 (d, J=8.0 Hz, 2H), 3.72 (t, J=6.4 Hz, 2H), 2.77 (t, J=7.6 Hz, 2H), 1.99~1.90 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 141.2, 141.1, 139.0, 129.0, 128.8, 127.3, 127.2, 127.1, 62.4, 34.3, 31.8.
Compounds 2b~2t were prepared according to the synthetic method of compound 2a.
2-Phenylethan-1-ol (2b):[11c] Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.36~7.30 (m, 2H), 7.27~7.21 (m, 3H), 3.87 (t, J=6.8 Hz, 2H), 2.88 (t, J=6.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 138.6, 129.1, 128.7, 126.6, 63.8, 39.3.
3-Phenylpropan-1-ol (2c):[11c] Colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.32~7.26 (m, 2H), 7.23~7.16 (m, 3H), 3.68 (t, J=6.4 Hz, 2H), 2.72 (t, J=7.6 Hz, 2H), 1.95~1.85 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 141.9, 128.55, 128.53, 126.0, 62.4, 34.3, 32.2.
5-Phenylbutan-1-ol (2d):[11d] Colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.31~7.26 (m, 2H), 7.21~7.16 (m, 3H), 3.66 (t, J=6.4 Hz, 2H), 2.65 (t, J=7.2 Hz, 2H), 1.76~1.66 (m, 2H), 1.66~1.57 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 142.5, 128.5, 128.4, 125.9, 63.0, 35.8, 32.5, 27.7.
5-Phenylpentan-1-ol (2e):[11e] Colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.31~7.25 (m, 2H), 7.21~7.16 (m, 3H), 3.64 (t, J=6.8 Hz, 2H), 2.63 (t, J=7.6 Hz, 2H), 1.71~1.56 (m, 4H), 1.47~1.36 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 142.7, 128.5, 128.4, 125.8, 63.0, 36.0, 32.7, 31.4, 25.5.
2-Phenylpropan-1-ol (2f):[11f] Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.38~7.31 (m, 2H), 7.27~7.22 (m, 3H), 3.71 (d, J=6.8 Hz, 2H), 3.01~2.91 (m, 1H), 1.28 (d, J=6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 143.8, 128.8, 127.6, 126.8, 68.9, 42.6, 17.7.
Methyl 4-(3-hydroxypropyl)benzoate (2g):[11g] Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.95 (d, J=8.0 Hz, 2H), 7.27 (d, J=8.0 Hz, 2H), 3.90 (s, 3H), 3.67 (t, J=6.4 Hz, 2H), 2.77 (t, J=8.0 Hz, 2H), 1.95~1.85 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 167.3, 147.6, 129.8, 128.6, 62.0, 52.1, 33.9, 32.2.
Methyl 4-(3-hydroxypropoxy)benzoate (2h):[11h] White solid, m.p. 52~53 ℃ (Lit.[11i] 54 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.97 (d, J=9.2 Hz, 2H), 6.91 (d, J=9.2 Hz, 2H), 4.16 (t, J=6.0 Hz, 2H), 3.87 (s, 3H), 3.86 (t, J=6.0 Hz, 2H), 2.10~2.02 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 167.0, 162.7, 131.7, 122.7, 114.2, 65.6, 59.9, 52.0, 32.0.
2-(Naphthalen-1-yl)ethan-1-ol (2i):[11j] White solid, m.p. 59~61 ℃ (Lit.[11j] 63~65 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.06 (d, J=8.0 Hz, 1H), 7.87 (d, J=8.0 Hz, 1H), 7.77 (d, J=8.0 Hz, 1H), 7.57~7.46 (m, 2H), 7.46~7.34 (m, 2H), 4.00 (t, J=6.8 Hz, 2H), 3.36 (t, J=6.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 134.5, 134.1, 132.2, 129.0, 127.5, 127.3, 126.2, 125.8, 125.6, 123.8, 63.2, 36.4.
2-(Thiophen-2-yl)ethan-1-ol (2j):[11k] Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.18 (d, J=5.2 Hz, 1H), 6.96 (dd, J=5.2, 3.2 Hz, 1H), 6.88 (d, J=3.2 Hz, 1H), 3.86 (t, J=6.4 Hz, 2H), 3.09 (t, J=6.4 Hz, 2H), 1.68 (brs, 1H); 13C NMR (100 MHz, CDCl3) δ: 140.9, 127.1, 125.7, 124.1, 63.6, 33.4.
6-((Tert-butyldiphenylsilyl)oxy)hexan-1-ol (2k):[11l] Colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.69~7.64 (m, 4H), 7.45~7.35 (m, 6H), 3.69~3.59 (m, 4H), 1.63~1.50 (m, 4H), 1.44~1.28 (m, 4H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 135.7, 134.3, 129.6, 127.7, 64.0, 63.1, 32.9, 32. 6, 27.0, 25.7, 25.6, 19.4.
4-Phenylbutan-2-ol (2l):[11m] Colorless oil. 1H NMR (400 MHz, CDCl3) δ: 7.32~7.26 (m, 2H), 7.23~7.17 (m, 3H), 3.89~3.78 (m, 1H), 2.82~2.62 (m, 2H), 1.87~1.71 (m, 2H), 1.24 (d, J=6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 142.2, 128.5, 125.9, 67.6, 41.0, 32.3, 23.8.
4-Phenylcyclohexan-1-ol (2m):[11n- 11q] cis-2m: White solid, m.p. 97~100 ℃ (Lit.[11r] 76~77 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.33~7.27 (m, 2H), 7.27~7.22 (m, 2H), 7.22~7.15 (m, 1H), 4.16~4.10 (m, 1H), 2.60~2.47 (m, 1H), 1.97~1.82 (m, 4H), 1.74~1.62 (m, 4H); 13C NMR (150 MHz, CDCl3) δ: 147.5, 128.5, 127.0, 126.1, 65.8, 44.0, 33.2, 27.9. trans-2m: White solid, m.p. 109~113 ℃ (Lit.[11r] 118~118.5 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.33~7.27 (m, 2H), 7.24~7.17 (m, 3H), 3.75~3.65 (m, 1H), 2.55~2.45 (m, 1H), 2.17~1.88 (m, 4H), 1.65~1.38 (m, 5H); 13C NMR (150 MHz, CDCl3) δ: 146.7, 128.5, 126.9, 126.2, 70.8, 43.6, 36.1, 32.6.
2, 3-Dihydro-1H-inden-2-ol (2n):[11s] White solid, m.p. 62~63 ℃ (Lit.[11t] 67~69 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.28~7.22 (m, 2H), 7.21~7.16 (m, 2H), 4.73~4.67 (m, 1H), 3.22 (dd, J=16.4, 6.0 Hz, 2H), 2.92 (dd, J=16.0, 3.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 140.9, 126.8, 125.1, 73.4, 42.8.
1-Tosylpiperidin-4-ol (2o):[11p] Yellow solid, m.p. 126~128 ℃ (Lit.[11u] 130~132 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.64 (d, J=8.0 Hz, 2H), 7.32 (d, J=8.0 Hz, 2H), 3.80~3.70 (m, 1H), 3.35~3.25 (m, 2H), 2.88~2.78 (m, 2H), 2.43 (s, 3H), 1.98~1.88 (m, 2H), 1.70~1.60 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 143.7, 133.3, 129.8, 127.7, 66.0, 43.3, 33.3, 21.6.
(4-Hydroxypiperidin-1-yl)(phenyl)methanone (2p):[11p] White solid, m.p. 83~86 ℃ (Lit.[11v] 82~84 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.43~7.35 (m, 5H), 4.35~4.08 (m, 1H), 4.00~3.92 (m, 1H), 3.80~3.57 (m, 1H), 3.50~3.28 (m, 1H), 3.28~3.08 (m, 1H), 2.06~1.90 (m, 1H), 1.90~1.73 (m, 2H), 1.70~1.40 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 170.5, 136.1, 129.7, 128.6, 126.8, 67.0, 45.0, 39.5, 34.6, 33.9.
2-Methyl-4-phenylbutan-2-ol (2q):[11w] Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.32~7.25 (m, 2H), 7.23~7.15 (m, 3H), 2.75~2.67 (m, 2H), 1.84~1.75 (m, 2H), 1.30 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 142.7, 128.5, 128.4, 125.9, 71.1, 45.9, 30.9, 29.5.
Adamantan-1-ol (2r):[11s] White solid, m.p. 210~211 ℃ (Lit.[11x] 220 ℃ sublimes); 1H NMR (400 MHz, CDCl3) δ: 2.18~2.09 (m, 3H), 1.71 (d, J=2.8 Hz, 6H), 1.67~1.55 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 68.3, 45.4, 36.2, 30.8.
Compound 2s: White solid. epicholesterol-2s:[11y] m.p. 119~120 ℃ (Lit.[11y] 135~136 ℃); 1H NMR (400 MHz, CDCl3) δ: 5.43~5.38 (m, 1H), 4.04~3.97 (m, 1H), 2.62~2.52 (m, 1H), 2.10~0.95 (m, 30H), 0.91 (d, J=6.4 Hz, 3H), 0.86 (d, J=6.4 Hz, 6H), 0.68 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 138.7, 124.2, 67.3, 56.9, 56.3, 50.5, 42.5, 40.0, 39.9, 39.7, 37.5, 36.3, 36.0, 33.4, 32.1, 32.0, 29.1, 28.4, 28.2, 24.4, 24.0, 23.0, 22.7, 21.0, 18.9, 18.8, 12.0. cholesterol-2s:[11z] m.p. 141~143 ℃ (Lit.[11aa] 147~152 ℃); 1H NMR (400 MHz, CDCl3) δ: 5.37~5.32 (m, 1H), 3.58~3.46 (m, 1H), 2.35~2.17 (m, 2H), 2.05~0.95 (m, 29H), 0.91 (d, J=5.6, 1.6 Hz, 3H), 0.86 (d, J=6.4 Hz, 6H), 0.67 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 140.9, 121.9, 72.0, 56.9, 56.3, 50.3, 42.48, 42.46, 39.9, 39.7, 37.4, 36.7, 36.4, 35.9, 32.07, 32.06, 31.8, 28.4, 28.2, 24.5, 24.0, 23.0, 22.7, 21.2, 19.6, 18.9, 12.0.
(1R, 2S, 5R)-2-Isopropyl-5-methylcyclohexan-1-ol (2t):[11ab] White solid. m.p. 40~41 ℃ (Lit.[11ac] 38~40 ℃); 1H NMR (400 MHz, CDCl3) δ: 3.41 (td, J=10.4, 4.4 Hz, 1H), 2.23~2.10 (m, 1H), 2.00~1.92 (m, 1H), 1.70~1.55 (m, 2H), 1.48~1.34 (m, 2H), 1.15~1.06 (m, 1H), 1.02~0.84 (m, 9H), 0.80 (d, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 71.7, 50.3, 45.2, 34.7, 31.8, 26.0, 23.3, 22.4, 21.1, 16.3.
Supporting Information Procedure for radical capturing experiment with TEMPO and the characterization data for compound 5. 1H NMR and 13C NMR spectra copies for alcohols 2. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Larock, R. C. Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, New York, 1999, p. 890.
(b) Smith, M. D.; March, J. Advanced Organic Chemistry, 6th ed., Wiley-Interscience, New York, 2007, p. 425.
(a) Harris, M.; Bull, M. J. Synth. Commun. 1985, 15, 1225.
(b) Alexander, J.; Renyer, M. L.; Veerapanane, H. Synth. Commun. 1995, 25, 3875.
(c) Ruddick, C. L.; Hodge, P.; Houghton, M. P. Synthesis 1996, 1359.
(d) Abad, A.; Agulló, C.; Cuñat, A.C.; Navarro, I. Synthesis 2005, 3355.
(e) Chougala, B. M.; Samundeeswari, S.; Holiyachi, M.; Shastri, L. A. ChemistrySelect 2017, 2, 1290.
(f) Liu, H.; Liu, J.; Cheng, X.; Jia, X.; Yu, L.; Xu, Q. ChemSusChem 2019, 12, 2994.
(a) Nakamura, E.; Inubushi, T.; Aoki, S.; Machii, D. J. Am. Chem. Soc. 1991, 113, 8980.
(b) Nakamura, E.; Sato, K.; Imanishi, Y. Synlett 1995, 525.
(c) Sawamura, M.; Kawaguchi, Y.; Nakamura, E. Synlett 1997, 801.
(d) Sawamura, M.; Kawaguchi, Y.; Sato, K.; Nakamura, E. Chem. Lett. 1997, 26, 705.
Cai, Y.-M.; Xu, Y.-T.; Zhang, X.; Gao, W.-X.; Huang, X.-B.; Zhou, Y.-B.; Liu, M.-C.; Wu, H.-Y. Org. Lett. 2019, 21, 8479. doi: 10.1021/acs.orglett.9b03317
(a) Itoh, A.; Kodama, T.; Inagaki, S.; Masaki, Y. Org. Lett. 2001, 3, 2653.
(b) Li, J.; Wang, H.; Liu, L.; Sun, J. RSC Adv. 2014, 4, 49974.
(c) Xu, J.; Liu, N.; Zou, L.; Cheng, F.; Shen, X.; Liu, T.; Lv, H.; Khan, R.; Fan, B. Asian J. Org. Chem. 2019, 8, 261.
(a) Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828.
(b) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322.
(c) Dai X.; Xu, X.; Li, X. Chin. J. Org. Chem. 2013, 33, 2046(in Chinese).
(戴小军, 许孝良, 李小年, 有机化学, 2013, 33, 2046.)
(d) Shaw, M. H.; Twilton, J.; MacMillan, D. W. C. J. Org. Chem. 2016, 81, 6898.
(e) Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075.
(f) Matsui, J. K.; Lang, S. B.; Heitz, D. R.; Molander, G. A. ACS Catal. 2017, 7, 2563.
(a) Chatgilialoglu, C. Acc. Chem. Res. 1992, 25, 188.
(b) Chatgilialoglu, C.; Ferreri, C.; Landais, Y.; Timokhin, V. I. Chem. Rev. 2018, 118, 6516.
Mills, I. N.; Porras, J. A.; Bernhard, S. Acc. Chem. Res. 2018, 51, 352. doi: 10.1021/acs.accounts.7b00375
(a) Lowry, M. S.; Goldsmith, J. I.; Slinker, J. D.; Rohl, R.; Pascal, R. A. Jr.; Malliaras, G. G.; Bernhard, S. Chem. Mater. 2005, 17, 5712.
(b) Nguyen, J. D.; Tucker, J. W.; Konieczynska, M. D.; Stephenson, C. R. J. J. Am. Chem. Soc. 2011, 133, 4160.
(c) Wallentin, C.-J.; Nguyen, J. D.; Finkbeiner, P.; Stephenson, C. R. J. J. Am. Chem. Soc. 2012, 134, 8875.
(a) Stien, D.; Gastaldi, S. J. Org. Chem. 2004, 69, 4464.
(b) Damont, A.; Médran-Navarrete, V.; Cacheux, F.; Kuhnast, B.; Pottier, G.; Bernards, N.; Marguet, F.; Puech, F.; Boisgard, R.; Dolle, F. J. Med. Chem. 2015, 58, 7449.
(c) Dudnik, A. S.; Fu, G. C. J. Am. Chem. Soc. 2012, 134, 10693.
(a) Lee, K. C.; Lee, S.-Y.; Choe, Y. S.; Chi, D. Y. Bull. Korean Chem. Soc. 2004, 25, 1225.
(b) Nicholson, J. S.; Richards, H. C.; Adams, S. S. GB 1030756, 1966.
(c) Osako, T.; Torii, K.; Hirata, S.; Uozumi, Y. ACS Catal. 2017, 7371.
(d) Itami, K.; Kamei, T.; Mitsudo, K.; Nokami, T.; Yoshida, J. J. Org. Chem. 2001, 66, 397.
(e) Khan, S. N.; Zaman, M. K.; Li, R.; Sun, Z. J. Org. Chem. 2020, 85, 5019.
(f) Sakai, N.; Kawana, K.; Ikeda, R.; Nakaike, Y.; Konakahara, T. Eur. J. Org. Chem. 2011, 2011, 3178.
(g) Reddy, A. G. K.; Krishna, J.; Satyanarayana, G. ChemistrySelect 2016, 1, 1151.
(h) Cavedon, C.; Madani, A.; Seeberger, P. H.; Pieber, B. Org. Lett. 2019, 21, 5331.
(i) Ishibashi, M. J. Polym. Sci. B Polym. Lett. 1964, 2, 789.
(j) Reddy, M. V.; Kang, S. M.; Yoo, S.; Woo, S. S.; Kim, D. W. RSC. Adv. 2019, 9, 9435.
(k) Rysak, V.; Descamps-Mandine, A.; Simon, P.; Blanchard, F.; Burylo, L.; Trentesaux, M.; Vandewalle, M.; Collière, V.; Agbossou-Niedercorn, F.; Michon, C. Catal. Sci. Technol. 2018, 8, 3504.
(l) Green, R. A.; Jolley, K. E.; Al-Hadedi, A. A. M.; Pletcher, D.; Harrowven, D. C.; De Frutos, O.; Mateos, C.; Klauber, D. J.; Rincón, J. A.; Brown, R. C. D. Org. Lett. 2017, 19, 2050.
(m) Xu, G.; Leloux, S.; Zhang, P.; Meijide Suárez, J.; Zhang, Y.; Derat, E.; Ménand, M.; Bistri-Aslanoff, O.; Roland, S.; Leyssens, T.; Riant, O.; Sollogoub, M. Angew. Chem., Int. Ed. 2020, 59, 7591.
(n) Ouali, A.; Majoral, J.-P.; Caminade, A.-M.; Taillefer, M. ChemCatChem 2009, 1, 504.
(o) Rösler, S.; Obenauf, J. Kempe, R. J. Am. Chem. Soc. 2015, 137, 7998.
(p) Soulard, V.; Villa, G.; Vollmar, D. P.; Renaud, P. J. Am. Chem. Soc. 2018, 140, 155.
(q) Zhong, R.; Wei, Z.; Zhang, W.; Liu, S.; Liu, Q. Chem 2019, 5, 1552.
(r) Ungnade, H. E. J. Org. Chem. 1948, 13, 361.
(s) Weng, W.-Z.; Liang, H.; Zhang, B. Org. Lett. 2018, 20, 4979.
(t) Polidano, K.; Williams, J. M. J.; Morrill, L. C. ACS Catal. 2019, 9, 8575.
(u) Tohma, H.; Maegawa, T.; Takizawa, S.; Kita, Y. Adv. Synth. Catal. 2002, 344, 328.
(v) Parshikov, I. A.; Modyanova, L. V.; Dovgilivich, E. V.; Terent'ev, P. B.; Vorob'eva, L. I.; Grishina, G. V. Chem. Heterocycl. Compd. (Engl. Transl.) 1992, 28, 159.
(w) Aguilar Troyano, F. J.; Ballaschk, F.; Jaschinski, M.; Ӧzkaya, Y.; Gómez-Suórez, A. Chem. Eur. J. 2019, 25, 14054.
(x) Bach, R. D.; Taaffee, T. H.; Holubka, J. W. J. Org. Chem. 1980, 45, 3439.
(y) Boonyarattanakalin, S.; Martin, S. E.; Dykstra, S. A.; Peterson, B. R. J. Am. Chem. Soc. 2004, 126, 16379.
(z) Matt, C.; Kern, C.; Streuff, J. ACS Catal. 2020, 10. 6409.
(aa) Pujari, N. S.; Kulkarni, M. R.; Large, M. C. J.; Bassett, I. M.; Ponrathnam, S. J. Appl. Polym. Sci. 2005, 98, 58.
(ab) Toogood, H. S.; Cheallaigh, A. N.; Tait, S.; Mansell, D. J.; Jervis, A.; Lygidakis, A.; Humphreys, L.; Takano, E.; Gardiner, J. M.; Scrutton, N. S. ACS Synth. Biol. 2015, 4, 1112.
(ac) Zengin, G. Chem. Nat. Compd. 2011, 47, 550.

Scheme 1 Strategies for the conversion of alkyl halides to alcohols via carbon radical processes
Table 1. Investigation of the impact of reaction conditions on the TTMSS/O2 promoted conversion of alkyl iodides to alcoholsa
|
||||
| Entry | Conditions | Yieldb/% | Recovered 1ab/% | |
| 2a | 3a | |||
| 1 | As shown | 94 | Trace | 0 |
| 2 | DMA instead of DMF | 86 | Trace | 7 |
| 3 | DMSO instead of DMF | 24 | 6 | 31 |
| 4 | No TTMSS | 4 | 0 | 95 |
| 5 | Under N2 atmosphere | 0 | 99 | 0 |
| 6 | No photoredox catalyst | 33 | 2 | 63 |
| 7 | No blue LEDs | 39 | 0 | 57 |
| 8 | No photocatalyst and no blue LED | 32 | 0 | 65 |
| 9 | No Na2CO3 | 32 | 6 | 15 |
| a The reaction was carried out with 1a (0.10 mmol), TTMSS (0.22 mmol), Na2CO3 (0.10 mmol), [Ir{dF(CF3)ppy}2(dtbbpy)]PF6 (0.0010 mmol) in DMF (1.0 mL) under 8 W blue LED light irradiation at room temperature for 24 h unless otherwise stated. b The NMR yields of 2a and 3a and the recovery percentage of 1a were determined by 1H NMR analysis of the crude reaction mixture. | ||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Substrate scope investigation for the transformation from alkyl iodides to alcoholsa
|
|
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

