引用本文:
杨阳, 李若梅, 王炜, 许子文, 谢光辉, 鲁郑全, 李靖靖, 宋力平, 李维实. 聚合物对单壁碳纳米管增溶及选择性分离的机理研究进展[J]. 有机化学,
2020, 40(10): 3249-3261.
doi:
10.6023/cjoc202006019 Citation:
Yang Yang, Li Ruomei, Wang Wei, Xu Zi-Wen, Xie Guanghui, Lu Zhengquan, Li Jingjing, Song Liping, Li Wei-Shi. Research Advances on the Mechanism of Polymer Solubilization and Selective Separation of Single-Wall Carbon Nanotubes[J]. Chinese Journal of Organic Chemistry,
2020, 40(10): 3249-3261.
doi:
10.6023/cjoc202006019
College of Science, Shanghai University, Shanghai 200444
b.
Key Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032
c.
College of Chemistry and Material Sciences, Shanghai Normal University, Shanghai 200034
d.
Engineering Research Center of Zhengzhou for High Performance Organic Functional Materials, Zhengzhou Institute of Technology, Zhengzhou 450044
Received Date:
11 June 2020 Revised Date:
29 July 2020 Available Online:
25 October 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21674125, 21672251, 51761145043), the Strategic Priority Research Program of Chinese Academy of Sciences (No. XDB20020000) and the Zhengzhou Institute of Technology
Abstract:
Single-walled carbon nanotubes (SWNTs) prepared by present methods are a mixture of semiconducting and metallic ones, which need to be separated and purified to give them full play with their excellent properties and for their attractive applications. Among numerous developed methods, the selective separation of semiconducting and metallic SWNTs by polymer non-covalent interactions is considered to be the most simple, efficient and no-damage to carbon nanotube structure and properties method. So far, a lot of works have been reported, and many polymer systems have been developed. Meanwhile, various separation and purification mechanisms have been proposed, but there are still no unified and convincing understandings. In this review, the reported SWNT solubilization and selective separation works are summarized, the interactions between polymer and SWNTs are analyzed, and the effects of polymer structures, molecular weights, side chain lengths, polymer/SMNT ratio, temperature, and solvent are focally discussed. After comparing various separating mechanisms, our own understandings and views are put forward.
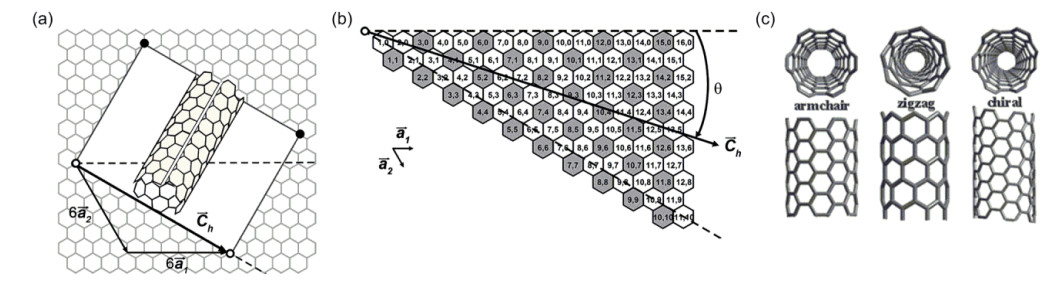
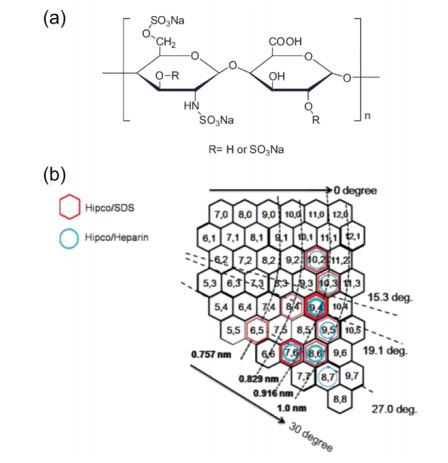
Figure 1.
(a) Illustration of the construction of a carbon nanotube from a graphene sheet using (6, 6) nanotube as an example; [2] (b) the possible SWNT (n, m) structures represented on a single graphene sheet; [2] (c) the structures of armchair, zigzag and chiral nanotubes[3]
(a) The detail construction is to roll up the white area graphene sheet and overlap the corresponding two sets of lattice points. (b) Semiconducting SWNTs are white and metallic ones are shaded[2]. Reprinted with permission from Ref. [2]. Copyright 2008 American Chemical Society. (c) Reprinted with permission from Ref. [3]. Copyright 2011 Royal Society of Chemistry
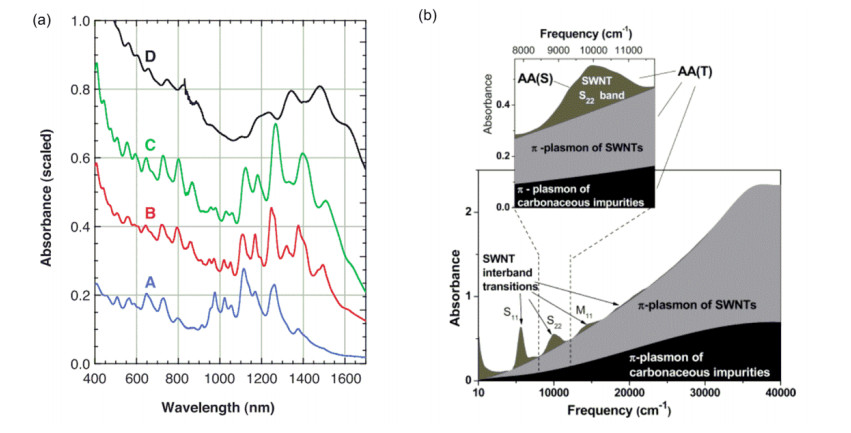
Figure 2.
(a) Absorption spectra of curves A, B and C dispersed individual carbon nanotubes and absorption spectrum of curve D aggregated small bundle carbon nanotubes; [13] (b) Electronic spectrum of a typical SWNT sample
(a) Reprinted with permission from Ref. [13]. Copyright 2002 the American Association for the Advancement of Science; (b) the inset shows the region of the S22 interband transition utilized for NIR purity evaluation. In the diagram, AA(S) area of the S22 spectral band after linear baseline correction and AA(T) total area of the S22 band including SWNT and carbonaceous impurity contributions. The NIR carbon tube relative purity is given by RP=(AA(S)/AA(T))/0.141[14]. Reprinted with permission from Ref. [14]. Copyright 2005 American Chemical Society
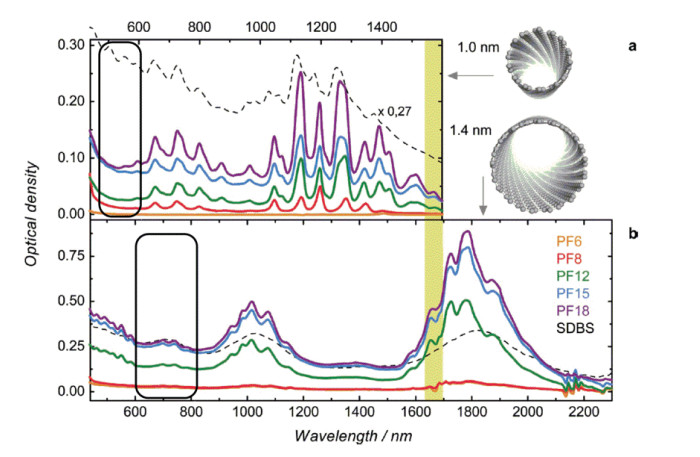
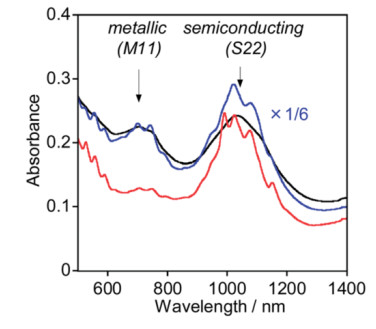
Figure 3.
(a) Absorption spectra of HiPCO SWCNTs (average diameter 1 nm) dispersed with polymers; (b) absorption spectra of SO SWCNTs (average diameter 1.4 nm) dispersed with polymers[16]
The black rectangle area is the typical absorption peak area of the metallic SWCNTs. Reprinted with permission from Ref. [16]. Copyright 2013 John Wiley and Sons
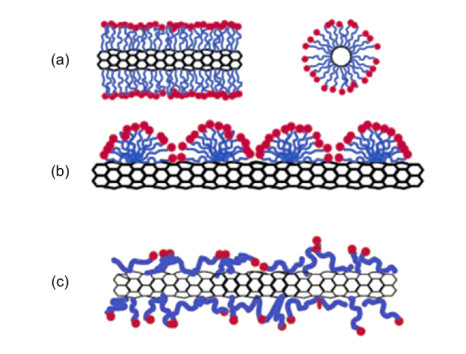
Figure 4.
(a) SWNT encapsulated in a cylindrical surfactant micelle (left: side view, right: cross section); (b) Hemimicellar adsorption of surfactant molecules on a SWNT; (c) Random adsorption of surfactant molecules on a SWNT[24]
Reprinted with permission from Ref. [24]. Copyright 2004 American Chemical Society
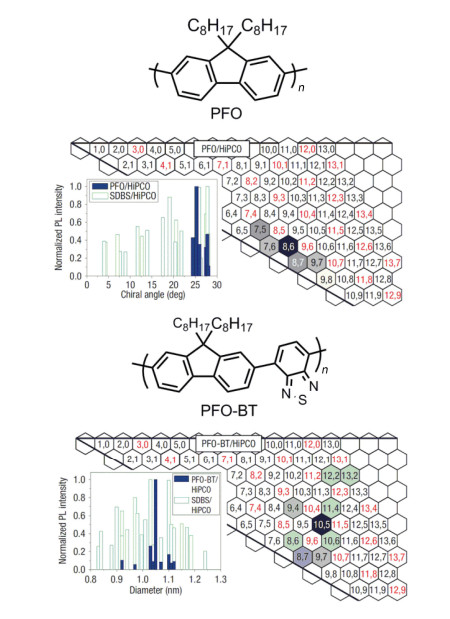
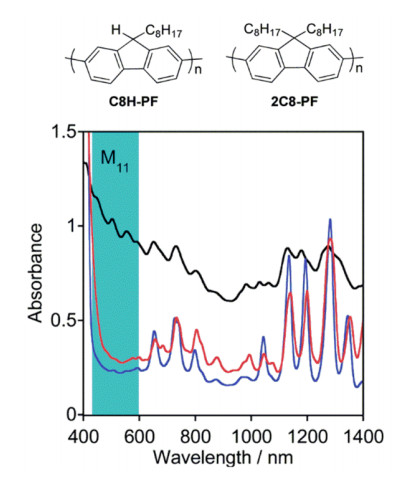
Figure 10.
Chirality map of SWCNTs selected by polyfluorene wrapping[16]
In yellow the SWNTs selected are underlined; the color of the dots inside the hexagons indicates which of the polyfluorene derivatives (color code used for the chemical structures) is able to select the nanotubes. Reprinted with permission from Ref. [16]. Copyright 2013 John Wiley and Sons
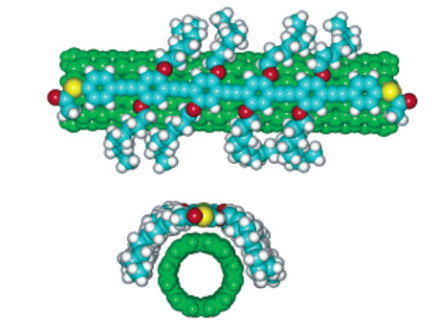
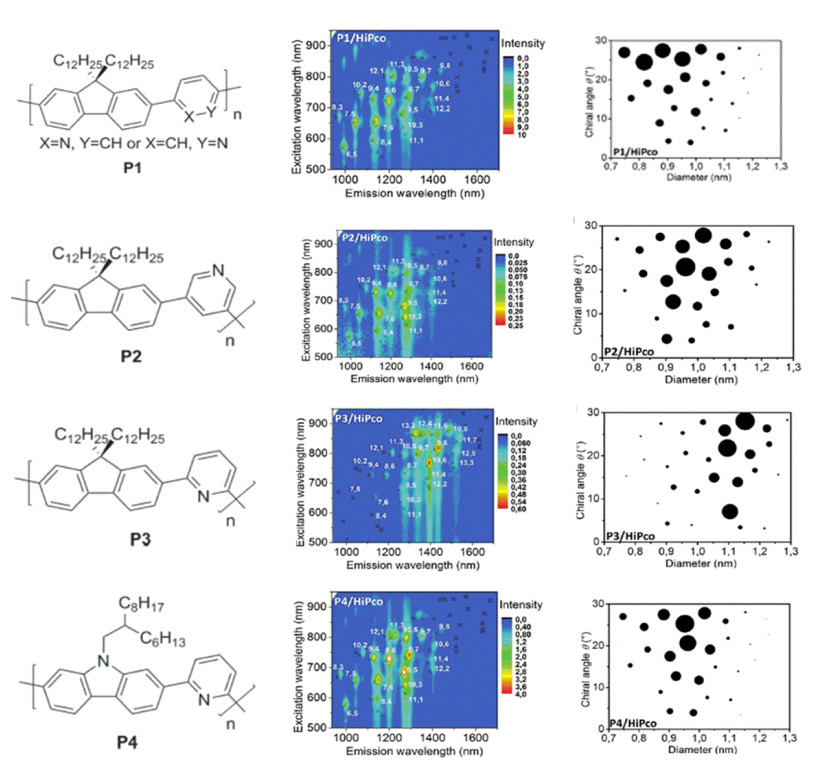
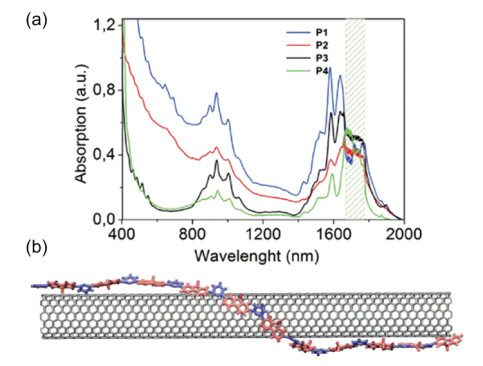
Figure 12.
(a) UV-vis-NIR light absorption spectra of the solutions of separated SWNTs by polymer P1, P2,
P3, P4, and (b) schematic diagram of P3 wrapping on the surface of SWNT[39]
In (b) Red part represents fluorene unit, while blue rings are pyridine units[39]. Reprinted with permission from Ref. [39]. Copyright 2013 John Wiley and Sons
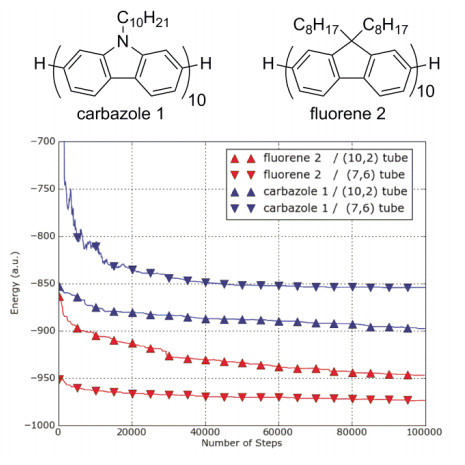
Figure 13.
Computing optimization on energies of polymer/SWNT complexes formed from binding (10, 2) and (7, 6) SWNTs with fluorene decamer and carbazole decamer[12]
Reprinted with permission from Ref. [12]. Copyright 2011 American Chemical Society<
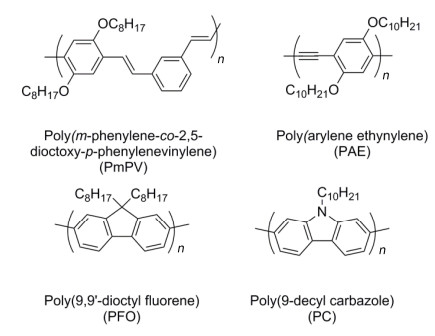
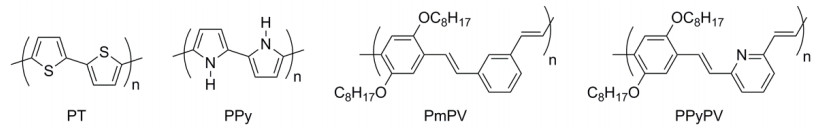
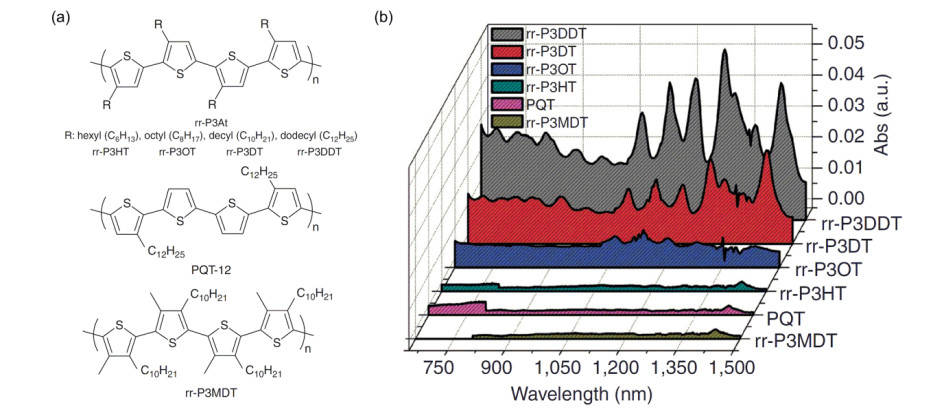
Figure 19.
(a) Chemical structures of polythiophenes bearing different alkyl side chains, and (b) light-absorption spectra of SWNT solutions sorted therewith[11]
Reprinted with permission from Ref. [11]. Copyright 2011 Springer Nature
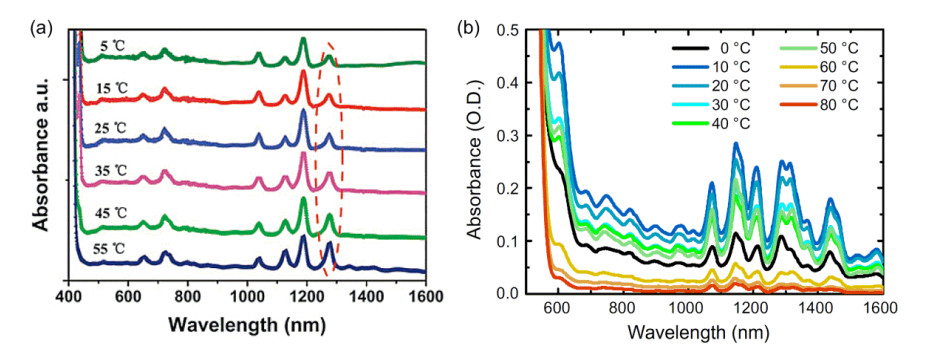
Figure 20.
(a) Light-absorption spectra of SWNT solutions obtained by PFO sorting at various temperatures, [56] and (b) light-absorption spectra of SWNT solutions obtained by P3DDT sorting at various temperatures[57]
(a) Reprinted with permission from Ref. [56]. Copyright 2015 Royal Society of Chemistry. (b) Reprinted with permission from Ref. [57]. Copyright 2015 Elsevier
Wang, H.; Wang, Y.; Tee, B. C. K.; Kim, K.; Lopez, J.; Cai, W.; Bao, Z. Adv. Sci.2015, 2, 1500103. doi: 10.1002/advs.201500103
[5]
Ha, M.; Xia, Y.; Green, A. A.; Zhang, W.; Frisbie, C. D. ACS Nano2010, 8, 4388.
[6]
Chae, S. H.; Yu, W. J.; Bae, J. J.; Dinh Loc, D.; Perello, D.; Jeong, H. Y.; Quang Huy, T.; Thuc Hue, L.; Quoc An, V.; Yun, M.; Duan, X.; Lee, Y. H. Nat. Mater.2013, 12, 403. doi: 10.1038/nmat3572
[7]
Arnold, M. S.; Green, A. A.; Hulvat, J. F.; Stupp, S. I.; Hersam, M. C. Nat. Nanotechnol.2006, 1, 60. doi: 10.1038/nnano.2006.52
[8]
Krupke, R.; Hennrich, F.; Lohneysen, H.; Kappes, M. M. Science2003, 301, 344. doi: 10.1126/science.1086534
[9]
Zheng, M.; Jagota, A.; Strano, M. S.; Santos, A. P.; Barone, P.; Chou, S. G.; Diner, B. A.; Dresselhaus, M. S.; Mclean, R. S.; Onoa, G. B.; Samsonidze, G. G.; Semke, E. D.; Usrey, M.; Walls, D. J. Science2003, 1545.
[10]
Nish, A.; Hwang, J. Y.; Doig, J.; Nicholas, R. J. Nat. Nanotechnol. 2007, 2, 640. doi: 10.1038/nnano.2007.290
[11]
Lee, H. W.; Yoon, Y.; Park, S.; Oh, J. H.; Hong, S.; Liyanage, L. S.; Wang, H.; Morishita, S.; Patil, N.; Park, Y. J.; Park, J. J.; Spakowitz, A.; Galli, G.; Gygi, F.; Wong, P. H.; Tok, J. B.; Kim, J. M.; Bao, Z. Nat. Commun.2011, 2, 541. doi: 10.1038/ncomms1545
[12]
Lemasson, F. A.; Strunk, T.; Gerstel, P.; Hennrich, F.; Lebedkin, S.; Barner-Kowollik, C.; Wenzel, W.; Kappes, M. M.; Mayor, M. J. Am. Chem. Soc.2011, 133, 652. doi: 10.1021/ja105722u
[13]
O'Connell, M. J.; Bachilo, S. M.; Huffman, C. B.; Moore, V. C.; Strano, M. S.; Haroz, E. H.; Rialon, K. L.; Boul, P. J.; Noon, W. H.; Kittrell, C.; Ma, J.; Hauge, R. H.; Weisman, R. B.; Smalley, R. E. Science2002, 297, 593. doi: 10.1126/science.1072631
[14]
Itkis, M. E.; Perea, D. E.; Jung, R.; Niyogi, S.; Haddon, R. C. J. Am. Chem. Soc.2005, 127, 3439. doi: 10.1021/ja043061w
Gomulya, W.; Costanzo, G. D.; de Carvalho, E. J.; Bisri, S. Z.; Derenskyi, V.; Fritsch, M.; Frohlich, N.; Allard, S.; Gordiichuk, P.; Herrmann, A.; Marrink, S. J.; dos Santos, M. C.; Scherf, U.; Loi, M. A. Adv. Mater.2013, 25, 2948. doi: 10.1002/adma.201300267
Dalton, A. B.; Stephan, C.; Coleman, J. N.; McCarthy, B.; Ajayan, P. M.; Lefrant, S.; Bernier, P.; Blau, W. J.; Byrne, H. J. J. Phys. Chem. B2000, 104, 10012. doi: 10.1021/jp002857o
[31]
Yi, W.; Malkovskiy, A.; Xu, Y.; Wang, X.-Q.; Sokolov, A. P.; Lebron-Colon, M.; Meador, M. A.; Pang, Y. Polymer2010, 51, 475. doi: 10.1016/j.polymer.2009.11.052
[32]
In het Panhuis, M.; Maiti, A.; Dalton, A. B.; van den Noort, A.; Coleman, J. N.; McCarthy, B.; Blau, W. J. J. Phys. Chem. B2003, 107, 478. doi: 10.1021/jp026470s
[33]
Yi, W.; Malkovskiy, A.; Chu, Q.; Sokolov, A. P.; Colon, M. L.; Meador, M.; Pang, Y. J. Phys. Chem. B2008, 112, 12263.
[34]
Chen, J.; Liu, H.; Weimer, W. A.; Halls, M. D.; Waldeck, D. H.; Walker, G. C. J. Am. Chem. Soc.2002, 124, 9034. doi: 10.1021/ja026104m
[35]
Kang, Y. K.; Lee, O.-S.; Deria, P.; Kim, S. H.; Park, T.-H.; Bonnell, D. A.; Saven, J. G.; Therien, M. J. Nano Lett.2009, 9, 1414. doi: 10.1021/nl8032334
[36]
Chen, Y.; Xu, Y.; Wang, Q.; Gunasinghe, R. N.; Wang, X. Q.; Pang, Y. Small2013, 9, 870. doi: 10.1002/smll.201202103
Wang, H.; Koleilat, G. I.; Liu, P.; Jiménez-Osés, G.; Lai, Y.-C.; Vosgueritchian, M.; Fang, Y.; Park, S.; Houk, K. N.; Bao, Z. ACS Nano2014, 8, 2609. doi: 10.1021/nn406256y
[46]
Lee, M.-H.; Lee, S.-H.; Kim, J.; Lee, S. Y.; Lim, D.-H.; Hwang, K.; Hwang, H.; Jung, Y. C.; Noh, Y.-Y.; Kim, D.-Y. Carbon2017, 125, 571. doi: 10.1016/j.carbon.2017.09.068
[47]
Gomulya, W.; Derenskyi, V.; Kozma, E.; Pasini, M.; Loi, M. A. Adv. Funct. Mater.2015, 25, 5858. doi: 10.1002/adfm.201502912
[48]
Lei, T.; Pitner, G.; Chen, X.; Hong, G.; Park, S.; Hayoz, P.; Weitz, R. T.; Wong, H.-S. P.; Bao, Z. Adv. Electron. Mater.2016, 2, 1500299. doi: 10.1002/aelm.201500299
[49]
Min, S. H.; Kim, H.-I.; Kim, K.-S.; Cha, I.; Ha, S.; Yun, W. S.; Kwak, S. K.; Kim, J.-H.; Kim, B.-S.; Song, C. Polymer2016, 96, 63. doi: 10.1016/j.polymer.2016.04.063
[50]
Zheng, M.; Jagota, A.; Semke, E. D.; Diner, B. A.; McLean, R. S.; Lustig, S. R.; Richardson, R. E.; Tassi, N. G. Nat. Mater.2003, 2, 338. doi: 10.1038/nmat877
[51]
Zaremba, O.; Goldt, A.; Ramirez-Morales, M.; Khabushev, E. M.; Shulga, E.; Eremin, T.; Prikazchikova, T.; Orekhov, A.; Grebenko, A.; Zatsepin, T. S.; Obraztsova, E. D.; Nasibulin, A. G. Carbon2019, 151, 175. doi: 10.1016/j.carbon.2019.05.076
[52]
Li, H.; Zhou, B.; Lin, Y.; Gu, L.; Wang, W.; Fernando, K. A. S.; Kumar, S.; Allard, L. F.; Sun, Y.-P. J. Am. Chem. Soc.2004, 126, 1014. doi: 10.1021/ja037142o
[53]
Yan, L. Y.; Li, W.; Fan, X. F.; Wei, L.; Chen, Y.; Kuo, J.-L.; Li, L.-J.; Kwak, S. K.; Mu, Y.; Chan-Park, M. B. Small2010, 6, 110. doi: 10.1002/smll.200900865
Gomulya, W.; Rios, J. M. S.; Derenskyi, V.; Bisri, S. Z.; Jung, S.; Fritsch, M.; Allard, S.; Scherf, U.; dos Santos, M. C.; Loi, M. A. Carbon2015, 84, 66. doi: 10.1016/j.carbon.2014.11.037
Figure 1
(a) Illustration of the construction of a carbon nanotube from a graphene sheet using (6, 6) nanotube as an example; [2] (b) the possible SWNT (n, m) structures represented on a single graphene sheet; [2] (c) the structures of armchair, zigzag and chiral nanotubes[3]
(a) The detail construction is to roll up the white area graphene sheet and overlap the corresponding two sets of lattice points. (b) Semiconducting SWNTs are white and metallic ones are shaded[2]. Reprinted with permission from Ref. [2]. Copyright 2008 American Chemical Society. (c) Reprinted with permission from Ref. [3]. Copyright 2011 Royal Society of Chemistry
Figure 2
(a) Absorption spectra of curves A, B and C dispersed individual carbon nanotubes and absorption spectrum of curve D aggregated small bundle carbon nanotubes; [13] (b) Electronic spectrum of a typical SWNT sample
(a) Reprinted with permission from Ref. [13]. Copyright 2002 the American Association for the Advancement of Science; (b) the inset shows the region of the S22 interband transition utilized for NIR purity evaluation. In the diagram, AA(S) area of the S22 spectral band after linear baseline correction and AA(T) total area of the S22 band including SWNT and carbonaceous impurity contributions. The NIR carbon tube relative purity is given by RP=(AA(S)/AA(T))/0.141[14]. Reprinted with permission from Ref. [14]. Copyright 2005 American Chemical Society
Figure 3
(a) Absorption spectra of HiPCO SWCNTs (average diameter 1 nm) dispersed with polymers; (b) absorption spectra of SO SWCNTs (average diameter 1.4 nm) dispersed with polymers[16]
The black rectangle area is the typical absorption peak area of the metallic SWCNTs. Reprinted with permission from Ref. [16]. Copyright 2013 John Wiley and Sons
Figure 4
(a) SWNT encapsulated in a cylindrical surfactant micelle (left: side view, right: cross section); (b) Hemimicellar adsorption of surfactant molecules on a SWNT; (c) Random adsorption of surfactant molecules on a SWNT[24]
Reprinted with permission from Ref. [24]. Copyright 2004 American Chemical Society
Figure 10
Chirality map of SWCNTs selected by polyfluorene wrapping[16]
In yellow the SWNTs selected are underlined; the color of the dots inside the hexagons indicates which of the polyfluorene derivatives (color code used for the chemical structures) is able to select the nanotubes. Reprinted with permission from Ref. [16]. Copyright 2013 John Wiley and Sons
Figure 12
(a) UV-vis-NIR light absorption spectra of the solutions of separated SWNTs by polymer P1, P2,
P3, P4, and (b) schematic diagram of P3 wrapping on the surface of SWNT[39]
In (b) Red part represents fluorene unit, while blue rings are pyridine units[39]. Reprinted with permission from Ref. [39]. Copyright 2013 John Wiley and Sons
Figure 13
Computing optimization on energies of polymer/SWNT complexes formed from binding (10, 2) and (7, 6) SWNTs with fluorene decamer and carbazole decamer[12]
Reprinted with permission from Ref. [12]. Copyright 2011 American Chemical Society<
Figure 19
(a) Chemical structures of polythiophenes bearing different alkyl side chains, and (b) light-absorption spectra of SWNT solutions sorted therewith[11]
Reprinted with permission from Ref. [11]. Copyright 2011 Springer Nature
Figure 20
(a) Light-absorption spectra of SWNT solutions obtained by PFO sorting at various temperatures, [56] and (b) light-absorption spectra of SWNT solutions obtained by P3DDT sorting at various temperatures[57]
(a) Reprinted with permission from Ref. [56]. Copyright 2015 Royal Society of Chemistry. (b) Reprinted with permission from Ref. [57]. Copyright 2015 Elsevier

 下载:
下载:

 下载:
下载:





















