图式 1.
代表性生物活性碳苷
Scheme 1.
Representative bioactive C-glycosides

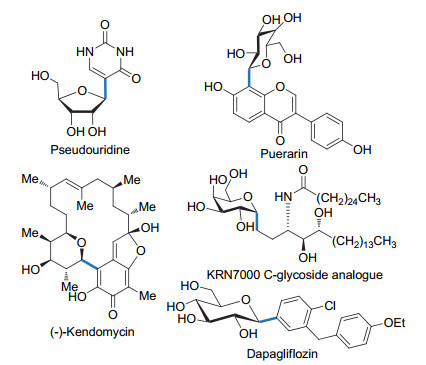
碳苷是指糖上端基碳原子和配糖体通过碳-碳键相连的糖类化合物.自然界存在的天然碳苷化合物多具有重要的生理活性.假尿嘧啶核苷[1](pseudouridine) (Scheme 1)是第一个被报道的碳核苷.它被认为是tRNA降解的最终产物, 在肿瘤患者体内含量会异常增高, 可以作为肿瘤标志物.从中药葛根中分离得到的异黄酮类碳苷葛根素[2](puerarin), 具有舒张血管、缓解心绞痛等多种活性.很多具有重要生理活性的天然产物也含有碳苷单元, 比如Kendomycin[3].与氧苷的C—O键相比, 碳苷端基的C—C键对酶和化学(酸)水解有更强的耐受性.例如, α-半乳糖神经酰胺KRN7000的碳苷类似物对小鼠B16黑色素瘤细胞的保护作用比其氧苷母体KRN7000强1000倍[4]; 由根皮苷(phlorizin)改造后的碳苷类似物可以用于糖尿病的治疗, 已有多个药物品种成功上市, 如达格列净(Dapagliflozin).由于碳苷良好的药用价值, 碳苷的合成研究越来越被人们所重视.但碳-碳键的构筑却是一项富有挑战性的工作.迄今为止, 已经有许多关于碳苷合成方法和策略的报道[5].本文主要总结基于糖烯的碳苷合成方法.
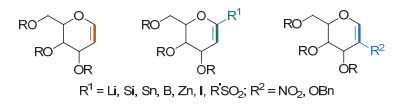
糖烯通常是指糖环的1, 2位含有双键的糖类化合物.对双键进行官能团化从而构筑新的碳-碳键是有机化学中常用的策略.糖烯具有与烯基醚和烯丙醇相似的烯键; 理论上, 烯基醚和烯丙醇底物能发生的碳-碳键构筑反应均可用于碳苷的合成.糖烯双键取代基的不同, 其构筑碳-碳糖苷键的方式也不一样.本文将按照反应类型, 从Ferrier Ⅰ型碳苷化反应、Heck偶联型碳苷化反应、1-取代糖烯的过渡金属催化偶联碳苷化反应、2-取代糖烯的Michael加成型和自由基加成型碳苷化反应(Scheme 2), 来总结基于糖烯的碳苷合成方法.
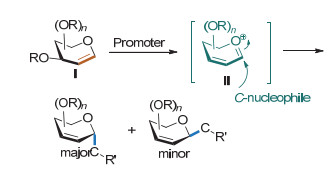
在Lewis酸等催化剂的作用下, 糖烯I的3位酰(烷)氧基离去, 产生离域的氧杂烯丙基碳正离子II继而与亲核试剂反应得到2, 3-不饱和糖苷化合物的反应称为Ferrier Ⅰ型反应.由于电子效应等因素影响, 产物一般以α-构型为主; 通过理论计算, 一般α-构型产物的半椅式构象能量较低(Scheme 3)[6]; 当使用含碳亲核试剂时能得到碳苷.根据含碳亲核试剂种类的不同, 可分为有机硅亲核试剂、有机金属亲核试剂和不饱和烃亲核试剂等.
有机硅试剂是一种理想的亲核试剂.利用有机硅亲核试剂和糖烯通过Ferrier Ⅰ型反应得到以α构型为主的2, 3-不饱和碳苷.有机硅试剂包括烯丙基硅、炔丙基硅以及烯醇硅醚等.根据使用Lewis酸的不同, 底物适用性和选择性略有差异.
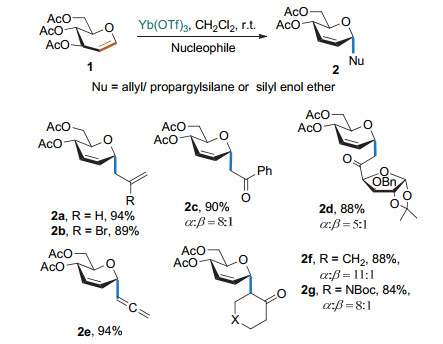
2001年, Schmidt课题组[7a]以Yb(OTf)3为Lewis酸, 乙酰基保护的葡萄糖烯为糖基供体, 烯丙基三甲基硅基/炔丙基三甲基硅基/烯醇三甲基硅醚为亲核试剂, 在二氯甲烷中进行反应得到不同的2, 3-不饱和烷基碳苷, 产物以α构型为主(Scheme 4). Grée课题组[7b]发现该反应在离子液中也可以很好地进行.
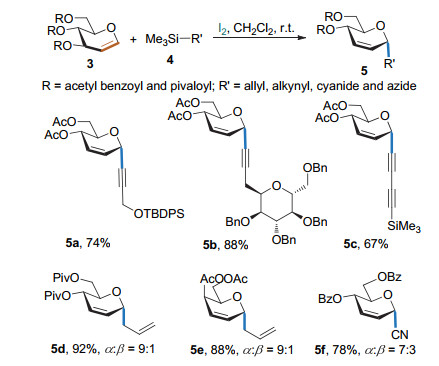
分子碘也可以作为催化剂使糖烯与有机硅试剂发生碳苷化反应(Scheme 5). Isobe课题组[8a]发现, 在I2的作用下全乙酰化的葡萄糖烯可以与不同取代的炔基三甲基硅烷发生Ferrier Ⅰ型碳苷化反应, 得到全α-构型的炔苷. Yadav课题组[8b]也通过碘分子完成了多种糖烯的烯丙基化和氰基化反应; 该反应只能得到以α-构型为主的混合物.
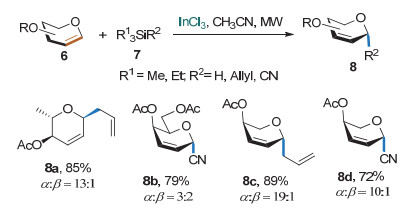
Sachwani课题组[9]利用InCl3作为Lewis酸, 在微波的作用下实现了糖烯与有机硅烷的碳苷化反应(Scheme 6).该反应对亲核试剂有限制, 必须是烯丙基三甲基硅和氰基三甲基硅, 生成的碳苷以α构型为主, 并不能保证为单一构型.
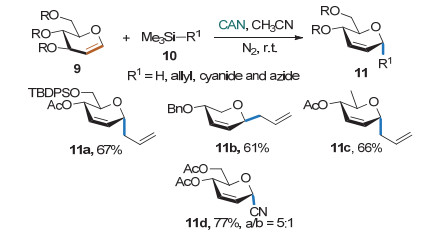
2014年, Vankar课题组[10]用硝酸铈铵(CAN)作为氧化剂, 以单电子氧化的方式使糖烯转化成离域的氧杂烯丙基碳正离子, 然后与烯丙基三甲基硅和氰基三甲基硅在乙腈中进行反应, 得到碳苷(Scheme 7).该方法适用于不同保护基的多种糖烯, 但亲核试剂只能是烯丙基三甲基硅和氰基三甲基硅.作者利用该方法进行了2-脱氧-2-氨基碳苷的合成.
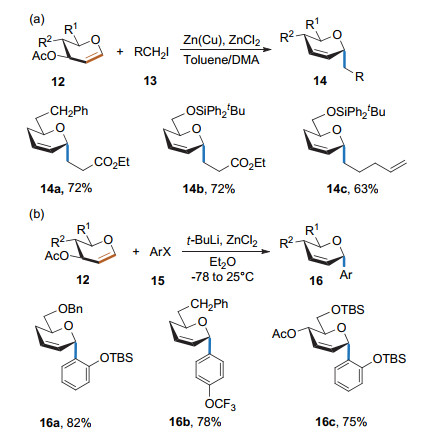
2001年, Bois课题组[11]在乙醚为溶剂和室温条件下, 实现了有机锌试剂参与的Ferrier Ⅰ型反应, 得到α构型为主的碳苷(Scheme 8a).该反应具有良好的底物适用性, 能同时适用于脂肪族和芳香族的有机锌试剂.有机锌化合物可由卤代烃在叔丁醇锂和氯化锌的存在下原位制得, 并直接用于碳苷合成(Scheme 8b).
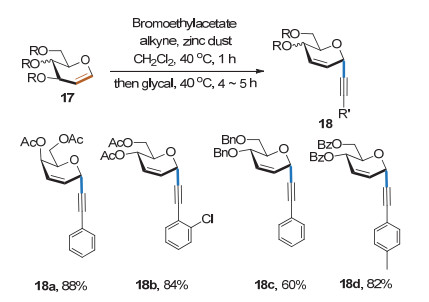
2014年, Mukherjee课题组[12]利用糖烯17、端炔、溴代乙酸乙酯和锌粉发展了一种新的炔基碳苷合成方法(Scheme 9).该方法以原位生成的炔基锌为亲核试剂, 具有良好的底物适用性和α立体选择性.值得一提的是, 该方法还适用于2-乙酰氧基取代的糖烯.
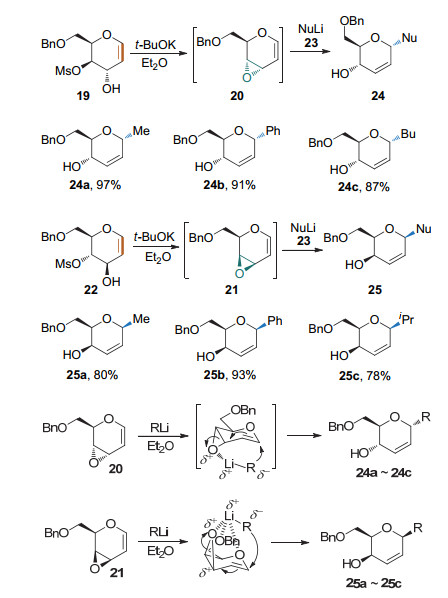
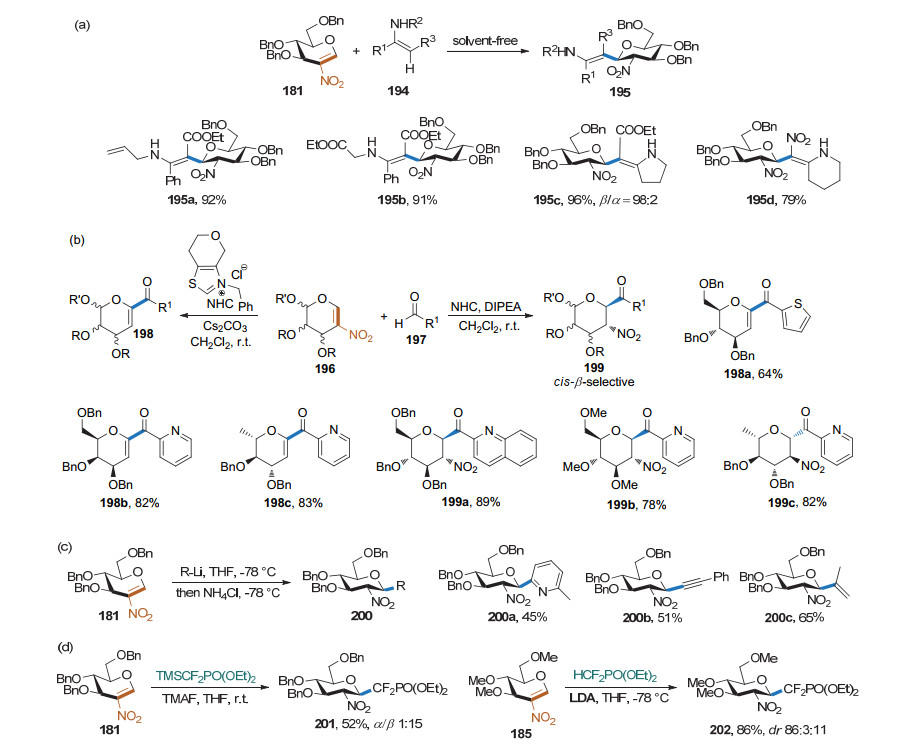
2003年, Crotti课题组[13]发现3-羟基-4-甲磺酰氧基糖烯在叔丁醇钾的作用下可得到烯基环氧中间体.该中间体可作为供体与有机锂亲核试剂反应得到碳苷(Scheme 10).产物的构型与生成的环氧中间体的构型有关, 有机锂试剂可通过与环氧协同配位, 生成与环氧同向的产物.简单的烷基锂和芳基锂都可顺利进行此反应.
2009年, Pohmakotr课题组[14]发现, 在Yb(OTf)3的作用下, 糖烯可与有机铝发生Ferrrier重排反应, 得到碳苷产物(Scheme 11).该方法对糖型和保护基有较好的耐受性, 收率良好, 但亲核试剂只有甲基铝和乙基铝, 立体选择性较差.
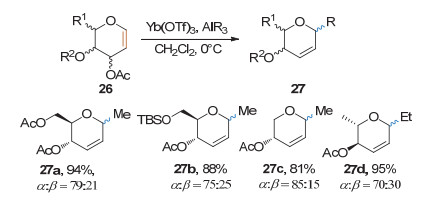
炔基本身很难作为Ferrier Ⅰ型反应的亲核试剂, 因此, 要制备炔苷通常需要先将端炔活化. 2007年, Auge课题组[15]发现金属铟可以原位与碘代炔反应, 生成炔基铟, 进而与糖烯发生Ferrier Ⅰ型反应, 生成炔基碳苷(Scheme 12).该方法对糖烯具有较好的底物适用性, 收率较好, 但反应时间长, 立体选择性中等.
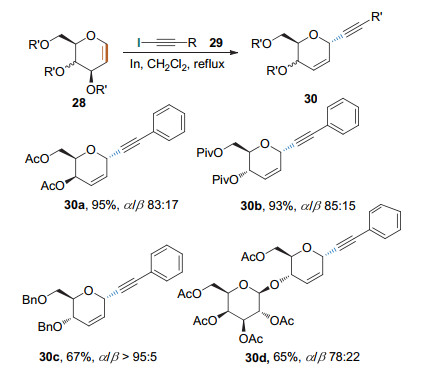
2008年, Stefani课题组[16]发现糖烯可以在三氟化硼乙醚的催化下与炔基三氟硼酸钾发生Ferrier Ⅰ型反应, 生成炔基碳苷.该反应条件温和, 在0 ℃条件下乙腈溶液中0.5 h即可反应完全.反应对亲核试剂有良好的底物适用范围.产物的立体选择性好, α/β比例大于98/7 (Scheme 13).
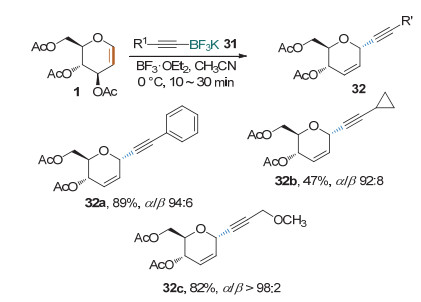
Mukherjee课题组[17]在2013年做过炔基碳苷的合成研究(Scheme 14).他们发现, 在抗坏血酸的存在下, Cu(OTf)2可以作为催化剂促进糖烯与不活泼的炔烃的快速偶联得到碳苷.该反应经历炔铜中间体, 反应收率高、反应时间短(2 min以内)、条件温和、α立体选择性大于96/4.
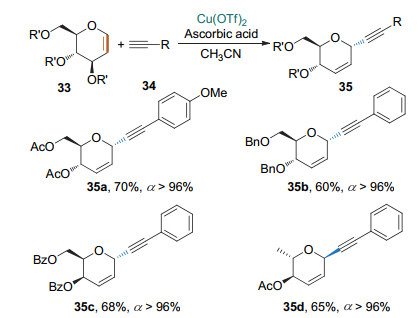
2016年, 史晓东课题组[18]用(ArO)3PAu(TA-Ph)OTf作为催化剂, 通过炔金中间体进一步将该反应拓展到不活泼糖烯同不活泼炔烃的Ferrier Ⅰ型反应.他们还将亲核试剂拓展到一些含天然产物片段的芳炔的合成, 进一步展示了该反应的实用性(Scheme 15).
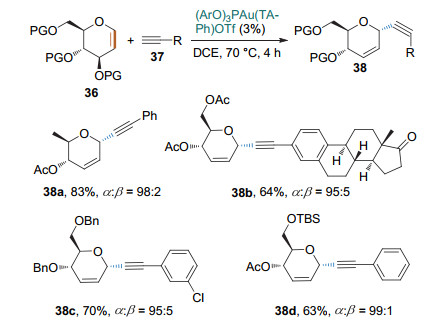
Shah课题组[19]提出一种新型的无金属催化策略(Scheme 16).他们以TMSOTf为催化剂, 原位生成炔基三甲基硅烷, 进而与糖烯发生Ferrier Ⅰ型反应, 生成高α立体选择性的炔基碳苷.张剑波课题组[20]也报道了类似的结果.
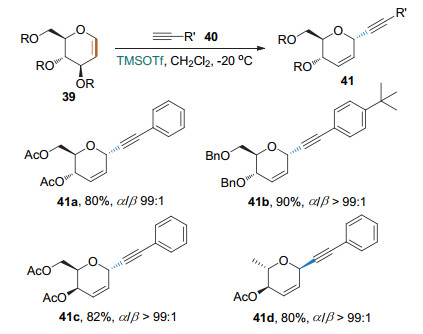
2014年, 刘学伟课题组[21]以三氯化铁为催化剂, 实现了β-酮酸的Ferrier Ⅰ型碳苷化反应(Scheme 17); 烯醇负离子为该反应的实际亲核试剂.该反应条件相对温和, 有较好的底物适用性, 但立体选择性并不是很好.
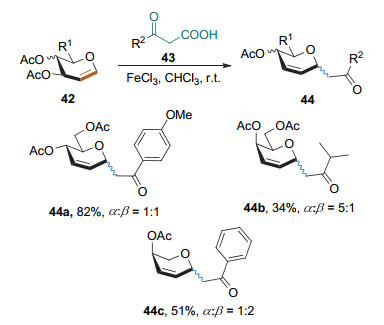
2017年, Mukherjee课题组[22]以TBSOTf为路易斯酸, DIPEA为碱, 实现了芳基乙酮与糖烯的Ferrier Ⅰ型碳苷化反应, 以较高的产率和α立体选择性得到了苯丙二氢吡喃碳苷(Scheme 18).该方法还适用于克级规模的制备.
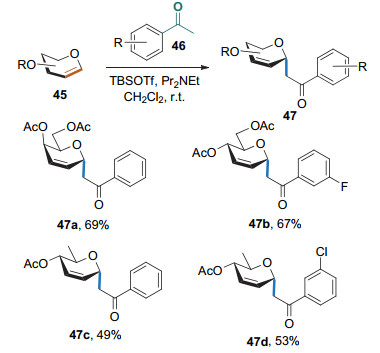
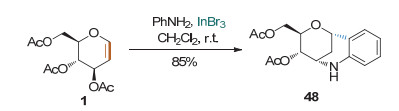
Yadav课题组[23]发现, 在InBr3的存在下, 苯胺上的邻位碳可与糖烯的1位相连, 而氮原子则与糖烯的3位相连, 生成高立体选择性的1-C, 3-N-双环化合物(Scheme 19).该反应条件温和, 具有较宽的底物适用性和高立体选择性; 但对于生成C—C和C—N键的先后顺序, 作者并没有进行深入研究.
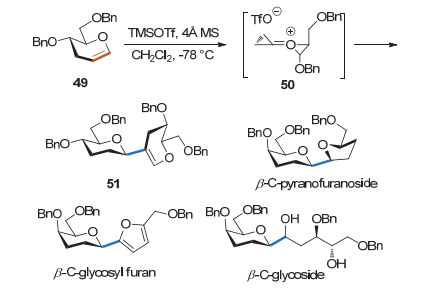
Sridhar课题组[24]发现在TMSOTf存在时, 3位脱氧的糖烯能两两相连生成β-1, 2连接的糖类化合物(Scheme 20).作者认为在酸的作用下糖烯可形成半椅式构象中间体50, 另一分子糖烯的双键可进攻该中间体的端位, 得到β-1, 2连接的假寡糖.产物具有很好的β立体选择性, 且可以进一步转化生成一系列天然碳苷片段.
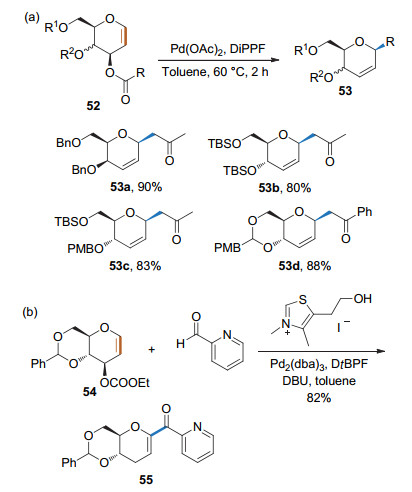
糖烯或其衍生物与钯生成的烯丙基钯络合物也可发生烯丙基碳正离子类似的反应[25a-25b]. 2013年, 刘学伟课题组[25c]利用3-烷/芳酰基乙酸酯保护的糖烯为底物, 在Pd(OAc)2/DiPPF的催化下生成烯丙基钯络合物, 继而发生脱羧偶联, 得到2, 3-不饱和的烷基碳苷(Scheme 21a).对于3, 6-顺式的底物, 该反应有非常好的立体选择性; 而对于3, 6-反式底物, 则生成αβ混合物.随后, 刘学伟课题组[25d]用NHC将醛进行极性反转后也可与烯丙基钯络合物进行反应, 得到相应的碳苷(Scheme 21b).
2017年, 刘学伟课题组[25e]用葡萄糖烯、磷叶立德和醛作为底物, Pd(OAc)2/1, 4-二苯基膦丁烷(DPPB)为催化剂, 实现了2, 3-不饱和的乙烯基碳苷的制备(Scheme 22).该反应的第一步也是形成烯丙基钯络合物, 继而发生磷叶立德的反面进攻和后续的Wittig反应.可根据加入醛基的不同得到不同构型的乙烯基碳苷.当加入含有吡啶基的醛时, 得到E构型的乙烯基碳苷; 加入不含有吡啶基的醛时, 则得到Z构型的乙烯基碳苷.
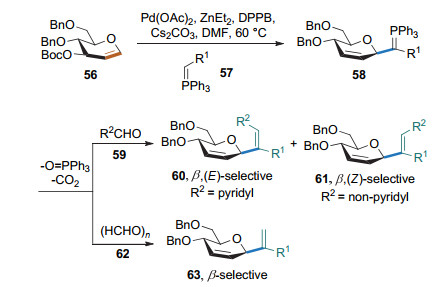
2019年, 张强课题组[26]以α, β-不饱和内酯和3, 4-碳酸酯保护的半乳糖烯为底物, 在Pd(OAc)2的催化下, 实现了α, β不饱和内酯的γ-位甲基与端基碳的偶联, 生成了2, 3-不饱和碳苷(Scheme 23).该反应是烯丙基钯络合物与α, β-不饱和内酯负离子的Ferrier Ⅰ型碳苷化反应.香豆素类化合物也可以作为该反应的底物.该反应得到的碳苷为β构型.
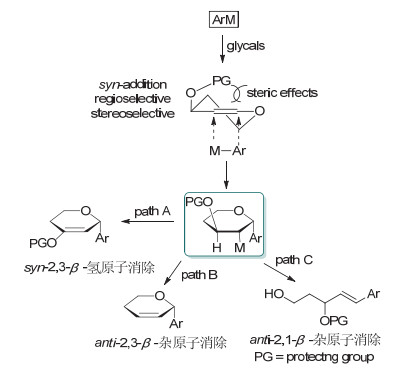
1968年, Heck[27]报道了卤代烃与碳碳双键在钯催化剂和碱的存在下进行偶联生成取代烯烃的反应, 被称为Heck反应. Heck反应已成为构筑C—C键最有效的方法之一.糖烯中的双键也能与M—Ar发生配位, 得到的中间体可继续发生β-氢消除或β-杂原子消除得到不同的碳苷产物, 这些反应被称之为Heck型碳苷化反应; 由于3位保护基的位阻效应, 一般都是生成α-构型的产物.在过去的五十年里, 化学家基于不同的偶联试剂, 发展了一系列Heck型碳苷化新方法(Scheme 24).
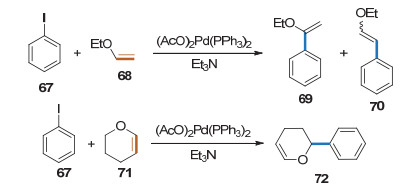
Heck型偶联反应在糖环上的应用是从简单烯醇醚开始的.早在1979年, Daves课题组[28]就对卤代芳烃和烯醇醚的Heck偶联反应进行了深入研究(Scheme 25).他们发现碘苯和乙基乙烯基醚发生偶联反应时得到1-乙氧基乙苯和两种构型的2-乙氧基乙苯, 而碘苯与二氢吡喃反应得到单一的苯基取代产物.
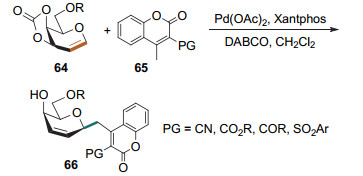
Heck型碳苷化反应最早用于一些具有特定生物活性的分子的合成, 硅基保护的呋喃型糖烯和吡喃型糖烯均可作为底物参与反应. 1990年, Daves课题组[29]利用碘代芳环衍生物和叔丁基二甲基硅烷基(TBS)保护葡萄糖烯的Heck型碳苷化反应成功合成了芳环类碳苷(Scheme 26a), 该类化合物具有抗菌活性. 1992年, Daves课题组[30]用5-碘脲醛和呋喃核糖烯作为底物进行Heck偶联反应(Scheme 26b), 得到的烯醇硅醚产物经脱硅和还原操作后得到2-脱氧假尿苷.该化合物具有艾滋病病毒(HIV)逆转录酶抑制活性.
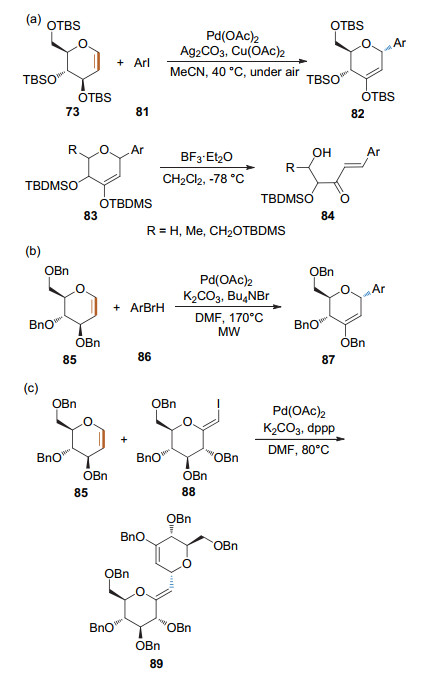
2009年, 本课题组和杨劲松课题组分别用芳基碘(Scheme 27a)[31]和芳基溴(Scheme 27b)[32]作为底物, 在Pd(OAc)2的催化下高效合成了α-芳基碳苷.其中, 当使用芳基溴作为底物时, 反应需要在微波照射条件下进行. 2019年, Savic课题组[33]利用本课题组的方法合成芳基碳苷, 并在此基础上发展了路易斯酸促进的开环反应, 立体选择性地合成了一些具有抗癌活性的α, β-不饱和酮. 2013年, 李英霞课题组[34]用碘乙烯糖底物与糖烯进行Heck偶联反应, 以较高的收率得到了碳苷假糖(Scheme 27c).
有机硼也可以作为Heck型碳苷化反应的底物. 2001年, Maddaford课题组[35a]以乙腈为溶剂, 在Pb- (OAc)2的催化下, 实现了全乙酰基保护的葡萄糖烯与苯硼酸的偶联反应, 以80%的收率得到了2, 3不饱和的α-苯基碳苷产物(Scheme 28).他们还以α, β-不饱和酮为反应物, 得到了酮式碳苷产物[35b].在此基础上, 2005年Figuera小组[36]观察到了使用芳基硼酸与全乙酰糖烯偶联反应时某些条件下会伴有anti-2, 1-β-杂原子消除的开环产物生成, 但未见可控生成开环产物的报道. 2020年, 姚辉课题组[37]利用White催化剂实现了3, 4-碳酸酯保护糖烯的anti-2, 3-β-杂原子消除反应, 得到了2, 3不饱和的α-苯基碳苷.
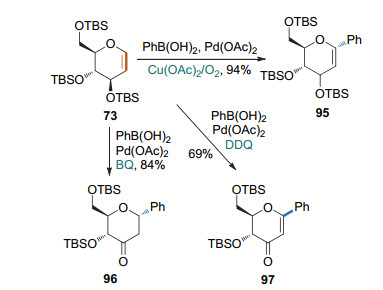
2009年, 本课题组[38]利用芳基硼酸与全TBS保护的1, 2-吡喃糖烯的氧化Heck偶联反应, 发展了一种syn-2, 3-β-氢原子消除反应制备芳基碳苷的方法(Scheme 29).该反应以醋酸钯为催化剂, 在乙腈溶剂中进行, 加入不同的氧化剂可实现催化剂的循环和产物的调控.以苯醌(BQ)为氧化剂时生成酮式碳苷96; 以Cu(OAc)2/O2为氧化剂时生成烯醇醚式碳苷95; 以2, 3-二氯-5, 6-二氰基-1, 4-苯醌(DDQ)为氧化剂时生成α, β-不饱和酮式碳苷97.该方法具有良好的立体选择性, 即苷元部分总是与3-位取代基在糖环上的朝向相反. 2018年, Dujardin和Collet课题组[39]进一步证实了该立体选择性规律. 2019年, Arisawa和Nimura课题组[40]利用本课题组的方法完成了Spliceostatin A碳苷类似物的合成.
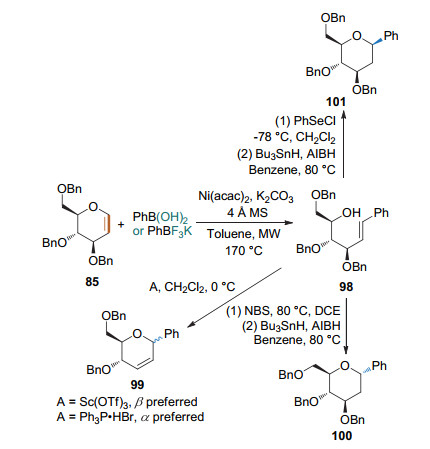
2014年, 本课题组[41]发现了可控的anti-2, 1-β-杂原子消除反应, 并利用开环-关环策略通过控制反应条件实现了2-脱氧碳苷的立体选择性合成(Scheme 30).糖烯和芳基硼酸在微波加热170 ℃条件下, 通过镍催化的Heck型偶联/anti-2, 1-β-杂原子消除反应得到开环产物; 该中间体在不同的条件下可以选择性关环得到不同的碳苷.当以Sc(OTf)3作为催化剂时得到以β构型为主的碳苷; 而以Ph3P•HBr为催化剂时则得到α构型为主的产物.通过NBS的关环脱溴可得到α构型的2-脱氧碳苷; 而利用PhSeCl的关环脱硒则得到β构型的2-脱氧碳苷.
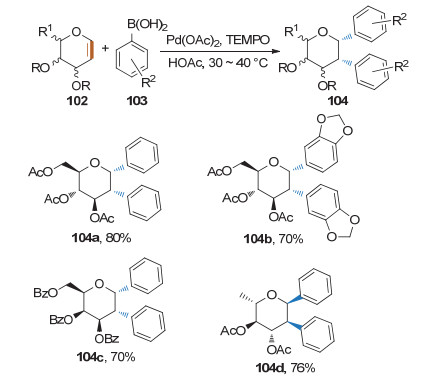
2015年, Mukherjee课题组[42]发现在2, 2, 6, 6-四甲基哌啶氮氧化物(TEMPO)存在下, 1 equiv.糖烯和2 equiv.硼酸在醋酸钯的催化下生成饱和的1, 2-双芳基碳苷.该方法对给电子硼酸和吸电子芳基硼酸都能耐受, 也能适用于其它糖型(Scheme 31).
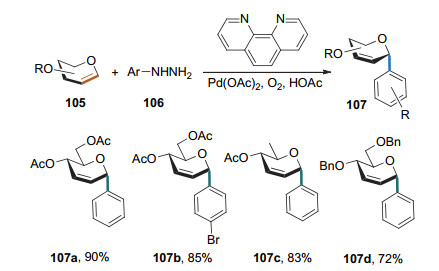
芳肼也可以作为偶联试剂. 2013年, 刘学伟课题组[43]发展了芳基肼与葡萄糖烯的偶联反应(Scheme 32).在Pd(OAc)2的催化和氧气的存在下, 该反应可高收率地实现2, 3-不饱和α-芳基碳苷的合成.他们还发现, 糖烯3位的取向不是1, 3反式立体选择性的唯一决定因素.因为, 将葡萄糖烯的3位异构化之后反应仍可顺利进行, 但得到的是α, β异构体的混合物, 且随着芳基取代基的不同立体异构比例稍有差异.
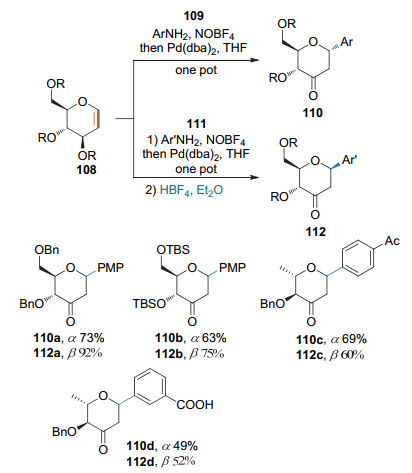
2018年, 本课题组[44]发展了一种以芳胺为偶联剂、立体可控的2-脱氧芳基碳苷的合成方法.用芳胺作为原料, 在NOBF4的作用下原位生成芳基重氮盐, 继而在Pd(dba)2的催化下与糖烯发生偶联反应, 生成α构型的3-羰基碳苷.如果在反应结束后加入四氟硼酸二乙醚溶液则得到β构型的产物.该方法从芳胺出发原位制备重氮盐, 比直接用重氮盐更加安全(Scheme 33).此外, 该方法仅通过简单条件的改变, 实现了立体选择性的调控, 操作简单、实用性高.该方法还可用于含有3-羰基碳苷骨架的天然产物的合成.
随后, Kandasamy课题组[45]也报道了在钯的催化下重氮盐和糖烯偶联生成α构型的酮式碳苷的反应. 2019年, 该课题组还从苯胺出发, 经亚硝酸叔丁酯/HBF4原位制得重氮盐, 然后根据3-位取代基取向的不同调控立体选择性.
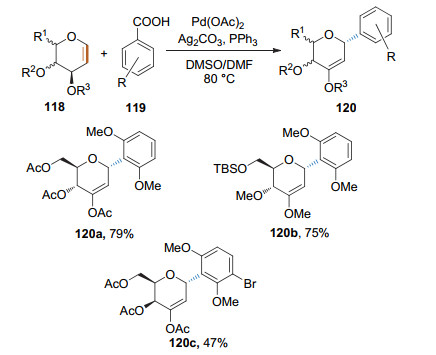
2011年, 刘学伟课题组将偶联试剂的范围扩大到了有机羧酸[46].他们以Pd(OAc)2/PPh3作为催化剂, Ag2CO3为氧化剂, 在二甲亚砜(DMSO)和DMF的混合溶液中实现了芳甲酸和葡萄糖烯的脱羧偶联反应, 得到了syn-2, 3-β-氢消除的2-脱氧-α-芳基糖苷(Scheme 34).该反应需要在加热条件下进行, 具有良好的选择性, 收率中等.
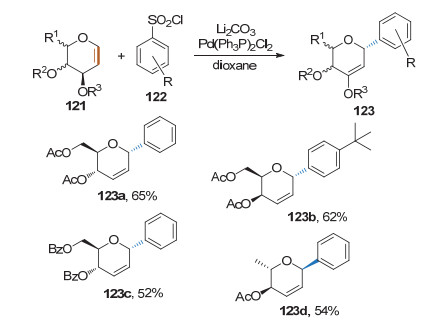
2015年, Mukherjee课题组[47]将偶联试剂的范围扩大至苯磺酰氯.糖烯和芳基磺酰氯在Pd(Ph3P)2Cl2的催化下, 经由anti-2, 3-β-杂原子消除反应, 生成2, 3-不饱和的芳基碳苷.产物均为α构型, 反应收率中等(Scheme 35).
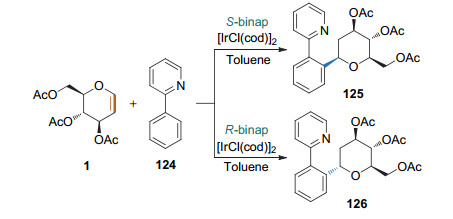
前述Heck类型碳苷化反应均通过改变偶联试剂中的离去基团来实现. 2019年, Nishimura课题组[48]在铱/ binap的催化下, 利用吡啶基团导向的碳氢键活化, 实现了2-苯基吡啶和糖烯的Heck类型碳苷化反应, 得到了饱和的2-脱氧碳苷(Scheme 36).更有趣的是, 不同的手性配体可实现立体选择性的调控. (S)-binap配体通常得到β-(为主)的碳苷.
除了Heck型碳苷化反应之外, 还可以在糖烯的1位引入活化、定位或极性反转基团, 使其更容易发生过渡金属催化的偶联反应, 生成1, 2-不饱和的碳苷.近二十年来, 化学家们发展了多种基于1-取代糖烯的碳苷化反应.
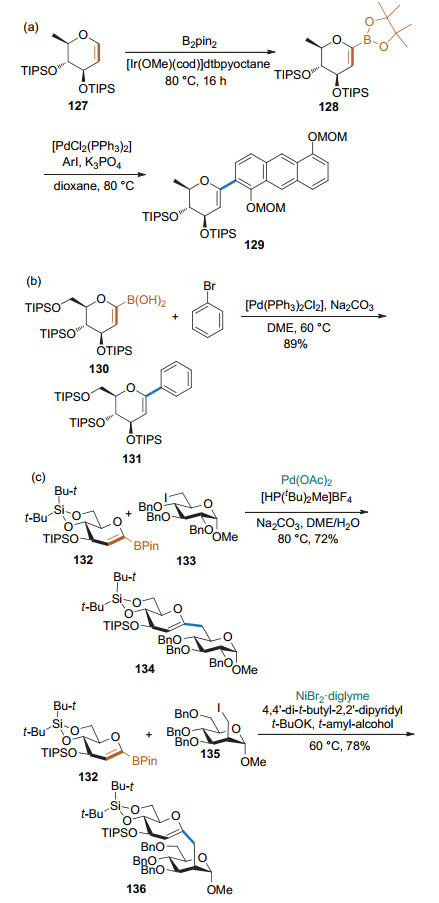
有机硼化合物是Suzuki反应的理想底物. Miyaura和Ishiyama课题组[49]发展了1-硼酸频那醇酯糖烯的制备方法, 并发现其可与芳基碘代物发生Suzuki-Miyaura偶联反应, 得到1, 2-不饱和碳苷(Scheme 37a). Parkan和Kotora[50]课题组进一步完善了1-硼酸频那醇酯糖烯与芳基/烯基碘或溴的偶联反应, 并将该反应拓展至1-硼酸糖烯(Scheme 37b).随后, Parkan课题组[51]用6-碘代糖代替溴代芳基作为反应底物进行反应, 实现了1-硼酸频那醇酯糖烯与sp3碳的偶联, 以较高的收率得到1→6连接的伪二糖(Scheme 37c).
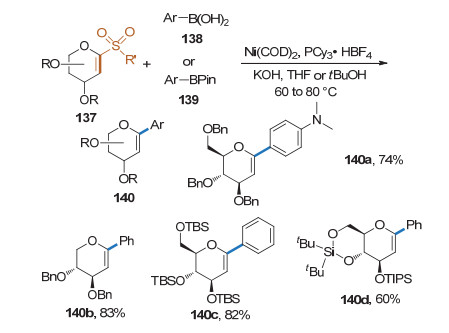
烯基砜也是理想的C—C键构筑前体. 2019年, 钮大文课题组[52]以1-砜代糖烯为糖基供体, 在镍催化剂的作用下与芳基硼酸发生Suzuki-Miyaura偶联反应, 得到了1, 2-不饱和的碳苷(Scheme 38).该反应有良好的底物适用性, 可用于伊格列净等药物或药物活性片段的合成.
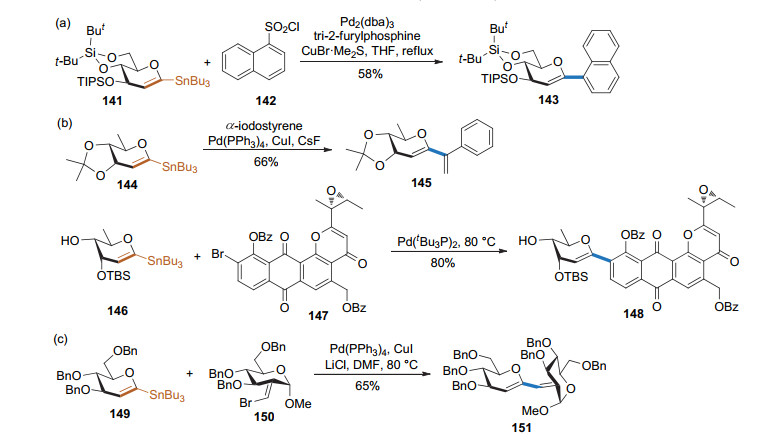
糖烯的1-位可以被锡取代, 进而通过Stille偶联反应制备碳苷. 2004年, Vogel课题组[53]发展了一种1-锡基取代的糖烯与芳基磺酰氯之间的Stille偶联反应(Scheme 39a), 得到了1, 2-不饱和的碳苷. 2005年, McDonald课题组[54]以1-锡取代的糖烯和α-碘代苯乙烯作为底物, 在Pd(PPh3)4/CuI/CsF的条件下进行Stille偶联反应, 得到嵌合1, 3-丁二烯的碳苷产物. Danishefsky课题组[55]用大位阻的芳基溴与端基被锡取代的糖烯进行Stille偶联, 得到芳基碳苷产物(Scheme 39b). 2013年, Werz课题组[56]以1-锡基取代的糖烯和含烯基溴取代的葡萄糖为底物, 在Pd(PPh3)4/CuI条件下进行Stille偶联反应, 以较高的收率得到1→2, 1→3, 1→4和1→6连接的伪二糖(Scheme 39c), 为碳连接的寡糖合成提供了通用的方法.
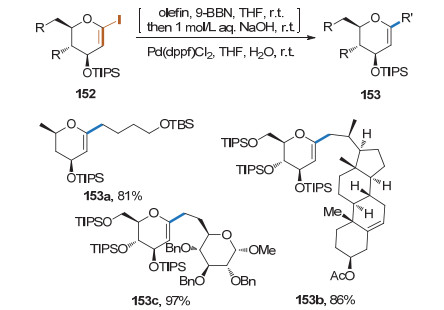
碘作为一种很好的离去基团也可以活化糖烯. 2004年, Tan课题组[57]利用烯烃和9-BBN原位生成的硼烷与1-碘糖烯的Suzuki偶联反应, 在Pd(dppf)Cl2的催化下, 实现了1, 2-不饱和烷基碳苷的合成(Scheme 40).该反应还可以结合羰基插入反应, 得到酰基碳苷.
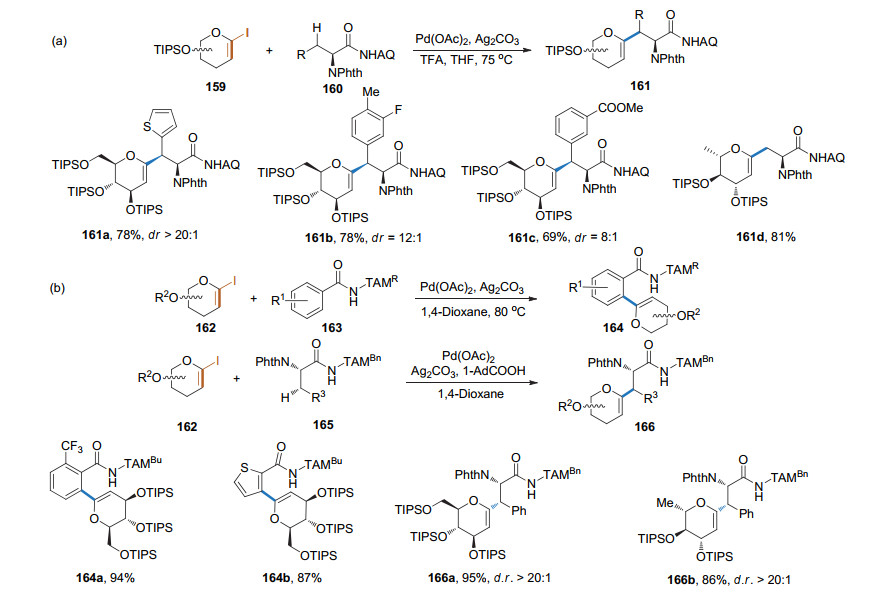
2016年, 本课题组[58]以端基碘取代的糖烯和N-喹啉苯甲酰胺类化合物为底物, Pd(OAc)2为催化剂和氨基酸为配体, 经由8-氨基喹啉(AQ)导向的碳氢键活化和偶联, 以较高的收率得到了1, 2-不饱和的芳基碳苷, 并通过简单的化学转化实现了螺环碳苷骨架和β-碳苷的合成. 2017年, 本课题组[59]以CuI和Pd(OAc)2为催化剂, 通过无导向基团的碳氢键活化方式, 在糖烯的1-位引入了杂环芳烃, 为杂环芳烃碳苷的合成提供了新的途径(Scheme 41).
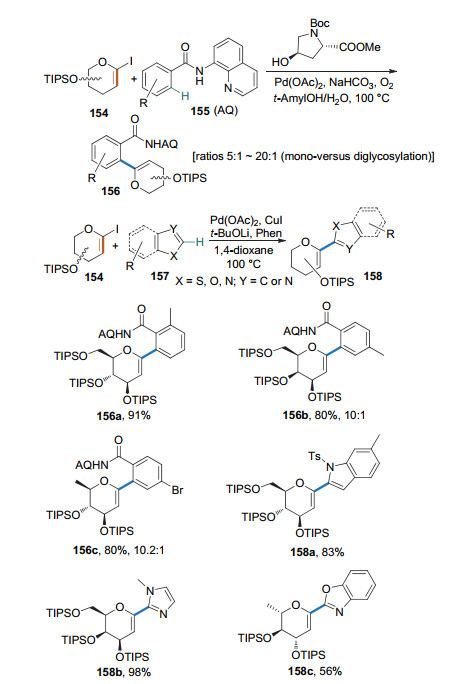
2019年, 柳红和李英霞课题组[60]以8-氨基喹啉(AQ)作为导向基团, 实现了氨基酸β-位sp3碳的碳氢键活化, 进而与1-碘糖烯进行偶联反应, 得到1, 2-不饱和的糖氨基酸(Scheme 42a). 2020年, Ackermann课题组[61]以三唑基二甲基甲基(triazolyldimethylmethyl, TAM)作为导向基团, 完成了类似的反应(Scheme 42b).这些方法有可能用于多肽的后修饰.
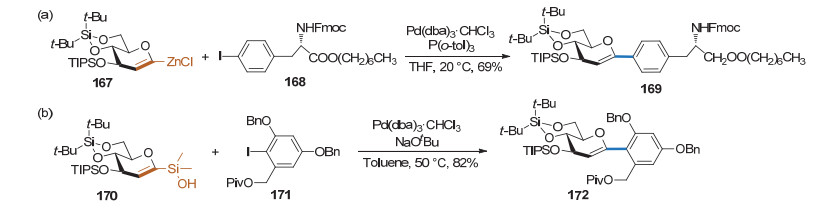
在糖烯合成碳苷的反应中, 1-锌代的糖烯也可以作为偶联试剂参与反应. 2003年, Auge课题组[62]利用1-锌代的糖烯和卤代芳烃进行Negishi偶联反应得到碳苷产物(Scheme 43a). 1-硅代的糖烯也可作为偶联试剂参与Hiyama反应. 2012年, Pieters课题组[63]用1-硅代的糖烯和卤代芳烃作为底物, 在Pd(dba)3催化下发生偶联得到1, 2-不饱和的芳基碳苷产物(Scheme 43b).
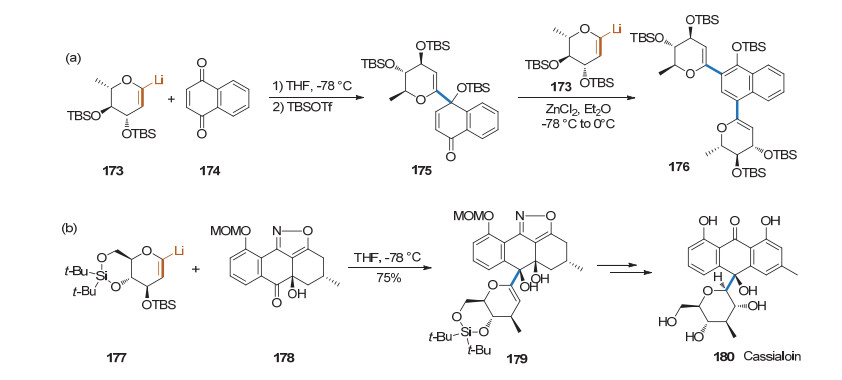
前述糖烯都是作为亲电试剂参与反应, 而端基用金属取代的糖烯也可以作为亲核试剂参与反应. 1994年, Parker课题组[64]利用1-锂代的糖烯, 通过糖与酮羰基的亲核加成反应, 实现了萘醌的双糖基化(Scheme 44a). 2008年, Suzuki课题组[65]以1-锂代的葡萄糖烯和异恶唑酮作为底物, 完成了决明子碳苷的合成(Scheme 44b).
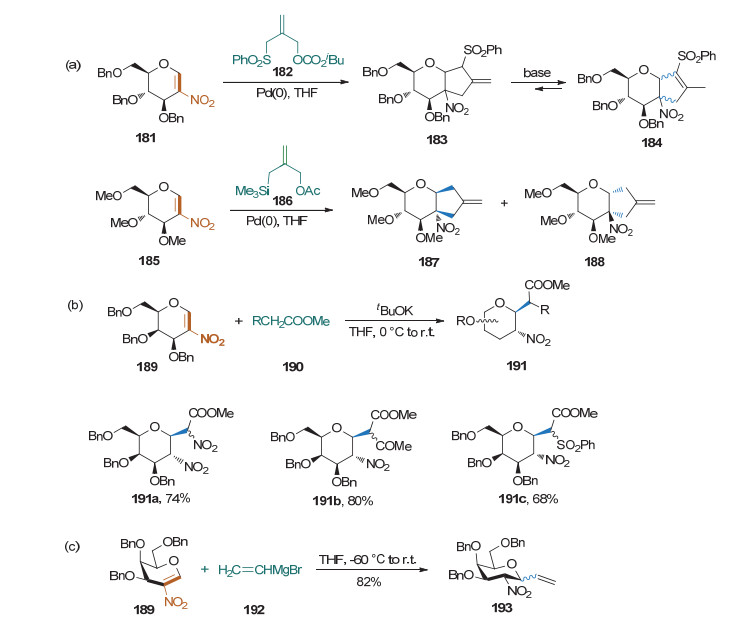
当糖烯的2-位有硝基存在时, 糖烯的双键和硝基构成了α, β-不饱和体系, 该体系可以在碱性条件下和碳负离子进行Michael加成反应, 生成相应的碳苷. 1996年Holzapfel课题组[66]利用烯丙负离子与2-硝基糖烯的Michael加成反应, 并结合烯丙基钯化学, 发展了含1, 2-并环结构的碳苷合成方法.该方法的立体选择性不是很好, 得到β-构型为主的产物(Scheme 45a). 2002年, Schmidt和Vankar课题组[67]以叔丁醇钾为碱, 实现了2-硝基半乳糖烯和丙二酸二甲酯等含活泼亚甲基底物的Michael加成反应, 以较高收率得到β构型为主的碳苷(Scheme 45b). 2005年, Vankar课题组[68]实现了乙烯基溴化镁等格式试剂底物的Michael加成反应, 并用该方法完成了一些具有糖苷酶抑制活性的杂糖的合成(Scheme 45c)[69].
2010年, 俞初一课题组[70]将底物扩展到了烯氨酯, 完成了一系列烯氨酯类化合物与2-硝基葡萄糖烯在碱性条件下的Michael加成反应(Scheme 46a). 2012年, 刘学伟课题组[71]利用NHC催化剂对醛的极性反转效应, 实现了醛与2-硝基糖烯的偶联反应.该方法有良好的β立体选择性(Scheme 46b). 2014年, Pannecoucke课题组[72]实现了有机锂与2-硝基糖烯的Michael加成反应, 得到β型的碳苷产物(Scheme 46c).随后, Pannecoucke课题组[73]和Ruijter课题组[74]分别实现了含氟的硅试剂和锂试剂与2-硝基糖烯的Michael加成反应, 得到了氟代碳苷(Scheme 46d).
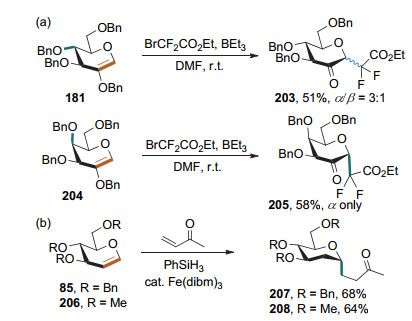
理论上, 碳自由基也可以对糖烯进行加成[75].但碳自由基通常会加成到糖烯的2-位.当糖烯的2-位有取代基的时候, 碳自由基可加成到端位, 生成碳苷. Leclerc课题组[76]以BEt3作为自由基引发剂, 使溴代物裂解得到CF2CO2Et和CF2Br自由基, 从而与2-苄氧基糖烯发生自由基加成反应, 生成相应的氟代碳苷.当用葡萄糖烯作为底物时, 得到α:β=3:1的混合物; 而用半乳糖烯作为底物时可得到α构型的产物(Scheme 47a). Baran课题组[77]利用铁催化剂, 在苯基硅烷的存在下, 通过氢自由基对葡萄糖烯2-位的加成使其形成糖自由基, 继而对丙烯酸甲酯进行自由基加成, 得到α-2-脱氧碳苷(Scheme 47b).
碳苷具有良好的生物活性和成药性, 一直是糖化学家和药物化学家研究的热点, 其独特的C—C糖苷键是化学合成的一个挑战.随着有机化学的飞速发展, 基于烯烃的C—C键生成方法也被逐渐引入到碳苷合成.本文从Ferrier Ⅰ型碳苷化反应、Heck偶联型碳苷化反应、1-取代糖烯的过渡金属催化偶联碳苷化反应、2-取代糖烯的Michael加成型和自由基加成型碳苷化反应等方面系统地总结了基于糖烯的碳苷合成方法和策略.从糖烯出发制备碳苷已成为碳苷合成最有效的策略之一, 极大地加速了碳苷天然产物及类似物的生物活性研究.尽管该研究领域已取得较大进展, 但仍存在进一步研究和发展的空间.首先, 碳苷通常有两种构型, 但现有大部分方法只能得到混合物或少部分方法能得到某单一构型的碳苷, 能通过配体或简单条件改变而实现立体选择性调控的体系仍然很少, 目前仅限于3-酮-2-脱氧芳基碳苷以及2-吡啶苯基碳苷的合成等极少数例子.因此发展立体可控的碳苷合成方法是该领域未来发展的主要方向之一.其次, 利用当前方法合成的多为非天然碳苷结构, 要得到天然碳苷结构通常需要进行多步化学转化; 大部分方法只适用于简单底物, 对于更加复杂的天然苷元的引入缺乏有效的途径.随着有机化学的发展和碳苷化学研究的深入, 相信未来会发展出更加实用和好用的合成新方法, 并被成功运用于碳苷天然产物合成和新药研发领域.
Štambaský, J.; Hocek, M.; Kočovský, P. Chem. Rev. 2009, 109, 6729. doi: 10.1021/cr9002165
Cao, X.; Tian, Y.; Zhang, T.; Li, X.; Ito, Y. J. Chromatogr. A 1999, 855, 709. doi: 10.1016/S0021-9673(99)00715-3
(a) Funahashi, Y.; Kawamura, N.; Ishimaru, T. JP 08231551, 1996.
(b) Funahashi, Y.; Kawamura, N.; Ishimaru, T. JP 08231552, 1996.
Franck, R. W. Angew. Chem., Int. Ed. 2004, 43, 3818. doi: 10.1002/anie.200454215
(a) Kitamura, K.; Ando, Y.; Matsumoto, T.; Suzuki, K. Chem. Rev. 2018, 118, 1495.
(b) Yang, Y.; Yu, B. Chem. Rev. 2017, 117, 12281.
(a) Gómez, A. M.; Lobo, F.; Uriel, C.; López, J. C. Eur. J. Org. Chem. 2013, 7221.
(b) Vieira, A. S.; Fiorante, P. F.; Hough, T. L. S.; Ferreira, F. P.; Ludtke, D. S.; Stefani, H. A. Org. Lett. 2008, 10, 5215.
(c) Huang, N.; Liao, H.; Yao, H.; Xie, T.; Zhang, S.; Zou, K.; Liu, X. W. Org. Lett. 2018, 20, 16.
(a) Takhi, M.; Rahman, A. H. A.; Schmidt, R. R. Tetrahedron Lett. 2001, 42, 4053.
(b) Anjaiah, S.; Chandrasekhar, S.; Grée, R. J. Mol. Catal. A: Chem. 2004, 214, 133.
(a) Saeeng, R.; Sirion, U.; Sahakitpichan, P.; Isobe, M. Tetrahedron Lett. 2003, 44, 6211.
(b) Yadav, J. S.; Reddy, B. V. S.; Rao, C. V.; Chand, P. K.; Prasad, A. R. Synlett 2001, 1638.
Das, S. K.; Reddy, K. A.; Abbineni, C.; Roy, J.; Rao, K. V. L. N.; Sachwani, R. H.; Iqbal, J. Tetrahedron Lett. 2003, 44, 4507. doi: 10.1016/S0040-4039(03)01012-8
Ansari, A. A.; Reddy, Y. S.; Vankar, Y. D. Beilstein J. Org. Chem. 2014, 10, 300. doi: 10.3762/bjoc.10.27
Steinhuebel, D. P.; Fleming, J. J.; Bois, J. D. J. Org. Lett. 2002, 4, 293. doi: 10.1021/ol010273e
Tatina, M. B.; Kusunuru, A. K.; Yousuf, S. K.; Mukherjee, D. Org Biomol. Chem. 2014, 12, 7900. doi: 10.1039/C4OB01405G
(a) Di Bussolo, V.; Caselli, M.; Pineschi, M.; Crotti, P. Org. Lett. 2003, 5, 2173.
(b) Bussolo, V. D.; Caselli, M.; Romano, M. R.; Pineschi, M.; Crotti, P. J. Org. Chem. 2004, 69, 7383.
Deelertpaiboon, P.; Reutrakul, V.; Jarussophon, S.; Tuchinda, P.; Kuhakarn, C.; Pohmakotr, M. Tetrahedron Lett. 2009, 50, 6233. doi: 10.1016/j.tetlet.2009.09.036
Lubin-Germain, N.; Hallonet, A.; Huguenot, F.; Palmier, S.; Uziel, J.; Augé, J. Org. Lett. 2007, 9, 3679. doi: 10.1021/ol701480x
Vieira, A. S.; Fiorante, P. F.; Hough, T. L.; Ferreira, F. P.; Lüdtke, D. S.; Stefani, H. A. Org. Lett. 2008, 10, 5215. doi: 10.1021/ol8022177
Kusunuru, A. K.; Tatina, M.; Yousuf, S. K.; Mukherjee, D. Chem Commun. 2013, 49, 10154. doi: 10.1039/c3cc44250k
Hosseyni, S.; Smith, C. A.; Shi, X. Org. Lett. 2016, 18, 6336. doi: 10.1021/acs.orglett.6b03228
Devari, S.; Kumar, M.; Deshidi, R.; Rizvi, M.; Shah, B. A. Beilstein J. Org. Chem. 2014, 10, 2649. doi: 10.3762/bjoc.10.277
Chen, H.; Luo, X.; Qiu, S.; Sun, W.; Zhang, J. Glycoconjugate J. 2017, 34, 13. doi: 10.1007/s10719-016-9718-7
Tan, H. Y.; Xiang, S.; Leng, W. L.; Liu, X.-W. RSC Adv. 2014, 4, 34816. doi: 10.1039/C4RA07429G
Dash, A. K.; Madhubabu, T.; Yousuf, S. K.; Raina, S.; Mukherjee, D. Carbohydr. Res. 2017, 438, 1. doi: 10.1016/j.carres.2016.11.018
Yadav, J. S.; Reddy, B. V. S.; Rao, K. V.; Saritha Raj, K.; Prasad, A. R.; Kiran Kumar, S.; Kunwar, A. C.; Jayaprakash, P.; Jagannath, B. Angew. Chem. Int. Ed. 2003, 115, 5356. doi: 10.1002/ange.200351267
Reddy, G. M.; Maheswara Rao, B. U.; Sridhar, P. R. J. Org. Chem. 2016, 81, 2782. doi: 10.1021/acs.joc.5b02879
(a) Moineau, C.; Bolitt, V.; Sinou, D. J. Org. Chem. 1998, 63, 582.
(b) Bertini, B.; Moineau, C.; Sinou, D.; Gesekus, G.; Vill, V. Eur. J. Org. Chem. 2001, 2001, 375.
(c) Zeng, J.; Ma, J.; Xiang, S.; Cai, S.; Liu, X.-W. Angew. Chem. Int. Ed. 2013, 125, 5238.
(d) Bai, Y.; Leng, W. L.; Li, Y.; Liu, X.-W. Chem. Commun. 2014, 50, 13391.
(e) Leng, W.-L.; Liao, H.; Yao, H.; Ang, Z.-E.; Xiang, S.; Liu, X.-W. Org. Lett. 2017, 19, 416.
Dai, Y.; Tian, B.; Chen, H.; Zhang, Q. ACS Catal. 2019, 9, 2909. doi: 10.1021/acscatal.9b00336
Heck, R. F. J. Am. Chem. Soc. 1968, 90, 5518. doi: 10.1021/ja01022a034
Arai, I.; Daves, G. D. J. Org. Chem. 1979, 44, 21. doi: 10.1021/jo01315a005
Farr, R. N.; Outten, R. A.; Cheng, J. C.-Y.; Daves, G. D., Jr. Organometallics 1990, 9, 3151. doi: 10.1021/om00162a029
Zhang, H.; Daves, G. D., Jr. J. Org. Chem. 1992, 57, 4690. doi: 10.1021/jo00043a029
Li, H.-H.; Ye, X.-S. Org. Biomol. Chem. 2009, 7, 3855. doi: 10.1039/b909248j
Lei, M.; Gao, L.; Yang, J.-S. Tetrahedron Lett. 2009, 50, 5135. doi: 10.1016/j.tetlet.2009.06.116
Jovanovic, P.; Petkovic, M.; Simic, M.; Jovanovic, M.; Tasic, G.; Crnogorac, M. D.; Zizak, Z.; Savic, V. Eur. J. Org. Chem. 2019, 2019, 4701. doi: 10.1002/ejoc.201900672
Tao, Y.; Ding, N.; Ren, S.; Li, Y. Tetrahedron Lett. 2013, 54, 6101. doi: 10.1016/j.tetlet.2013.08.118
(a) Rammauth, J.; Poulin, O.; Rakhit, S.; Maddaford, S. P. Org. Lett. 2001, 3, 2013.
(b) Ramnauth, J.; Poulin, O.; Bratovanov, S. S.; Rakhit, S.; Maddaford, S. P. Org. Lett. 2001, 3, 2571.
Figuera, N.; Forns, P.; Fernandez, J. C.; Fiol, S.; Fernandez-Forner, D.; Albericia, F. Tetrahedron Lett. 2005, 46, 7271. doi: 10.1016/j.tetlet.2005.07.128
Lai, M.; Othman, K. A.; Yao, H.; Wang, Q.; Feng, Y.; Huang, N.; Liu, M.; Zou, K. Org. Lett. 2020, 22, 1144. doi: 10.1021/acs.orglett.9b04665
Xiong, D.-C.; Zhang, L.-H.; Ye, X.-S. Org. Lett. 2009, 11, 1709. doi: 10.1021/ol900273d
Mabit, T.; Siard, A.; Legros, F.; Guillarme, S.; Martel, A.; Lebreton, J.; Carreaux, F.; Dujardin, G.; Collet, S. Chem.-Eur. J. 2018, 24, 14069. doi: 10.1002/chem.201803674
Yoshikawa, Y.; Ishibashi, A.; Murai, K.; Kaneda, Y.; Nimura, K.; Arisawa, M. Tetrahedron Lett. 2019, 60, 151313. doi: 10.1016/j.tetlet.2019.151313
Liu, C.-F.; Xiong, D.-C.; Ye, X.-S. J. Org. Chem. 2014, 79, 4676. doi: 10.1021/jo500730y
Kusunuru, A. K.; Jaladanki, C. K.; Tatina, M. B.; Bharatam, P. V.; Mukherjee, D. Org. Lett. 2015, 17, 3742. doi: 10.1021/acs.orglett.5b01722
Bai, Y.; Kim, L. M. H.; Liao, H.; Liu, X.-W. J. Org. Chem. 2013, 78, 8821. doi: 10.1021/jo401032r
(a) Tang, S.; Zheng, Q.; Xiong, D.-C.; Jiang, S.; Li, Q.; Ye, X.-S. Org. Lett. 2018, 20, 3079.
(b) Xiong, D.-C.; Gao, C.; Li, W.-M.; Wang, Y.; Li, Q.; Ye, X.-S. Org. Chem. Front. 2014, 1, 798.
(c) Liu, M.; Li, B.-H.; Li, T.; Liu, M.; Xiong, D.-C.; Ye, X.-S. Org. Biomol. Chem. 2020, 18, 3043.
(d) Zheng, Q.; Tang, S.; Xiong, D.-C.; Li, Q.; Ye, X.-S. J. Org. Chem. 2020, 85, 9339.
(a) Singh, A. K.; Kandasamy, J. Org. Biomol. Chem. 2018, 16, 5107.
(b) Singh, A. K.; Venkatesh, R.; Kandasamy, J. Synthesis 2019, 51, 4215.
Xiang, S.; Cai, S.; Zeng, S.; Liu, X.-W. Org. Lett. 2011, 13, 4608. doi: 10.1021/ol201820m
Kusunuru, A. K.; Yousuf, S. K.; Tatina, M.; Mukherjee, D. Eur. J. Org. Chem. 2015, 2015, 459. doi: 10.1002/ejoc.201403195
Sakamoto K.; Nagai M.; Ebe Y.; Yorimitsu H.; Nishimura k. ACS Catal. 2019, 9, 1347. doi: 10.1021/acscatal.8b04686
Kikuchi, T.; Takagi, J.; Isou, H.; Ishiyama, T.; Miyaura, N. Chem. Asian J. 2008, 3, 2082. doi: 10.1002/asia.200800157
Parkan, K.; Pohl, R.; Kotora, M. Chem.-Eur. J. 2014, 20, 4414. doi: 10.1002/chem.201304304
Oroszova, B.; Choutka, J.; Pohl, R.; Parkan, K. Chem.-Eur. J. 2015, 21, 7043. doi: 10.1002/chem.201406591
Gong, L.; Sun, H.-B.; Deng, L.-F.; Zhang, X.; Liu, J.; Yang, S.; Niu, D. J. Am. Chem. Soc. 2019, 141, 7680. doi: 10.1021/jacs.9b02312
Dubbaka, S. R.; Steunenberg, P.; Vogel, P. Synlett 2004, 1235.
Koo, B.; E. McDonald, F. Org. Lett. 2005, 7, 3621. doi: 10.1021/ol050975u
Hartung, J.; Wright, B. J. D.; Danishefsky, S. J. Chem.-Eur. J. 2014, 20, 8731. doi: 10.1002/chem.201402254
Koester, D. C.; Kriemen, E.; Werz, D. B. Angew. Chem. Int. Ed. 2013, 52, 2985. doi: 10.1002/anie.201209697
Potuzak, J. S.; Tan, D. S. Tetrahedron Lett. 2004, 45, 1797. doi: 10.1016/j.tetlet.2003.12.006
(a) Liu, M.; Niu, Y.-H.; Wu, Y.-F.; Ye, X.-S. Org. Lett. 2016, 18, 1836.
(b) Wang, H.; Niu, Y.-H.; Zhang, G.; Ye, X.-S. Tetrahedron Lett. 2016, 57, 4544.
Zhang, S.; Niu, Y.-H.; Ye, X.-S. Org. Lett. 2017, 19, 3608. doi: 10.1021/acs.orglett.7b01583
Liu, Y.; Wang, Y.; Dai, W.; Huang, W.; Li, Y.; Liu, H. Angew. Chem. Int. Ed. 2020, 59, 3491. doi: 10.1002/anie.201914184
Wu, J.; Kaplaneris, N.; Ni, S.; Kaltenhauser, F.; Ackermann, L. Chem. Sci. 2020, 11, 6521. doi: 10.1039/D0SC01260B
Boucard, V.; Larrieu, K.; Lubingermain, N.; Uziel, J.; Augé, J. Synlett 2003, 1834.
Marjolein, V. D. K.; Eefjan, B.; Pieters, R. Beilstein J. Org. Chem. 2012, 8, 732. doi: 10.3762/bjoc.8.82
Parker, K. A.; Koh, Y. H. J. Am. Chem. Soc. 1994, 116, 11149. doi: 10.1021/ja00103a037
Yasuhito, K.; Ryo, Y.; Keisuke, S. Angew. Chem. Int. Ed. 2008, 120, 1100. doi: 10.1002/ange.200704625
Holzapfel, C. W.; Merwe, T. L. V. D. Tetrahedron Lett. 1996, 37, 2307. doi: 10.1016/0040-4039(96)00250-X
Pachamuthu, K.; Gupta, A.; Das, J.; Schmidt, R. R.; Vankar, Y. D. Eur. J. Org. Chem. 2002, 1479.
Reddy, B. G.; Vankar, Y. D. Angew. Chem. Int. Ed. 2005, 44, 2001. doi: 10.1002/anie.200462413
Jayakanthan, K.; Vankar, Y. D. Tetrahedron Lett. 2006, 47, 8667. doi: 10.1016/j.tetlet.2006.10.024
Zhang, T.; Yu, C.-Y.; Huang, Z.-T.; Jia, Y.-M. Synlett 2010, 2174.
Vedachalam, S.; Shi, M. T.; Hui, P. T.; Cai, S.; Liu, X.-W. Org. Lett. 2012, 14, 174. doi: 10.1021/ol202959y
(a) Delaunay, S.; Poisson, T.; Jubault, P.; Pannecoucke, X. Eur. J. Org. Chem. 2014, 3341.
(b) Verma, A. K.; Chennaiah, A.; Dubbu, S.; Vankar, Y. D. Carbohydr. Res. 2019, 473, 57
Delaunay, T.; Poisson, T.; Jubault, P.; Pannecoucke, X. J. Fluorine. Chem. 2015, 171, 56. doi: 10.1016/j.jfluchem.2014.10.001
Bouwman, S.; Orru, R. V. A.; Ruijter, E. Org. Biomol. Chem. 2015, 13, 1317. doi: 10.1039/C4OB02317J
(a) Lopez, J. C.; Fraser-Reid, B. J. Am. Chem. Soc. 1989, 111, 3450.
(b) Gomez, A. M.; Casillas, M.; Valverde, S.; Lopez, J. C. Tetrahedron: Asymmetry 2001, 12, 2175.
(c) Li, G.; Xiong, D.-C.; Ye, X.-S. Synlett 2001, 2410.
(a) Moreno, B.; Quehen, C.; Rose-Helene, M.; Leclerc, E.; Quirion, J.-C. Org. Lett. 2007, 9, 2477.
(b) Colombel, S.; Van Hijfte, N.; Poisson, T.; Leclerc, E.; Pannecoucke, X. Chem.-Eur. J. 2013, 19, 12778.
(a) Lo, J. C.; Gui, J.; Yabe, Y.; Pan, C.-M.; Baran, P. S. Nature 2014, 516, 343.
(b) Lo, J. C.; Kim, D.; Pan, C.-M.; Edwards, J. T.; Yabe, Y.; Gui, J.; Qin, T.; Gutiérrez, S.; Giacoboni, J.; Smith, M. W.; Holland, P. L.; Baran, P. S. J. Am. Chem. Soc. 2017, 139, 2484.

图式 8 烷基/芳基锌作为Ferrier Ⅰ型反应的亲核试剂
Scheme 8 Alkylzinc/arylzinc as the nucleophile for Ferrier Ⅰ reaction

图式 9 炔基锌作为Ferrier Ⅰ型反应的亲核试剂
Scheme 9 Alkynylzinc as the nucleophile for Ferrier Ⅰ reaction

图式 10 有机锂作为Ferrier Ⅰ型反应的亲核试剂
Scheme 10 Organolithium as the nucleophile for Ferrier Ⅰ reaction

图式 11 三烷基铝作为Ferrier Ⅰ型反应的亲核试剂
Scheme 11 Trialkyl aluminum as the nucleophile for Ferrier Ⅰ reaction

图式 12 炔基铟作为Ferrier Ⅰ型反应的亲核试剂
Scheme 12 Alkynyl indium as the nucleophile for Ferrier Ⅰ reaction

图式 13 炔基三氟硼酸钾作为Ferrier Ⅰ型反应的亲核试剂
Scheme 13 Potassium alkynyltrifluoroborate as the nucleophile for Ferrier Ⅰ reaction

图式 14 炔基铜作为Ferrier Ⅰ型反应的亲核试剂
Scheme 14 Copper(Ⅰ) alkyne as the nucleophile for Ferrier Ⅰ reaction

图式 15 炔基金作为Ferrier Ⅰ型反应的亲核试剂
Scheme 15 Gold alkyne as the nucleophile for Ferrier Ⅰ reaction

图式 16 炔基三甲基硅烷作为Ferrier Ⅰ型反应的亲核试剂
Scheme 16 Alkynyltrimethylsilane as the nucleophile for Ferrier Ⅰ reaction

图式 17 β-酮酸作为Ferrier Ⅰ型反应的亲核试剂
Scheme 17 β-Keto acid as the nucleophile for Ferrier Ⅰ reaction

图式 18 芳基乙酮作为Ferrier Ⅰ型反应的亲核试剂
Scheme 18 Aryl ethyl ketone as the nucleophile for Ferrier Ⅰ reaction

图式 22 膦叶立德作为Ferrier Ⅰ型反应的亲核试剂
Scheme 22 Ylide as the nucleophile for Ferrier Ⅰ reaction

图式 23 α, β不饱和内酯作为Ferrier Ⅰ型反应的亲核试剂
Scheme 23 α, β-Unsaturated lactone as the nucleophile for Ferrier Ⅰ reaction

图式 28 硼酸参与的Heck偶联反应和1, 4-加成反应
Scheme 28 Heck coupling reaction and 1, 4-addition reaction with arylboronic acid

图式 30 开环-关环策略合成碳苷
Scheme 30 "Ring opening-ring closure" strategy for the synthesis of C-glycoside

图式 41 1-碘糖烯和芳烃的碳苷化反应
Scheme 41 C-Glycosylation of 1-iodoglycals with aromatic hydrocarbon

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:





























