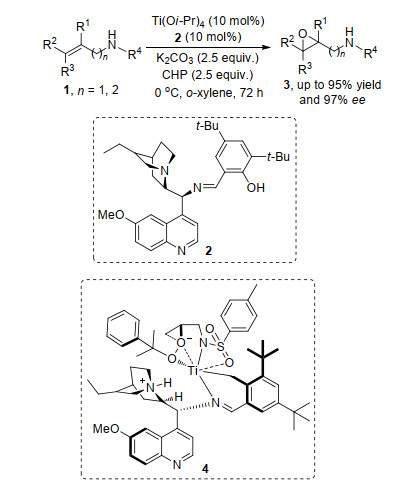
图式 1.
Ti催化烯基酰胺的不对称环氧化反应
Scheme 1.
Ti-catalyzed asymmetric epoxidation of enamide

不对称氧化反应(Asymmetric oxidation reaction)是指有机化合物分子在引入电负性原子过程中产生手性中心的一类反应, 是不对称合成的重要分支之一.在催化剂和氧化剂参与下, 底物分子氧化数升高, 通常表现为脱氢或者杂原子官能团的定向引入, 实现底物分子的不对称性官能团富集化过程, 为手性分子合成提供最直接的途径, 在天然产物和药物分子的合成方面显得尤为重要[1].
在不对称氧化反应中, 对映选择性控制是难点之一, 而过渡金属与配体的协同作用为难点的解决提供了一种有效途径.过渡金属和配体的协同作用过程中, 金属促使反应发生, 而配体决定产物的对映选择性.例如Sharpless报道的催化剂——钛和酒石酸的络合物在实现烯丙醇类化合物的不对称环氧化反应中, 配体酒石酸的构型改变氧原子的进攻方向, 调控产物的空间构型[2].烯烃的不对称氧化反应[3-4]的实现, 为不对称合成开辟出一条新路径, 为精准合成提供更多可能的策略.该方法的成功报道, 促进了该领域的发展.人们不断开发新的配体和中心过渡金属的种类, 发展新型不对称氧化反应, 引入光化学等途径在更深层次研究不对称氧化过程, 从而促使不对称氧化反应朝着更加高效和更好对映选择性的方向发展, 形成具有绿色、精准、实用等特征的不对称氧化反应合成方法学研究体系.
本文将重点介绍近年来利用热化学或者光化学的过渡金属介入的不对称氧化反应及其合成方法学研究, 包括过渡金属参与的烯烃不对称环氧化反应、C—H键不对称氧化反应、不对称BV氧化反应和硫醚不对称氧化反应等几个方面.
烯烃是重要的大宗基础化工原料, 其不对称氧化产物作为化学工业、材料工业和制药工业的重要前体, 是现代合成工业不可或缺的物质基础.本部分将主要介绍过渡金属参与的烯烃不对称氧化反应, 包括烯烃的不对称氧化Heck反应、不对称Wacker氧化反应、不对称双官能团化反应及不对称环氧化反应等.
烯烃分为官能团化和非官能团化烯烃, 在官能团化烯烃的对映选择性的环氧化过程中, 官能团发挥着重要的作用, 它与催化剂形成中间体, 控制反应的发生和产物的对映选择性. 2016年, 何炜等[5]报道了烯基磺酰胺的不对称环氧化反应, 反应中底物烯基磺酰胺1与Ti和配体2络合形成中间体4, 是该反应产物对映选择性的决定步.通过钛络合物催化剂与底物官能团形成中间体实现手性中心的构筑, 证明了催化剂与底物官能团作用这种因素在手性中心构筑过程中的重要作用(Scheme 1).
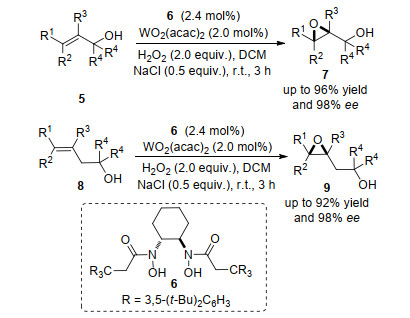
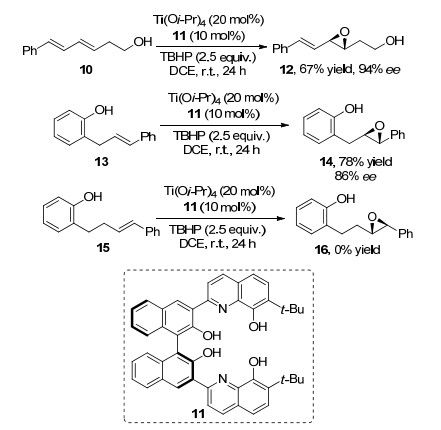
2014年, Yamamoto等[6]报道了多取代烯基醇类化合物5和配体6的不对称环氧化反应, 反应中金属钨和配体6的作用经过了与催化模型4类似的过程, 不同的是该方法中不涉及R4基团的络合作用.值得注意的是, 该方法使用双氧水为氧化剂, 室温条件下仅3 h就能高效得到环氧化产物(最高达96%产率和98% ee) (Scheme 2).该方法是通过中心金属与底物的氧原子和氧化剂中氧原子的孤电子对络合形成中间体, 该中间体的空间取向决定着氧原子插入双键的方向, 从而实现产物的对映选择性.由上述研究报道可知, 酰胺基或者羟基与烯基双键间隔一个或者两个碳原子的底物均可以得到对应的环氧化产物, 两者之间的空间距离对反应的进行和产物对映选择性较大的影响, 这在Yamamoto等[7]报道的钛酸酯与轴手性配体催化的这类反应中, 得到了很好的证实.多烯基醇或者烯基取代苯酚中双键的环氧化反应取决于羟基的位置, 随着双键与羟基或者酚羟基距离延长, 环氧化产物消失, 很好地证明了羟基与作用部分的距离在反应中的作用(Scheme 3).
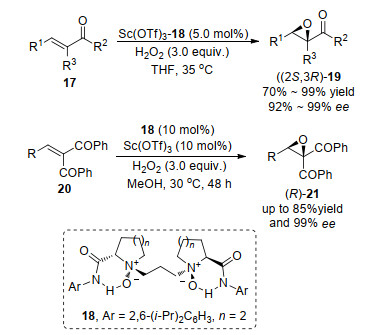
官能团化烯烃的不对称环氧化反应的成功实现, 促进了此类烯烃底物的应用, 一系列合成方法得以发展[1].近几年, 冯小明课题组[8]利用自己课题发展的手性氮氧配体, 制备了一系列手性N, N'-二氧化物-Sc(Ⅲ)催化剂, 实现了α, β-不饱和羰基化合物17或者20的不对称环氧化反应.在作者提出的催化模型中, Sc分别和配体、底物中羰基的氧原子络合, 在氧化剂的氧原子与中心金属键合作用下, 完成了对映选择性的环氧化过程.该方法对于α, β-不饱和单羰基或者双羰基化合物的不对称环氧化反应, 均得到了很好的反应结果.底物拓展实验中, 化合物17中的R1可以拓展为强吸电子基团(酯基、三氟甲基和三氯甲基)或者大空间位阻基团, 其中三氟甲基(或者三氯甲基)具有较大的药物应用意义, 利用化合物20的α-双羰基官能团特性, 可以进一步拓展其官能团化反应(Scheme 4).
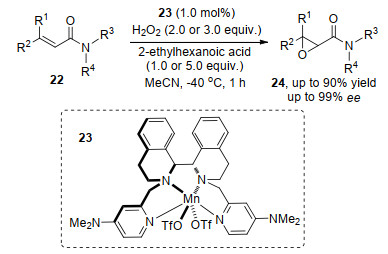
在冯小明课题组[8]和何炜课题组[5]的研究工作中, 羰基氧原子和胺基氮原子的配位作用是产物对映选择性的决定性因素, 同时此类基团也影响着反应的效果. 2019年, Clarasó等[9]利用Mn(Salan)催化剂23和氧化剂, 实现了β, β-二取代酰胺22的对映选择性环氧化反应, 将胺基换成酯基, 产物的选择性和收率明显下降, 证明了胺基对实现高对映选择性的重要性(Scheme 5).
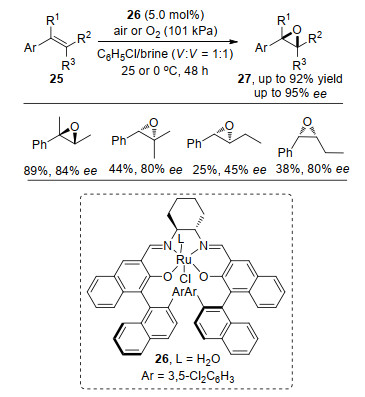
非官能团化的烯烃不对称环氧化反应的难点在于中间体空间取向的控制, Mn(Salen)催化剂在非官能团化烯的不对称环氧化反应中的应用很好地解决了这个的问题, 但是反应对于烯烃的不同构型也存在着差异.如顺式(反式)构型烯烃在Mn(Salen)催化剂的作用下, 得到的环氧化产物的对映选择性较好, 但反式(顺式)的烯烃在同等反应条件下其环氧化对映选择性一般, 导致这种现象的原因是烯烃取代基之一与Salen配体相互作用, 使氧进入烯烃的所需取向不稳定造成的[10].在Koya等[11]报道中, 利用Ru(Salen)配合物26作为催化剂, 实现了芳基烯烃25的高对映选择性环氧化反应, 顺式和反式烯烃均能得到目标产物, 但是受取代基的影响比较大.随着R2和R3的碳链延长, 反应产率均明显降低, 且顺式烯烃的对映选择性更好; 在三取代的烯烃的反应中, R1基团引入取代基有利于反应的发生(Scheme 6).
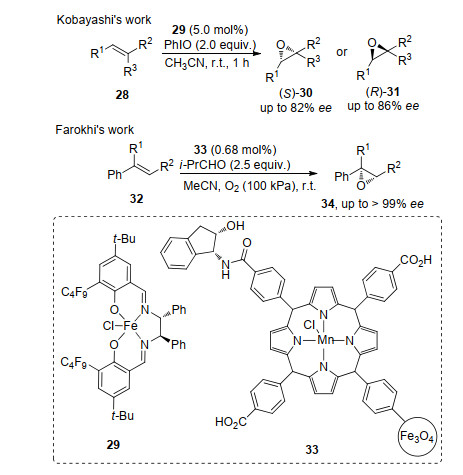
在三取代烯烃的反应中, 受取代基的影响, 实现其环氧化产物的对映选择性存在较大限制. Kobayashi等[12]利用以三价Fe作为中心金属、手性Salen为配体的络合物29实现了烯烃28的不对称环氧化反应.在该Fe(Salen)催化体系中, 通过改变手性配体的骨架, 得到了单一构型的对映体, 整个反应对E式的烯烃表现出更好的对映选择性.而Farokhi等[13]报道的Mn卟啉催化剂33在芳基烯烃32的环氧化反应上表现出了极高的对映选择性(最高>99% ee)(Scheme 7).
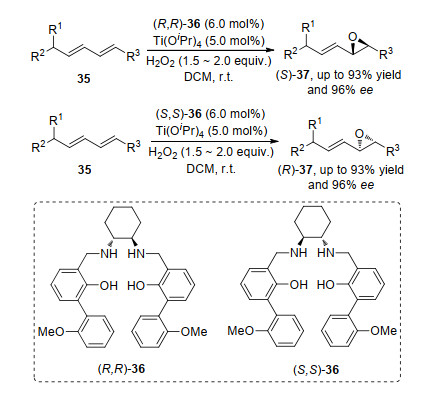
共轭烯烃的环氧化反应一般是单个双键被氧化, 反应位点取决于双键的空间位阻效应, 空间位阻较小的优先反应.如Jat等[14]在钛和Salan 36的络合物催化剂和双氧水氧化剂作用下, 实现了共轭二烯35的选择性单环氧化反应.反应中, 位阻较小的碳碳双键优先反应.在底物实验中, 发现通过改变配体36的构型, 可以高对映选择性地得到单一构型的非对映异构体, 这也表明底物原手性中心对产物最终构型没有影响(Scheme 8).
由此可见, 在官能团化烯烃中, 反应中催化剂与官能团络合形成中间体, 控制氧原子的进攻方向从而控制其产物的手性; 而在非官能团烯烃中, 最为常见的是催化剂与底物π键络合, 形成具有一定空间取向的中间体, 从而控制氧原子的进攻方向.在已有的报道中, 取代基团的空间效应对反应有比较大的影响, 通过取代基团的修饰可以改善不对称氧化反应的选择性, 并将反应拓展到多取代烯烃底物.
烯烃不对称双羟基化和羟胺化反应推进了烯烃氧化双官能化反应的应用[4], 近年来, 过渡金属催化体系的不断开发, 促进了不对称氧化双官能化的极大发展.
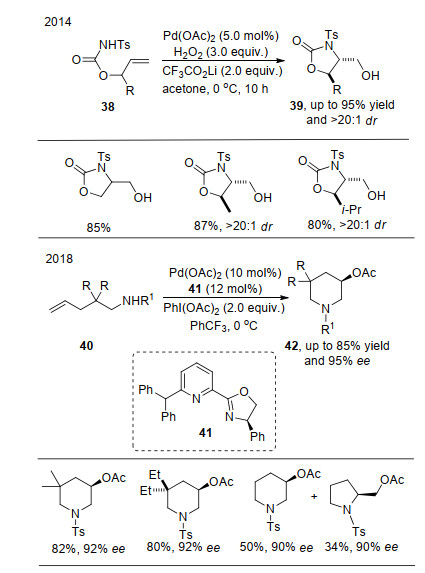
在烯烃的不对称环氧化反应中, 官能团化的烯烃易于发生此类反应, 官能团的存在不仅会影响烯烃中双键的活性, 也会在与催化剂作用时控制产物的手性, 这种影响因素在烯烃的不对称氧化双官能团化反应中也同样存在. 2014年, 刘国生课题组[15]报道了烯醇类化合物38的分子内氧化胺化反应.该反应借助钯催化剂与底物官能团的作用, 在氧化剂的作用下, 以高的产率和对映选择性得到多种烷基醇.该反应中, 首先钯催化剂与N原子以及双键络合, 随后进行迁移插入形成烷基钯, 烷基钯物种经过一步氧化后变成四价钯, 最后经历H2O2对烷基C—Pd键的氧化裂解, H2O对氧化裂解的碳进行亲核进攻得到目标产物.值得注意的是R基团是构筑产物手性中心的重要因素, 产物的非对映选择性是通过反应中环状中间体直接形成的, 并未涉及外加手性诱导因素, 去掉R基团非对映选择性就不能实现.随后, 他们[16]将反应拓展到烷基烯胺40的分子内不对称氧化胺化反应, 该反应在钯络合物催化剂和氧化剂作用下, 得到了很好的反应结果, R基团空间位阻效应可以控制产物中环的大小, 但是对产物的映选择性影响不大(Scheme 9).
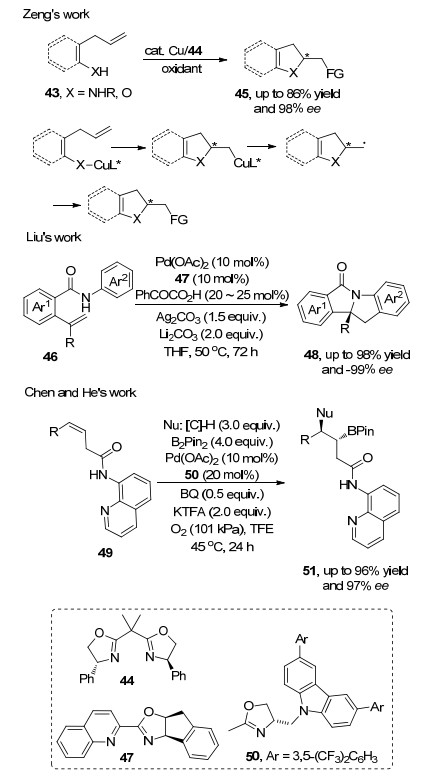
Chemler等[17]首次报道了胺铜化、氧铜化启动的烯烃不对称氧化双官能团化反应.该方法的发现促进了金属参与的烯烃双官能化不对称氧化反应的研究. 2015年, 曾伟课题组[18]利用铜和二氧化锰的氧化体系实现了烯烃43的分子内不对称双胺化反应, 产物ee值高达95%以上.反应涉及铜的迁移插入和碳铜键均裂, 最后经过高价铜的还原消除得到产物, 该方法为手性单环二胺45的合成提供了一个有效的途径. 2017年, 刘国生课题组[19]在烯烃分子内不对称氧化胺化反应的研究基础上, 发展了钯和手性配体喹啉-噁唑啉47催化体系, 在氧化剂Ag2CO3的作用下, 烯烃46发生分子内不对称环氧化胺化反应, 得到了多种二氢吲哚48, 反应具有高产率和优异的对映选择性.酮酸在反应过程中与催化剂络合参与反应, 故而酮酸的加入可加速反应和提高产物对映选择性, 不足的是反应时间较长(基本都在72 h左右).烯烃的官能团可以参与反应构建新化学键, 也可以仅作为辅助基团以活化特定位置的化学键.如在陈弓和何刚课题组[20]报道的烯烃双官能化反应中, 辅助基团喹啉是构筑其分子内叔碳和季碳中心的对映选择性的关键, 但不参与产物的成键.该方法以钯和手性配体50的络合物为催化剂, 在氧化剂的作用下, 可以得到较高的反应产率和对映选择性.在作者给出的机理中, 钯催化剂与底物形成络合物, 随后进行迁移插入形成烷基钯中间体, 接着中间体与硼酸进行转金属化反应, 最后还原消除得到产物51.底物实验中, 在众多亲核反应底物中, N-酰亚胺类底物在该反应中有高的收率和对映选择性(Scheme 10).
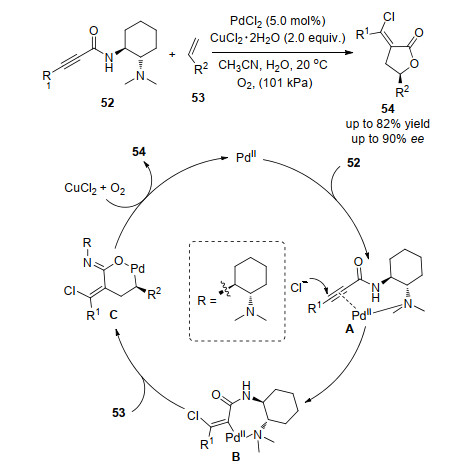
上述烯烃的双官能团化反应中, 烯烃都有一个共同的特点, 均借助分子内辅助基团对双键进行活化, 然后实现其分子内双键双官能团化反应, 对于分子间的反应, 具有辅助基团的底物是反应发生的关键.在我们课题组[21]报道的烯烃53与炔酰胺52的分子间不对称氧化环化反应中, 胺基作为辅助基团既是构筑手性中心的关键, 又是反应发生的关键.该反应在温和的反应条件下, 以高的收率和对映选择性得到系列产物.反应中, 首先, 二价钯和炔的叁键络合形成中间体A, 使得碳碳叁键活化, 氯离子反向进攻活化的叁键形成乙烯基钯中间体B (B空间效应影响决定产物的非对映选择性); 随后B完成对烯烃迁移插入得到中间体C, C接着水解和还原消除得到目标产物, 催化剂经过氧化再生, 完成催化循环(Scheme 11).
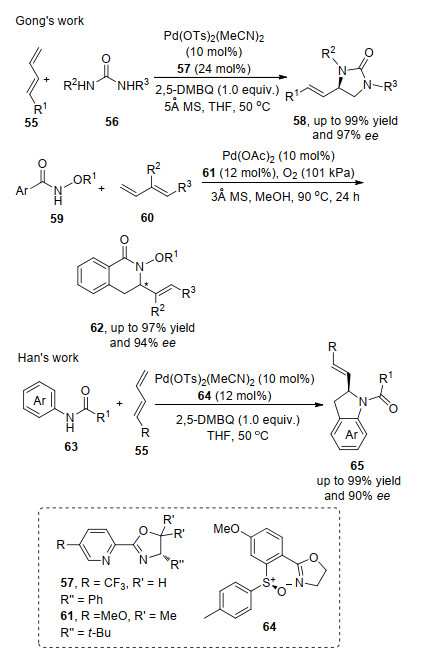
2017年, 韩志勇课题组[22c]报道了芳基脲63与共轭二烯55的胺化环化反应, 一步合成了吲哚啉并在其位实现了手性中心的构筑; 2018年, 龚流柱课题组[22a]报道了二烯与脲在钯盐和手性吡啶噁唑啉配体的络合物的催化作用下, 实现了共轭二烯55的3号位双键的不对称胺化反应.反应中与配体络合物和端烯双键络合脲中, 氮原子进攻活化双键完成催化剂的迁移插入形成烯丙基钯, 最后烯丙基钯还原消除得到目标产物; 随后, 龚流柱课题组[22b]在芳基脲的研究基础上, 把反应拓展到烷氧基芳基酰胺.首次实现了共轭二烯60与其的不对称胺化反应.该反应中钯和配体络合物催化剂与形成中间体芳基钯物种, 然后芳基钯物种对双键进行迁移插入形成烯丙基钯, 最后通过烯丙基钯的还原消除, 实现目标产物62的合成(Scheme 12).
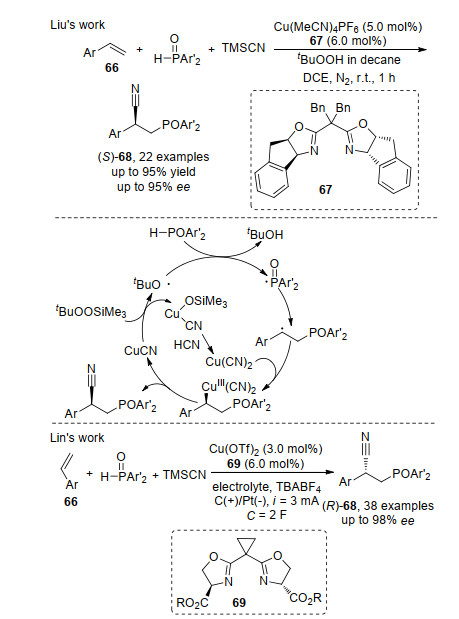
2019年, 刘国生课题组[23]在该课题组发展的铜催化的研究基础上, 首次实现了芳基烯烃66的不对称膦氰基化反应.作者认为该反应首先是一价铜与叔丁基过氧化物作用, 得到二价铜以及叔丁氧基自由基; 叔丁氧基自由基与亲核试剂亚磷酸进行单电子转移过程(SET)传递过程, 得到的相应膦自由基随后对烯烃进行自由基加成生成苄基自由基; 接着, 二价铜捕获苄基自由基得到三价铜物种; 最后三价铜物种还原消除得到目标产物(S)-68.同年, Lin等[24]发展了铜催化的电化学氧化烯烃的不对称膦氰基化反应.作者利用电化学合成方法, 在铜和N-位酯基的双噁唑啉配体69作用下, 以高的反应对映选择性得到(R)-68产物.通过实验证明二价CuCN物种是膦自由基产生的关键(Scheme 13).
Heck反应是烯烃反应中的一个经典反应类型, 氧化Heck反应为烯烃官能团化提供更多可能路径, 因此烯烃不对称氧化反应中, 不对称氧化Heck反应的研究也成为研究热点.
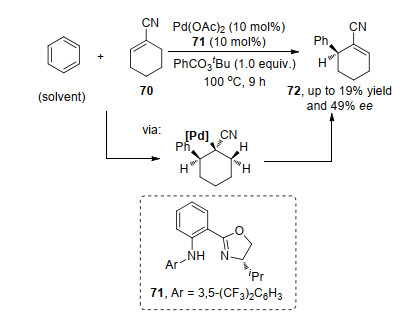
Mikami课题组[25]报道了首例不对称氧化Heck反应, 以醋酸钯为催化剂, 手性磺酰胺-噁唑啉71为配体, 在过氧苯甲酸的存在下得到中等ee值.反应中首先通过钯催化苯环的碳-氢键活化, 得到的芳基钯物种迁移插入到烯烃, 随后β-氢消除得到目标产物.产物的手性中心构筑是由1, 2-迁移插入决定的(Scheme 14).
随后, Oestreich课题组[26]发展了吲哚的2号位的碳-氢键活化与分子内烯烃的氧化Heck反应.反应利用醋酸钯和配体74的络合物为催化剂和对苯醌为氧化剂, 可以得到良好的反应收率及中等ee值(Scheme 15). 2010年该课题组将分子内烯烃片段引入吲哚的3号位, 通过相似的反应条件得到相应的手性环化产物.
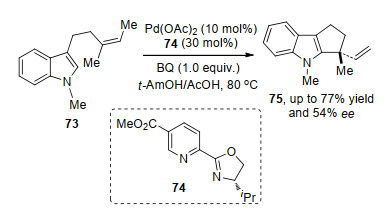
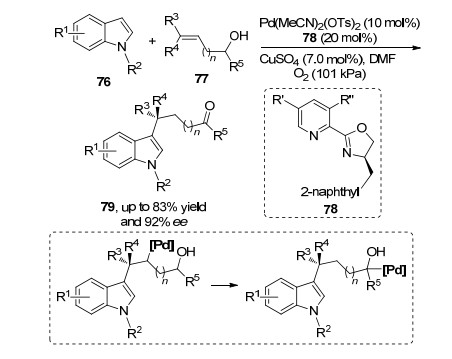
2015年, Sigman等[27]应用该课题组发展起来的不对称Heck反应体系, 成功地实现了吲哚76的3位碳氢键与多取代烯烃77的分子间氧化Heck反应.通过碳氢键的活化与迁移插入得到的烷基钯中间体, 经过链迁移得到羟基α-位烷基钯物种, 随后中间体钯不断进行插入与反插入, 最后发生β-氢消除得到相应的产物酮79 (Scheme 16).
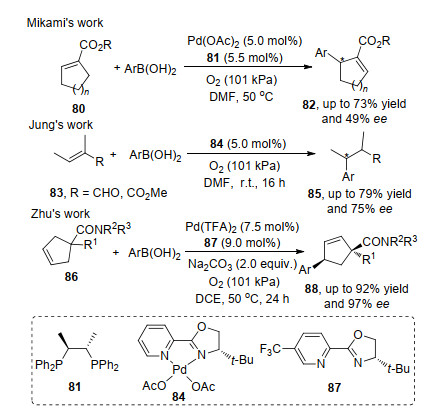
硼酸因其特殊性质从而在不对称氧化Heck反应中得到系列研究和应用. Mikami课题组[28]发展了硼酸与α, β-不饱和环烯羧酸酯80的2号碳的不对称氧化Heck反应.反应中, 首先是钯催化剂和硼酸的转金属化反应, 接着进行双键的迁移插入形成烷基钯物种, 最后通过β-氢消除得到产物82.随后, Jung课题组[29]将此类型的反应拓展到多取代链状α, β-不饱和类化合物83, 在系列的底物实验中, 均能够得到较好的ee值.值得注意的是, 该反应最后没有涉及β-氢消除的过程, 而是经过烷基钯的还原消除得到目标产物. 2020年, 祝介平课题组[30]将此类反应应用到α, β-不饱和环烯羧酸脂86的4号碳的不对称氧化Heck反应, 反应历程与上述反应基本相似, 不同的是酰胺的羰基氧与钯催化剂发生配位, 从而实现产物的动态动力学拆分过程(Scheme 17).
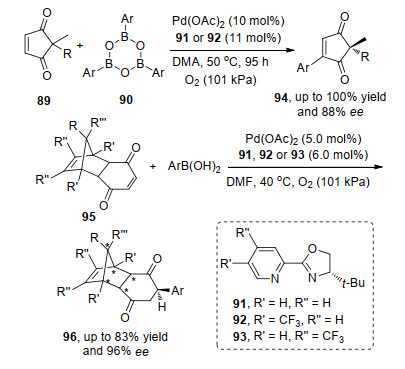
对于共轭洗酮类化合物, Lee等[31a]在2015年首次报道了环烯二酮的不对称氧化Heck反应.在钯催化剂和氧气条件下, 以高产率和对映选择性得到产物.随后,他们[31b]将反应底物发展到刚性结构的烯酮底物中,与之不同的是, 在硼酸的转金属化和对烯烃双键的迁移插入完成后, 最后没有进行反插入过程, 而是通过还原消除得到产物; 值得注意的是在构筑手性叔碳中心的同时实现了刚性结构中五个碳原子的非对映选择性(Scheme 18).
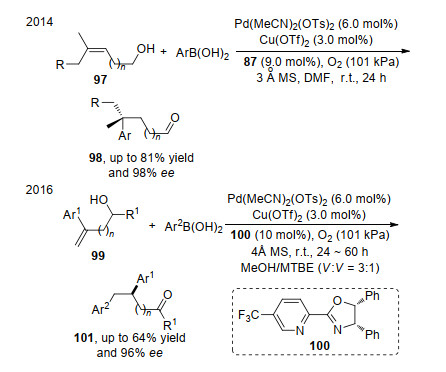
在早些时候, Sigman课题组[32a]利用多取代的烯醇化合物97为底物, 在钯和手性吡啶噁唑啉配体87的络合物作用下, 实现了其与硼酸的分子间不对称氧化Heck反应.反应通过硼酸和钯催化剂的转金属化启动, 然后对双键迁移插入形成烷基钯, 随后经过中间体钯的反复插入和反插入, 最后通过β-氢消除完成目标产物 98的合成. 2016年, 该课题组[32b]在上述研究基础上发展了端基芳基烯醇99的氧化Heck反应, 以高度的对映选择性得到一系列产物, 反应机理同前面反应类似, 通过反复的插入、反插入和β-氢消除得到手性酮类化合物101 (Scheme 19).
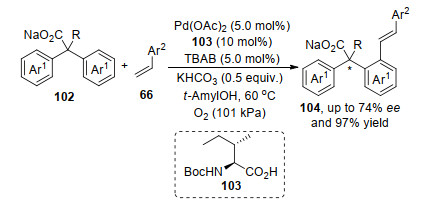
Yu等[33]报道的二芳基乙酸102, 在钯催化剂和氧化剂作用下, 实现了其与端烯的不对称氧化Heck反应.反应首先是钯络合物促进邻位苯基C—H活化, 随后催化剂钯络合物与底物羧酸形成中间体钯物种, 然后该钯物种对烯烃双键进行迁移插入得到烷基钯物种, 最后反插入得到目标产物104 (Scheme 20).
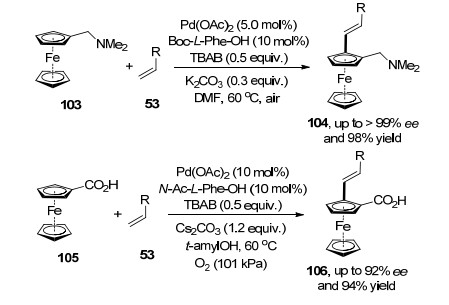
2013年, 吴养洁和崔秀灵课题组[34a]报道了二茂铁103与端烯53在钯催化剂和氧化剂的作用下的不对称氧化Heck反应.该方法通过底物中环戊烯中氨基官能团来实现其邻位C—H活化, 从而促进反应的发生.在底物实验中, 带吸电子取代基的烯烃和芳基烯烃均可以得到较高的产率和ee值, 含吸电子取代基的不饱和烯酸脂的反应效果优于芳基烯烃.随后, 他们[34b]把二茂铁取代官能团发展到羧酸官能团底物105, 在醋酸钯和氧气的作用下, 以极高的收率和对映选择性得到一系列目标产物.底物实验表明, 基团效应对于该方法影响较大, 芳基烯烃反应产率明显低于不饱和烯酸脂, 原因是苯环的空间效应不利于烯丙基钯物种对双键迁移插入(Scheme 21).
Heck反应不仅可以实现面手性分子的构筑, 也同样可以实现轴手性分子的构筑.如2016年, 游书力课题组[35]报道了Rh络合物催化的构筑轴手性分子的氧化Heck反应.在催化剂和氧化剂的作用下, 多数底物以优异的对映选择性得到产物.反应中首先钯催化剂促进C—H活化, 后与底物中氮原子络合, 随后对烯烃双键进行迁移插入得到烷基铑中间体, 最后烷基铑通过反插入得到产物(Scheme 22).
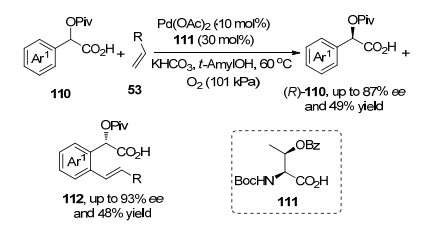
不对称氧化Heck反应在化合物动态拆分中可以取得很好的拆分效果. 2016年, Yu课题组[33b]利用芳基乙酸110的Heck反应, 实现了其动力学拆分.在醋酸钯、手性氨基酸配体和氧气作用下, 分子中单一构型(S)-110发生Heck反应, 以高的对映选择拆分出另一种构型的(R)-110分子(Scheme 23).
近年来, 金属催化的不对称Aza-Wacker氧化反应取得了一系列的进展, 成为构建手性含氮杂环化合物一类重要合成策略.
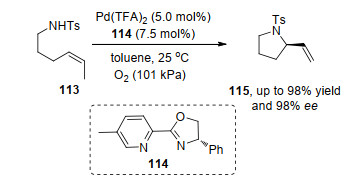
自首例不对称Aza-Wacker氧化反应被报道以来, 经过近年来的发展, 开发出了一系列高效的合成路径.在McDonald等[36]报道的烯烃113的对映选择性分子内氧化酰胺化反应中, 利用Pd和手性吡啶噁唑啉114络合物为催化剂, 能够得到中等及以上产率和较高的对映选择性.反应通过钯催化剂与底物的双键和氨基氮原子络合, 然后发生迁移插入过程形成烷基钯物种, 最后通过β-氢消除得到产物115 (Scheme 24).
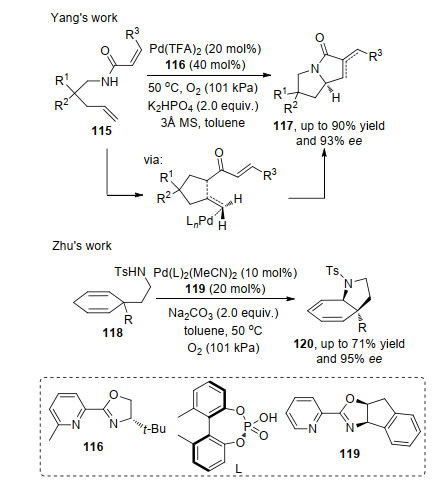
随着研究的深入, 不对称Aza-Wacker氧化反应从简单的烯胺底物拓展到复杂或者多氮杂烯胺的研究.杨丹课题组[37]以双烯115分子为底物, 在Pd和手性吡啶噁唑啉116络合物催化剂和氧化剂的作用下, 实现了手性双环产物的构建.作者提出的催化模型, 很好地解释了控制其对映选择性的方式.从机理来看, 反应是通过钯催化剂与底物中π键和氮原子络合, 然后对双键进行迁移插入形成烷基钯物种, 最后通过二次的迁移插入和还原消除得到目标产物117.随后, 祝介平课题组[38]发展了2, 5-环己二烯胺118的不对称Wacker氧化反应, 反应在钯络合物催化剂和氧气条件下, 得到中等的产率以及高的ee值, 而且在构建了新的手性中心同时得到高的非对映选择性(Scheme 25).
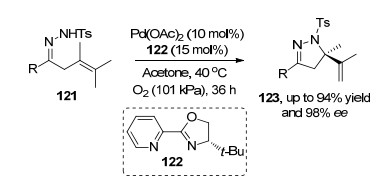
2018年, 张万斌课题组[39]报道了腙类化合物121的不对称Wacker氧化反应, 该反应在钯和手性吡啶噁唑啉122络合物催化剂作用下, 通过对分子内双键迁移插入得到的烷基钯, 最后发生β-氢消除得到具有光化学活性的吡唑啉123.该反应成功地将此类反应应用于多氮杂环的产物合成(Scheme 26).
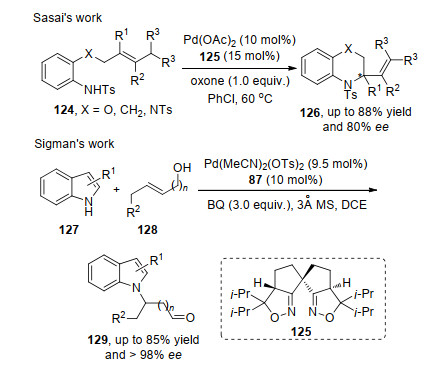
2018年, Sasai等[40]报道了钯与手性氮氧配体, 在氧化剂的作用下实现不对称Wacker氧化反应.该反应机理同样是钯络合物催化剂与底物络合后完成迁移插入产生烷基钯物种, 最后通过β-H消除得到产物. 2019年, Sigman等[41]借助其发展的不对称氧化Heck催化反应体系, 利用吲哚N—H键的活性, 发展了吲哚1号位的不对称Wacker反应.该反应利用钯和手性吡啶噁唑啉络合物为催化剂, 在氧化剂的作用下, 系列产物具有极高的对映选择性.底物实验中, 吲哚3位为取代手性氨基酸类底物会降低反应效果, 而3号位换成同碳数胺类取代基则有利于产物对映选择性的提高(Scheme 27).
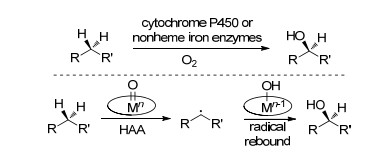
碳-氢键的直接不对称官能团化为手性官能团的引入提供了最直接与有效的途径.近年来碳-氢官能团化领域得到了极大的发展, 但是相应的C(sp3)—H的不对称官能团化反应仍极具挑战[42].然而, 天然生物催化剂, 如细胞色素P450和非血红素铁酶在C(sp3)—H键的高区域与对映选择性官能团化中已经显示良好的活性[43](Scheme 28).
在这些过程中, 常见的机理是在氧化剂的作用下形成的金属-氧物种对烷基碳氢键发生氢原子攫取, 得到相应的烷基自由基与价态降低一价的羟基金属中间体, 最后经过自由基的重新组合得到相应的目标产物.在酶催化的激励下, 化学家们设计了一系列仿生催化剂, 如卟啉和四配位胺基吡啶类金属络合物(Mn、Fe等)[44].这里将不进行展开讨论.
在C(sp2)—H键的反应中, 辅助基团实现分子内C—H键活化是重要合成策略, 底物分子内C—H键活化后, 金属催化剂与底物中辅助官能团作用得到金属中间体, 然后通过转金属化和还原消除完成产物的生成[45].利用此合成策略已经实现卤代烃、不饱和烃以及硼酸类化合物的不对称碳碳氧化偶联反应.
在碳氢键活化领域中, Yu课题组有着丰富研究经验.如在Yu[45]报道的分子内芳基与硼酸的不对称氧化偶联反应中, 利用分子内吡啶为辅助基团, 在醋酸钯催化剂和氧化剂的作用下, 反应可以得到优异的收率和ee值.该反应是通过辅助基团与催化剂络合实现其芳基邻位C—H键活化形成中间体钯络合物(该中间体决定着反应的对映选择性), 然后与硼酸发生转金属化反应, 最后还原消除得到目标产物, 系列底物都具有很好的反应结果(Scheme 29).利用此合成策略, 该课题组[46]在钯催化剂和氧化剂作用下, 利用苄胺类化合物与硼酸分子间的反应实现了苄胺类化合物的动力学拆分.
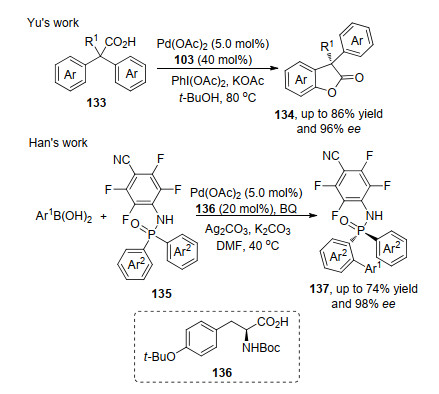
2013年, Yu和Wang等[47]首次利用二价钯到四价钯的催化体系, 实现了苯乙酸133分子内的不对称氧化反应.反应涉及钯催化剂与羧酸根络合以及对芳基上C—H活化, 随后形成分子内C—O键, 得到手性苯并呋喃酮134, 该反应通过分子内羧酸环化实现C—O的构建.随后, 韩福社课题组[48]利用芳基亚磷酰胺化合物135, 在催化剂与氧化剂的条件下, 实现了芳基亚磷酰胺类化合物与硼酸的不对称氧化反应, 与上述不同是该方法是一个二价钯到零价钯的催化过程.底物实验中, Ar2中取代基团位置影响反应进行, 间位基团能够促进反应的发生, 但底物取代基效应对产物的ee值影响不大, 该方法中由于酰胺基团的特殊性, 在一定程度限制此类方法的应用.两者反应均是利用辅助基团, 通过有机反应实现苄为手性中心的动力学拆分过程(Scheme 30).
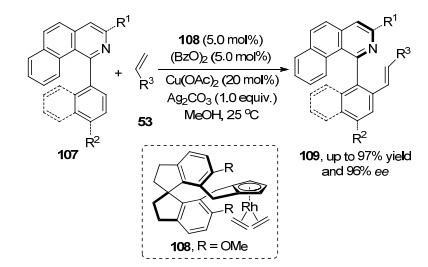
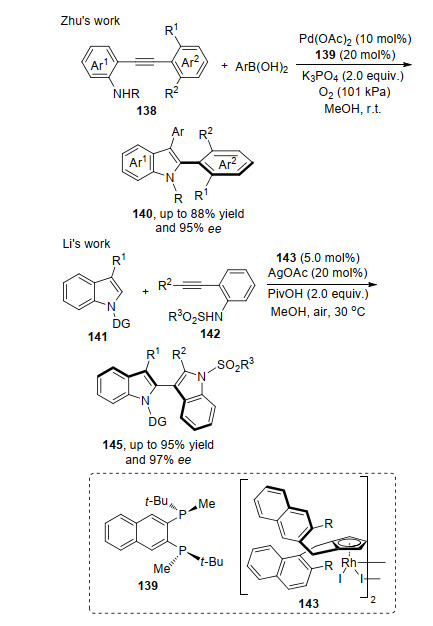
2017年, 石枫课题组[49]首次报道了在2-吲哚基甲醇的3位碳构建轴手性联芳基骨架的方法, 该方法是利用有机催化剂实现的. 2020年, 祝介平课题组[50]首次在金属钯催化作用下, 实现了吲哚2位碳轴手性联芳基骨架的构建.该方法是在Pd(OAc)2和配体139与氧气作用下, N-芳基(烷基)磺酰基-2-炔基苯胺138与芳基硼酸反应, 得到的2, 3-二取代吲哚140具有高产率和对映选择性.反应中钯配合物催化剂与芳基硼酸发生转金属化反应得到芳基钯络合物, 然后与底物中叁键络合经过迁移插入得到烯基钯中间体, 最后还原消除得到目标产物. 2019年, 李兴伟课题组[51]报道了轴手性分子2, 3'-联吲哚基145合成方法, 该方法是在Rh催化剂143和氧化剂的作用下实现, 产物具有优异的ee值.反应首先是催化剂完成吲哚2号碳氢键活化后与导向基团作用得到中间体铑物种, 随后与炔烃络合进行迁移插入环化过程得到烯基铑中间体, 最后烯基铑还原消除得到产物.在控制实验中, 作者分离出了手性的Rh环化中间体, 为机理提供了直接证据(Scheme 31).
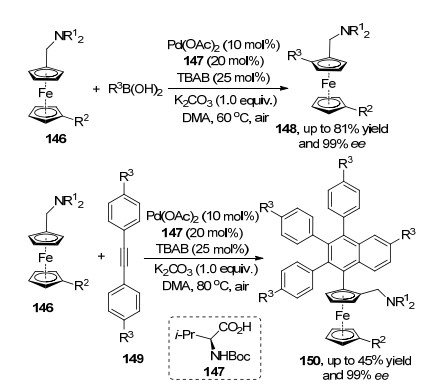
通过C(sp2)—H活化的策略构筑叔碳、季碳或者实现具有手性中心的底物动力学拆分的研究工作非常成功, 其中构筑面手性分子的研究以二茂铁为骨架较为典型.如游书力课题组[52]在前期研究基础上发展了取代二茂铁分别与硼酸和内炔149的C(sp2)—H氧化偶联反应, 该方法在钯催化剂和氧化剂的作用下, 系列产物均可以得到高的ee值.两者均借助于二茂铁辅助基团实现其碳氢活化, 反应启动方式相似.前者是催化剂与二茂铁辅助基团络合促进其环戊烯C—H键活化得到钯中间体, 随后与硼酸完成转金属化过程, 最后通过还原消除得到目标产物148; 后者则是先钯催化剂与底物作用完成C—H活化过程, 再和两分子内炔进行迁移插入形成烯基钯, 最后通过内炔芳基C—H键的反应进行还原消除得到目标产物[53]150 (Scheme 32).
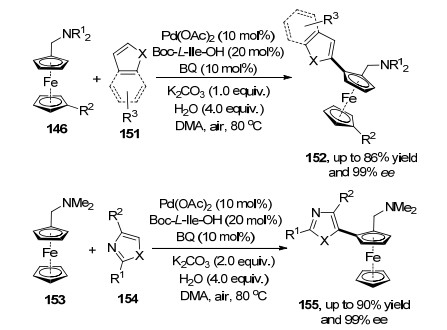
随后, 游书力课题组[54]在二茂铁构建面手性产物研究基础上, 发展了二茂铁146和杂芳烃151之间的钯催化的不对称氧化交叉偶联反应.该反应在醋酸钯催化剂和氧化剂的作用下进行, 具有中等反应收率以及高的对映选择性.反应通过芳杂环和二茂铁上碳氢活化后得到五元芳基钯中间体, 最后发生还原消除构建具有面手性的目标化合物152.接着他们把芳杂环拓展到芳二杂环154, 实现了芳二杂环与二茂铁的不对称氧化偶联反应.反应在醋酸钯催化剂和氧化剂的作用下实现, 得到的系列产物具有高对映选择性和收率.底物实验中, 杂环类底物、多取代噁唑和噻唑均能得到较高的ee值(Scheme 33).
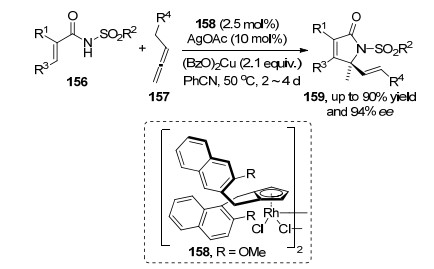
钯催化的C(sp2)—H不对称氧化反应的不断发展, 促进了其他过渡金属催化体系在该类型的反应中应用.如2019年, Cramer等[55]报道了在Rh络合物158催化剂作用下, 链状烯酰胺156与联烯157进行[4+1]环化反应.该方法是通过Rh催化剂进行链状烯烃的C(sp2)—H键活化, 然后对联烯双键进行迁移插入形成一个环状Rh中间体, 最后该中间体发生异构化和还原消除得到产物159.该反应条件温和, 具有广泛的基团耐受性和高的对映选择性(Scheme 34).
C(sp3)—H的活性高于C(sp2)—H的活性, 其不对称氧化反应的研究较为深入, 发展出了系列较为成熟的合成策略, 其底物适用性也较为广泛, 而且将传统的热反应发展到光反应, 使得合成朝着绿色、精准的路线发展.
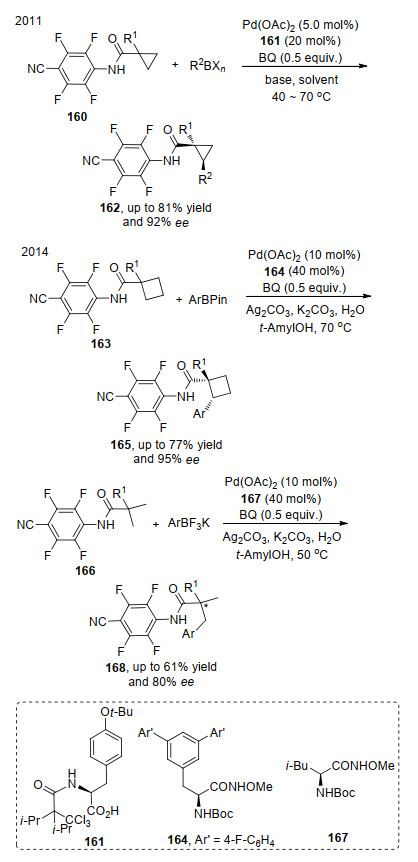
2011年, Yu等[56]报道了酰胺烷基的不对称氧化反应, 该反应在醋酸钯络合物催化剂和氧化剂作用下, 可以得到中等及以上的ee值.随后, 他们[57]将反应发展到环己烷的2号碳以及链状烷烃的C—H键活化, 两者都是利用硼酸类化合物为偶联分子, 均可以得到较好的反应结果, 反应的不足是受氨基取代基的限制, 无法进一步拓展其他酰胺类底物的应用(Scheme 35).
2019年, Yu等[58]发展了羧酸底物的C—H键不对称反应, 简化了之前反应底物的导向官能团, 突破了底物在此类反应中局限.该反应在钯络合物催化剂和氧化剂的作用下, 可以实现三元环取代羧酸和四元环取代羧酸环上的C—H活化, 整个反应过程中不改变原有底物的对映选择性, 且具有较好的ee值(Scheme 36).
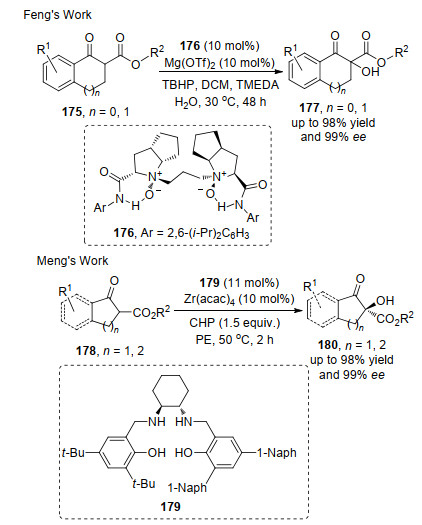
活泼氢的反应在C(sp3)—H的研究比较成熟, 如利用β-酮酸酯中两个羰基之间的C—H键的活性构建新的碳碳和碳杂键, 进一步转化为更有价值的分子骨架.在β-酮酸酯的不对称羟化反应研究中, 冯小明课题组[59]和孟庆伟课题组[60]报道的合成策略, 有效地实现了β-酮酸酯175和178的不对称α-羟基化.冯小明的报道中以Mg和N, N'-二氧化物的络合物176为催化剂, 过氧化氢叔丁基(TBHP)为氧化剂; 孟庆伟的报道中以Zr(Salan)络合物179为催化剂, 过氧化氢异丙苯(CHP)为氧化剂.两种方法均以较高收率和优异的对映选择性得到系列的目标产物(Scheme 37).
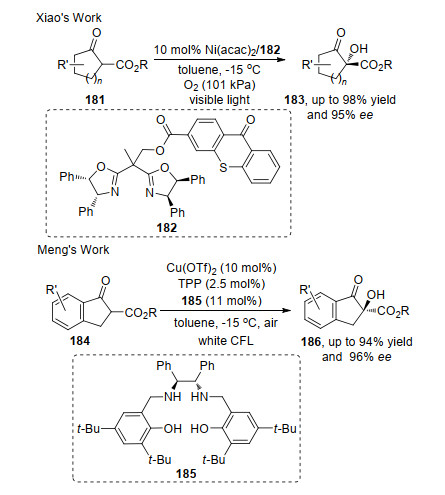
随后, 人们利用光化学反应绿色环保、条件温和及操作简单的特点, 将β-酮酸酯的不对称α-羟基化发展到光化学反应中.如肖文精课题组[61]报道了可见光诱导下, 在182与Ni(acac)2形成的络合物作用下β-酮酸酯181的不对称α-羟基化反应; 孟庆伟[62]报道了可见光诱导Cu(Salan)催化β-酮酸酯184的不对称α-羟基化反应.两种方法均使用可见光驱动反应, 氧气或空气作为氧化剂, 获得的产物具有较高收率和对映选择性(Scheme 38).不同的是, 前者是将配体修饰在光敏剂中, 在启动反应的同时, 配体和中心金属的作用控制了产物的对映选择性; 后者则是通过外加手性催化剂控制产物的手性.
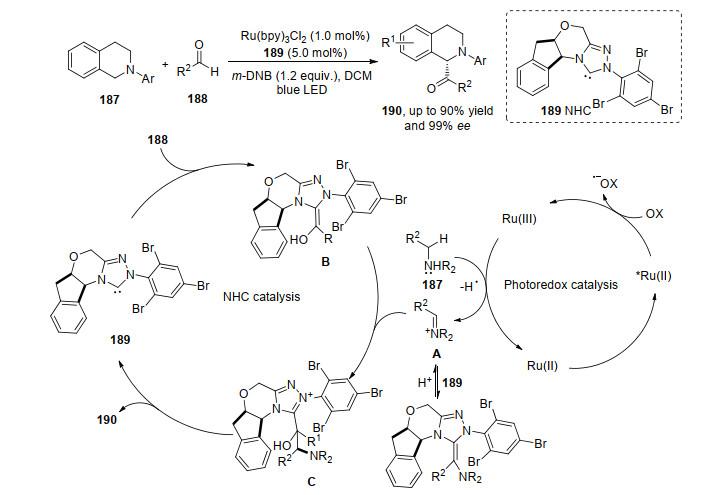
在有机光反应中, 依赖底物的手性诱导因素实现偶联反应的对映选择性, 是一种常见的合成策略[63].直接构建手性中心较为有效的方法是通过底物与共催化剂形成中间体, 该中间体进一步控制偶联反应产物的对映选择性[64]. 2012年, Rovis等[64]报道了手性有机催化剂N-杂环卡宾189和光催化剂组成的共催化体系, 实现了叔胺187与醛188的不对称α-酰化反应.醛188与有机催化剂189形成的中间体是决定产物对映选择性的关键.从机理可以看出, 反应涉及两个循环过程:光循环启动和有机催化剂循环生产过程.在光催化过程中, 光催化剂金属钌吸收光后激发成为激发态, 激发态的光敏剂与电子受体进行电子转移, 其氧化数升高变为三价钌, 底物胺187与三价钌作用, 失去一个氢自由基后胺异构化形成亚胺正离子A, 而三价钌在此反应过程中, 被还原成二价钌, 完成体系内光催化剂金属钌的循环; 在光催化发生的同时, 有机催化剂189与另一底物醛188形成中间体B, 而A与该过程中生成的中间体B进行胺化反应生成C或者直接与189作用; C解离出190生成189完成有机催化剂的循环再生(Scheme 39).
烯丙基的碳氢官能团化反应为功能烯烃分子的构建提供了快捷方法.过渡金属与手性配体的不对称催化碳-氢键氧化官能团化的反应中, Kharasch-Sosnovsky反应是典型的代表[65].烯丙基碳氢反应发展出了系列催化过程, 但是其手性构建发展却是缓慢的, 开发合适的催化体系实现烯丙基位或苄位碳氢键的不对称氧化官能团化, 仍然是一个研究热点.
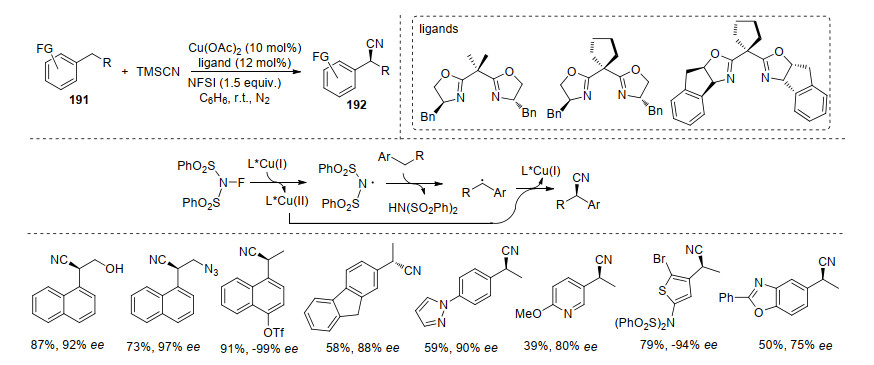
刘国生课题组在该领域的研究成就显著, 主要发展的是Cu催化的自由基反应过程, 如他们课题组[66]报道的Cu和手性双噁唑啉配体催化剂, 在不对称氧化碳-氢键氰基化反应中得到良好的收率和ee值.作者认为是一价铜催化N-氟代双苯磺酰胺(NFSI)的N—F键的均裂, 得到相应的氮自由基与二价铜.氮自由基与底物191作用得到苄基自由基, 随后被手性二价铜捕获形成中间体铜物种, 最后还原消除构建目标产物192.该反应中, 底物的基团可以控制氰基的进攻方向, 改变产物的构型; 在底物实验中, 大位阻和杂环类底物对反应的发生不利, 但是对其对映选择性影响不大(Scheme 40).
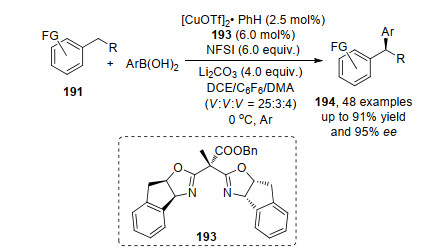
2019年, 刘国生课题组[67]又成功实现了在二氯乙烷、全氟苯、N, N-二甲基乙酰胺(25:3:4)的混合溶剂中, 芳基苯硼酸与苄基碳-氢键的不对称氧化偶联, 得到手性1, 1-二芳香化合物194.该反应成功的关键是在手性双噁唑啉193配体上引入羧酸酯基, 加速了芳基硼酸的转金属作用, 避免了其他副反应的发生(Scheme 41).
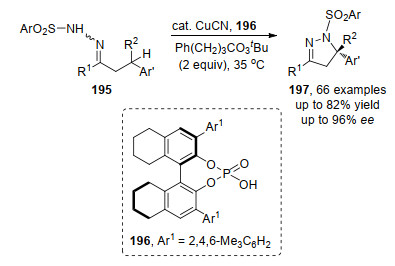
2020年, 刘心元课题组[68]利用铜催化剂与手性磷酸196的组合成功实现了分子内N—H与苄基C—H的不对称氧化偶联.该方法以高对映选择性合成了五元杂环化合物197 (Scheme 42).
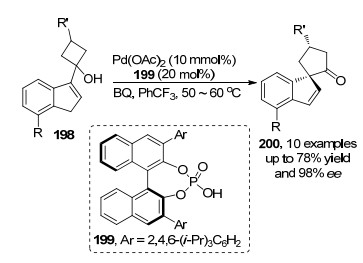
Chai和Rainey[69]于2012年报道了钯/手性布朗斯特酸199催化烯丙基碳氢键与分子内四元环醇的氧化偶联.反应涉及烯丙基碳氢活化后得到的烯丙基钯物种, 然后在手性磷酸协同作用下, 实现四元环醇的开环构建目标螺环化合物200 (Scheme 43).
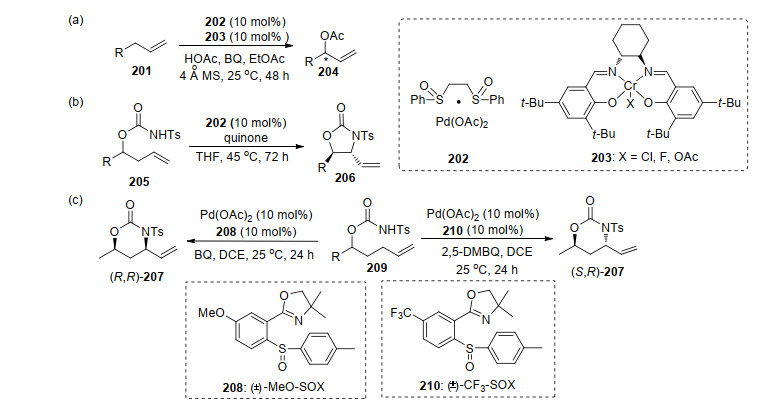
2008年, White课题组[70a]报道了首例钯催化的末端烯烃的不对称烯丙基碳氢键乙酰氧基化反应.该方法中使用双金属催化剂Pd(SOX) 202和Gr(Salen) 203, 手性路易斯酸为助催化剂, 实现了烯丙基的C—H键不对称氧化反应(Scheme 44, a). Pd和SOX配体络合的催化剂, 在实现反应的对映选择性方面最为成功, 是烯丙基C—H键不对称氧化反应的一种有效合成策略[70].该方法的立体选择性控制是通过手性路易斯酸与有机金属中间体相互作用的中间过程实现, 这一点在不加入手性路易斯酸或者加入无手性路易斯酸控制实验中, 其产物的对映选择性变化情况得到证实.随后借助此类催化剂, 他们发展了活泼亚甲基类型的底物与烯丙基的不对称氧化偶联反应.利用Pd(SOX)络合物催化剂, 实现了烯丙基C—H不对称胺化, 反应具有较好的反应效果.该方法中改变亚砜配体的取代基和改变氧化剂结构, 能够控制胺化产物的构型.产生该现象的原因是氧化剂参与反应中间体的形成, 改变了两者的取代基, 影响中间体的空间取向, 从而改变最终产物构型.该方法能够实现手性五元和六元的产物的合成, 且具有良好的对映选择性和产率, 其产物能够应用于多取代官能团的手性1, 2-氨基醇的合成(Scheme 44).
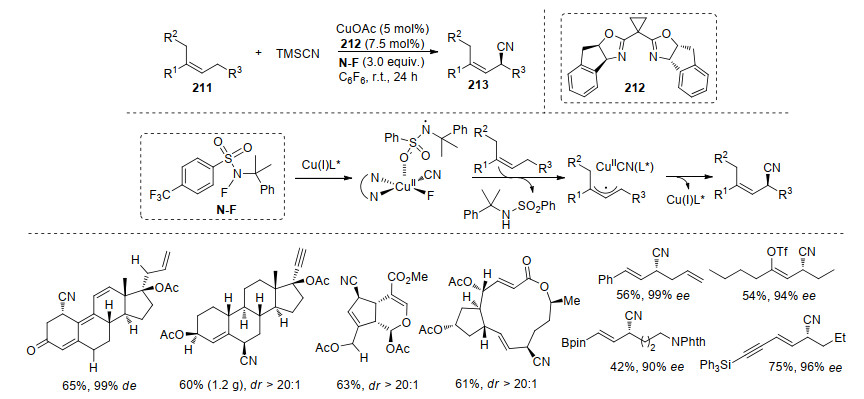
2018年, 我们课题组[71]报道了UiO型手性锆-金属硅铝构建的金属有机骨架化合物(MOFs)催化剂, 实现了苄基碳的不对称叠氮化反应.该方法通过Zr6O8团簇组成的封闭手性笼与金属盐络合, 这是控制产物对映选择性的关键. 2019, 刘国生课题组[72]在其前期研究基础上, 发展了烯丙基碳氢键的不对称氧化氰基化反应.该方法中N—F氧化剂与铜催化剂的络合物决定着烯丙基区域选择性.接着手性铜络合物发生迁移插入(该过程是产物的对映选择性的提高的关键), 最后发生还原消除得到目标产物213.底物实验中, 一些特殊的官能团化烯烃(烯基三氟甲磺酸酯和烯基硼等)也能够得到较好的反应结果, 这些官能团为产物的其进一步官能团化提供了可能(Scheme 45).
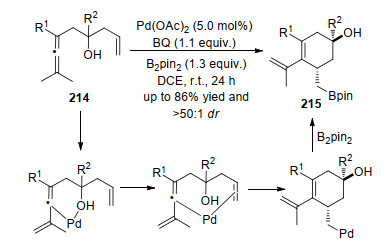
2018年, Bäckvall等[73]报道了在催化剂醋酸钯和氧化剂苯醌的作用下, 实现烯醇分子内的不对称氧化反应.该方法中, 对于含氧基团(酮、醇盐、乙酸盐)均可以实现环化反应生成环己烯215, 该反应关键在于含氧基团与催化剂的络合作用, 通过含氧基团完成催化剂对双键的迁移插入得到烷基钯物种, 此过程烯烃双键的面选择性配位是实现羟基的非对映选择性的关键.最后与硼酸酯发生系列转化得到目标产物.值得注意的是含氧官能团的配位作用对于反应的发生是必要的, 除去该官能团则反应就不发生(Scheme 46).
BV氧化反应是由酮类合成酯或内酯的一种氧化方法.通过不对称BV氧化反应获得的手性内酯在药物合成和天然产物合成具有重要的应用价值[74].
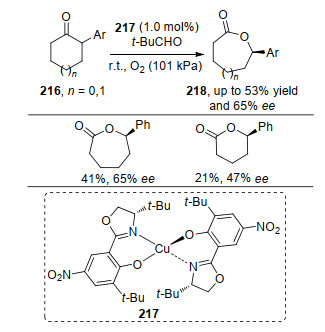
1994年, Bolm等[75]报道以铜络合物217为催化剂, 实现了2-取代环戊酮和环己酮等环酮类216的不对称BV氧化反应, 环己酮类底物相比环戊酮类底物表现出了更好的反应效果(Scheme 47).该方法的不足之处是产率和对映选择性不太理想(最高53%的收率和65% ee).
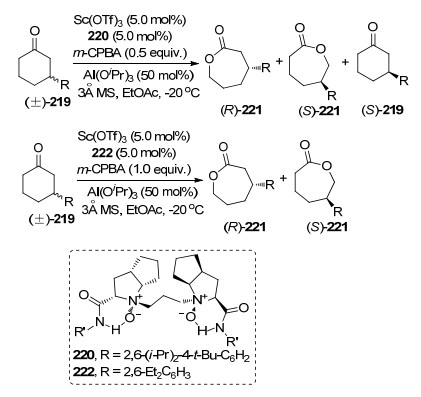
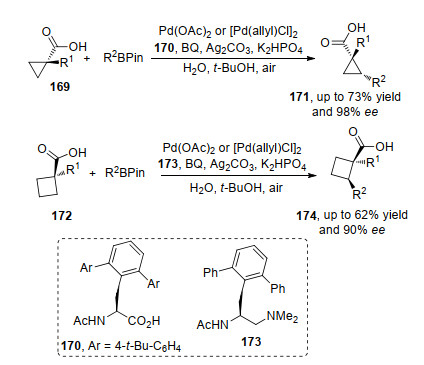
在金属与配体络合物催化剂促进的不对称BV氧化反应中, 对于2-或3-取代的己酮和2-取代的环戊酮, 能够较好地获得BV氧化产物; 对于二取代的环酮类化合物, 只有一些特定的官能团(如羟基和卤原子)存在时能够以较好的对映选择性得到目标产物, 其他取代基底物反应效率很差[76]; 而并环酮在反应条件下表现出极好的对映选择性[77].这些实验现象结果归结于环的特定的空间取向, 有利于反应的发生. 2019年, 冯小明课题组[78]基于前期研究, 利用Sc和N, N'-二氧化物络合的催化剂, 实现了3-取代外消旋环己酮的不对称BV氧化反应.该方法是一个动力学拆分的案例, 通过外消旋底物中单一构型参与BV氧化过程, 以较高的收率和拆分效果得到手性BV氧化产物和S构型的底物.值得注意的是通过改变配体骨架可以使S-219也发生BV氧化过程, 但也因此得不到单一构型的手性BV氧化产物.底物实验中, 取代基团的基团效应对反应的对映选择性影响较大, R基为芳基和脂肪族取代基有利于产物对映选择性的提高, 同等条件下S构型产物具有很好反应效果(Scheme 48).
在金属参与的不对称BV反应中, 已经实现2-或3-取代的环己酮和2-取代的环戊酮的BV氧化产物高对映选择性, 不足的是3-取代的环己酮底物无法得到单一构型的BV氧化产物; 再者是多取代的环酮类反应研究报道比较少, 而且反应效果较差.寻找新的合成策略解决这些问题具有重要的研究意义.
手性亚砜是天然产物和药物中普遍存在的结构单元, 目前获得手性亚砜化合物的主要途径是亚砜化合物的动力学拆分[79]和硫醚化合物的不对称氧化反应.
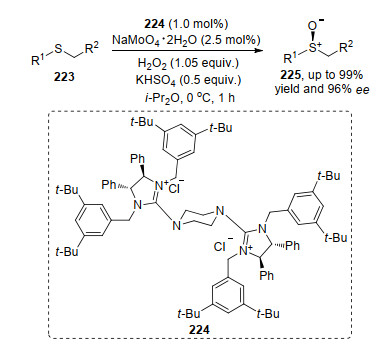
芳基、脂肪以及长链取代的硫醚化合物的不对称氧化反应研究报道中存在一些问题:一是过度氧化产生副产物砜, 导致主产物收率较低; 二是有些反应虽然产率高, 但无法控制其对映选择性. Barman等[80]报道的金属Ti和配体络合的催化剂在实现硫醚不对称氧化反应中, 反应产物的收率极佳(均大于90%), 但对映选择性比较差(小于50% ee).目前解决方法是在反应体系中加入一定量的添加剂. Tan等[81]报道了利用过氧钼酸盐224为催化剂的硫醚类化合物223的不对称氧化反应.他们在反应体系中加入0.5 equiv.的KHSO4, 能极大改善反应的效果.在底物实验中, 当R2为吸电子基团、R1为苄基或者芳基时, 在标准反应条件下产率和对映选择性都极好, 但是R1为烷基时, 反应收率较高, 对映选择性却较差(Scheme 49).
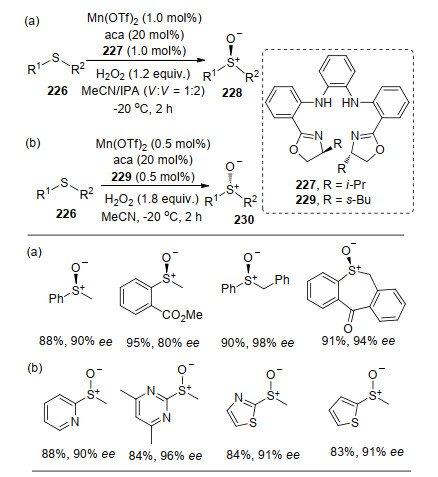
在众多反应的添加剂里, 羧酸作为添加剂的反应效果较好[79].在高爽课题组[82]报道中, 以Mn和卟啉衍生物络合物为催化剂, 以1-金刚烷甲酸(aca)为添加剂, 实现了芳基和芳香杂环硫化物226的不对称氧化反应.该反应在锰和配体络合物催化剂和H2O2氧化剂作用下, 底物的反应快速, 高效地得到目标产物(芳基硫化物反应需2 h, 而杂环硫化物仅需0.5 h).该方法中, 改变配体的骨架, 高收率和高对映选择性地得到不同构型的产物.在底物实验中, 不同的芳基底物以及R1和R2基团的改变对反应的影响不大, 只有当芳基R1的邻位为吸电子基团时反应的对映选择性稍微降低.对于环状硫化物的反应, 也能很好地得到相应的目标产物, 芳香杂环硫化物可以拓展至六元二氮、三氮杂环、五元噻吩、咪唑和吡咯等化合物(Scheme 50).
近些年来, 过渡金属催化的不对称氧化方法在烯烃不对称环氧化反应、C—H键不对称氧化反应、不对称BV氧化反应和硫醚不对称氧化反应等几个方面取得了较大的发展, 有些方法为获取天然产物和药物分子提供了简便高效的合成途径, 在合成和应用上展示了重要价值.值得注意的是, 伴随着资源高效利用和环境保护的必然要求, 金属参与的氧化反应正逐步迈向绿色、高效、精准的新时代, 包括氧气、双氧水、内源氧化剂以及MOFs催化剂在内的新型氧化体系不断涌现, 日臻成熟[83].从本文列举的研究实例中, 我们不难发现, 与新型氧化体系对应的不对称氧化反应的实例依然不多.展望未来的发展, 要实现绿色、高效和精准的不对称氧化反应, 首先, 与新型氧化体系匹配的手性配体和催化剂的设计、制备是实现高对映选择性氧化反应中的关键; 其次, 引入光化学等因素的共催化体系对于发展新型不对称氧化反应具有重要的作用; 最后, 实现不对称氧化反应在天然产物和药物分子合成中的应用是必然要求.
(a) Saisaha, P.; de Boerb, J. W.; Browne, W. R. Chem. Soc. Rev. 2013, 42, 2059.
(b) Zhu, Y.; Wang, Q.; Cornwall, R. G.; Shi, Y. Chem. Rev. 2014, 114, 8199.
(c) Liu, C.; Wen, K.-G.; Zeng, X.-P.; Peng, Y.-Y. Adv. Synth. Catal. 2020, 362, 1015.
(f) Bryliakov, K. P. Chem. Rev. 2017, 117, 11406.
Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1980, 102, 5974. doi: 10.1021/ja00538a077
(a) Zhang, W.; Loebach, J. L.; Wilson, S. R.; Jacobsen, E. N. J. Am. Chem. Soc. 1990, 112, 2801.
(b) Irie, R.; Noda, K.; Ito, Y.; Matusumoto, N.: Katsuki, T. Tetrahedron Lett. 1990, 31, 7345.
Jacobsen, E. N.; Markó, I.; Mungall, W. S.; Schroder, G.; Sharpless, K. B. J. Am. Chem. Soc. 1988, 110, 1968.
(b) Li, G.; Chang, H. T.; Sharpless, K. B. Angew. Chem., Int. Ed. 1996, 35, 451.
Ji, N.; Yuan, J.; Liu, M.; Lan, T.; He, W. Chem. Commun. 2016, 52, 7731. doi: 10.1039/C6CC02852G
Wang, C.; Yamamoto, H. J. Am. Chem. Soc. 2014, 136, 1222. doi: 10.1021/ja411379e
Bhadra, S.; Akakura, M.; Yamamoto, H. J. Am. Chem. Soc. 2015, 137, 15612. doi: 10.1021/jacs.5b11429
(a) Chu, Y.; Liu, X.; Li, W.; Hu, X.; Lin, L.; Feng, X. Chem. Sci. 2012, 3, 1996.
(b) Chu, Y.; Hao, X.; Lin, L.; Chen, W.; Li, W.; Tan, F.; Liu, X.; Feng, X. Adv. Synth. Catal. 2014, 356, 2214.
Clarasό, C.; Vicens, L.; Polo, A.; Costas, M. Org. Lett. 2019, 21, 2430. doi: 10.1021/acs.orglett.9b00729
Zhang, W.; Jacobsen, E. N. J. Org. Chem. 1991, 56, 2296. doi: 10.1021/jo00007a012
Koya, S.; Nishioka, Y.; Mizoguchi, H.; Uchida, T.; Katsuki, T. Angew. Chem., Int. Ed. 2012, 51, 8243. doi: 10.1002/anie.201201848
Kobayashi, Y.; Obayashi, R.; Watanabe, Y.; Miyazaki, H.; Miyata, I.; Suzuki, Y.; Yoshida, Y.; Shioiri, T.; Matsugi, M. Eur. J. Org. Chem. 2019, 2019, 2401. doi: 10.1002/ejoc.201900146
Farokhi, A.; Berijani, K.; Hosseini-Monfared, H. Catal. Lett. 2018, 148, 2608. doi: 10.1007/s10562-018-2447-8
Jat, J. L.; De, S. R.; Kumar, G.; Adebesin, A. M.; Gandham, S. K.; Falck, J. R. Org. Lett. 2015, 17, 1058. doi: 10.1021/acs.orglett.5b00281
Zhu, H.; Chen, P.; Liu, G. J. Am. Chem. Soc. 2014, 136, 1766. doi: 10.1021/ja412023b
Qi, X.; Chen, C.; Hou, C.; Fu, L.; Chen, P.; Liu, G. J. Am. Chem. Soc. 2018, 140, 7415. doi: 10.1021/jacs.8b03767
(a) Sherman, E. S.; Chemler, S. R.; Tan, T. B.; Gerlits, O. Org. Lett., 2004, 6, 1573.
(b) Chemler, S. R.; Karyakarte, S. D.; Khoder, Z. M. J. Org. Chem. 2017, 82, 11311.
Fu, S.; Yang, H.; Li, G.; Deng, Y.; Jiang, H.; Zeng, W. Org. Lett. 2015, 17, 1018. doi: 10.1021/acs.orglett.5b00131
Zhang, W.; Chen, P.; Liu, G. Angew. Chem., Int. Ed. 2017, 56, 5336. doi: 10.1002/anie.201700889
Bai, Z.; Zheng, S.; Bai, Z.; Song, F.; Wang, H.; Peng, Q.; Chen, G.; He, G. ACS Catal. 2019, 9, 6502. doi: 10.1021/acscatal.9b01350
(a) Huang, L.; Wang, Q.; Liu, X.; Jiang, H. Angew. Chem., Int. Ed. 2012, 51, 5696.
(b) Zhang, Z.; Wu, W.; Liao, J.; Li, J.; Jiang, H. Chem.-Eur. J. 2015, 21, 6708.
(a) Wu, M.-S.; Fan, T.; Chen, S.-S.; Han, Z.-Y.; Gong, L.-Z. Org. Lett. 2018, 20, 2485.
(b) Zhang, T.; Shen, H.-C.; Xu, J.-C.; Fan, T.; Han, Z.-Y.; Gong, L.-Z. Org. Lett. 2019, 21, 2048.
(c) Chen, S.-S.; Wu, M.-S.; Han, Z.-Y. Angew. Chem., Int. Ed. 2017, 56, 6641.
Zhang, G.; Fu, L.; Chen, P.; Zou, J.; Liu, G. Org. Lett. 2019, 21, 5015. doi: 10.1021/acs.orglett.9b01607
Fu, N.; Song, L.; Liu, J.; Shen, Y.; Siu, J. C.; Lin, S. J. Am. Chem. Soc. 2019, 141, 14480. doi: 10.1021/jacs.9b03296
Mikami, K.; Hatano, M.; Terada, M. Chem. Lett. 1999, 28, 55. doi: 10.1246/cl.1999.55
Schiffner, J. A.; Machotta, A. B.; Oestreich, M. Synlett 2008, 15, 2271.
Zhang, C.; Santiago, C. B.; Crawford, J. M.; Sigman, M. S. J. Am. Chem. Soc. 2015, 137, 15668. doi: 10.1021/jacs.5b11335
Akiyama, K.; Wakabayashi, K.; Mikami, K. Adv. Synth. Catal. 2005, 347, 1569 doi: 10.1002/adsc.200505154
Yoo, K. S.; Park, C. P.; Yoon, C. H.; Sakaguchi, S.; O'Neill, J.; Jung, K. W. Org. Lett. 2007, 9, 3933. doi: 10.1021/ol701584f
Chen, G.; Cao, J.; Wang, Q.; Zhu, J. Org. Lett. 2020, 22, 322. doi: 10.1021/acs.orglett.9b04357
(a) Walker, S. E.; Lamb, C. J. C.; Beattie, N. A.; Nikodemiak, P.; Lee, A.-L. Chem. Commun. 2015, 51, 4089.
(b) Lamb, C. J. C.; Vilela, F.; Lee, A.-L. Org. Lett. 2019, 21, 8689.
(a) Mei, T.; Patel, H.; Sigman, M. S. Nature 2014, 508, 340.
(b) Chen, Z.-M.; Hilton, M. J.; Sigman, M. S. J. Am. Chem. Soc. 2016, 138, 11461.
(a) Shi, B. F.; Zhang, Y. H.; Lam, J. K.; Wang, D. H.; Yu, J. Q. J. Am. Chem. Soc. 2010, 132, 460.
(b) Xiao, K.-J.; Chu, L.; Yu, J.-Q. Angew. Chem., Int. Ed. 2016, 55, 2856.
(a) Pi, C.; Li, Y.; Cui, X. L.; Zhang, H.; Han, Y. B.; Wu, Y. J. Chem. Sci. 2013, 4, 2675.
(b) Huang, Y.; Pi, C.; Cui, X.; Wu, Y. Adv. Synth. Catal. 2020, 362, 1385.
Zheng, J.; Cui, W.-J.; Zheng, C.; You, S.-L. J. Am. Chem. Soc. 2016, 138, 5242. doi: 10.1021/jacs.6b02302
McDonald, R. I.; White, P. B.; Weinstein, A. B.; Tam, C. P.; Stahl, S. S. Org. Lett. 2011, 13, 2830. doi: 10.1021/ol200784y
(a) Yip, K.-T.; Yang, M.; Law, K.-L.; Zhu, N.-Y.; Yang, D. J. Am. Chem. Soc. 2006, 128, 3130.
(b) He, W.; Yip, K.-T.; Zhu, N.-Y.; Yang, D. Org. Lett. 2009, 11, 5626.
(c) Du, W.; Gu, Q.; Li, Y.; Lin, Z.; Yang, D. Org. Lett. 2017, 19, 316.
Bao, X.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2018, 57, 1995. doi: 10.1002/anie.201712521
Kou, X.; Shao, Q.; Ye, C.; Yang, G.; Zhang, W. J. Am. Chem. Soc. 2018, 140, 7587. doi: 10.1021/jacs.8b02865
Sen, A.; Takenaka, K.; Sasai, H. Org. Lett. 2018, 20, 6827. doi: 10.1021/acs.orglett.8b02946
Allen, J. R.; Bahamonde, A.; Furukawa, Y.; Sigman, M. S. J. Am. Chem. Soc. 2019, 141, 8670. doi: 10.1021/jacs.9b01476
(a) Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861.
(b) Giri, R.; Shi, B.-F.; Engle, K. M.; Maugel, N.; Yu, J. Q. Chem. Soc. Rev. 2009, 38, 3242.
(c) Liao, K.; Negretti, S.; Musaev, D. G.; Bacsa, J.; Davies, H. M. L. Nature 2016, 533, 230.
(d) Wu, Q.-F.; Shen, P.-X.; He, J.; Wang, X.-B.; Zhang, F.; Shao, Q.; Zhu, R.-Y.; Mapelli, C.; Qiao, J. X.; Poss, M. A.; Yu, J.-Q. Science 2017, 355, 499.
(a) Meunier, B.; de Visser, S. P.; Shaik, S. Chem. Rev. 2004, 104, 3947.
(b) Butler, A.; Sandy, M. Nature 2009, 460, 848.
Milan, M.; Bietti, M.; Costas, M. ACS Cent. Sci. 2017, 3, 196.
(b) Sun, W.; Sun, Q. Acc. Chem. Rev. 2019, 52, 2370.
Shi, B. F.; Maugel, N.; Zhang, Y. H.; Yu, J. Q. Angew. Chem., Int. Ed. 2008, 47, 4882. doi: 10.1002/anie.200801030
Xiao, K.-J.; Chu, L.; Chen, G.; Yu, J.-Q. J. Am. Chem. Soc. 2016, 138, 7796. doi: 10.1021/jacs.6b04660
Cheng, X. F.; Li, Y.; Su, Y. M.; Yin, F.; Wang, J. Y.; Sheng, J.; Vora, H. U.; Wang, X. S.; Yu, J. Q. J. Am. Chem. Soc. 2013, 135, 1236. doi: 10.1021/ja311259x
Du, Z. J.; Guan, J.; Wu, G. J.; Xu, P.; Gao, L. X.; Han, F. S. J. Am. Chem. Soc. 2015, 137, 632. doi: 10.1021/ja512029x
Zhang, H.-H.; Wang, C.-S.; Li, C.; Mei, G.-J.; Li, Y.; Shi, F. Angew. Chem., Int. Ed. 2017, 56, 116. doi: 10.1002/anie.201608150
He, Y.-P.; Wu, H.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2020, 59, 2105. doi: 10.1002/anie.201914049
Tian, M.; Bai, D.; Zheng, G.; Chang, J.; Li, X. J. Am. Chem. Soc. 2019, 141, 9527. doi: 10.1021/jacs.9b04711
(a) Gao, D. W.; Shi, Y. C.; Gu, Q.; Zhao, Z. L.; You, S. L. J. Am. Chem. Soc. 2013, 135, 86.
(b) Shi, Y. C.; Yang, R. F.; Gao, D. W.; You, S. L. Beilstein J. Org. Chem. 2013, 9, 1891.
Zhang, H.; Cui, X. L.; Yao, X. N.; Wang, H.; Zhang, J. Y.; Wu, Y. J. Org. Lett. 2012, 14, 3012. doi: 10.1021/ol301063k
(a) Gao, D.-W.; Gu, Q.; You, S.-L. J. Am. Chem. Soc. 2016, 138, 2544.
(b) Cai, Z.-J.; Liu, C.-X.; Gu, Q.; Zheng, C.; You, S.-L. Angew. Chem., Int. Ed. 2019, 58, 2149.
Wang, S.-G.; Liu, Y.; Cramer, N. Angew. Chem., Int. Ed. 2019, 58, 18136. doi: 10.1002/anie.201909971
Wasa, M.; Engle, K. M.; Lin, D. W.; Yoo, E. J.; Yu, J. Q. J. Am. Chem. Soc. 2011, 133, 19598. doi: 10.1021/ja207607s
Xiao, K. J.; Lin, D. W.; Miura, M.; Zhu, R. Y.; Gong, W.; Wasa, M.; Yu, J. Q. J. Am. Chem. Soc. 2014, 136, 8138. doi: 10.1021/ja504196j
Hu, L.; Shen, P.-X.; Shao, Q.; Hong, K.; Qiao, J. X.; Yu, J.-Q. Angew. Chem., Int. Ed. 2019, 58, 2134. doi: 10.1002/anie.201813055
Yin, C.; Cao, W.; Lin, L.; Liu, X.; Feng, X. Adv. Synth. Catal. 2013, 355, 1924. doi: 10.1002/adsc.201300335
Yang, F.; Zhao, J.; Tang, X.; Zhou, G.; Song, W.; Meng, Q. Org. Lett. 2017, 19, 448. doi: 10.1021/acs.orglett.6b03554
Ding, W.; Lu, L.-Q.; Zhou, Q.-Q.; Wei, Y.; Chen, J.-R.; Xiao, W.-J. J. Am. Chem. Soc. 2017, 139, 63. doi: 10.1021/jacs.6b11418
Yang, F.; Zhao, J.; Tang, X.; Wu, Y.; Yu, Z.; Meng, Q. Adv. Synth. Catal. 2019, 361, 1673. doi: 10.1002/adsc.201801263
Banerjee, A.; Yamamoto, H. Org. Lett. 2017, 19, 4363. doi: 10.1021/acs.orglett.7b02076
DiRocco, D. A.; Rovis, T. J. Am. Chem. Soc. 2012, 134, 8094. doi: 10.1021/ja3030164
Kharasch, M. S.; Sosnovsky, G. J. Am. Chem. Soc. 1958, 80, 756.
Zhang, W.; Wang, F.; McCann, S. D.; Wang, D.; Chen, P.; Stahl, S. S.; Liu, G. Science 2016, 353, 1014. doi: 10.1126/science.aaf7783
Zhang, W.; Wu, L.; Chen, P.; Liu, G. Angew. Chem., Int. Ed. 2019, 58, 6425. doi: 10.1002/anie.201902191
Yang, C.; Zhang, C.; Gu, Q.-S.; Fang, J.-H.; Su, X.-L.; Ye, L.; Sun, Y.; Tian, Y.; Li, Z.-L.; Liu, X.-Y. Nat. Catal. 2020, 3, 539. doi: 10.1038/s41929-020-0460-y
Chai, Z.; Rainey, T. J. J. Am. Chem. Soc. 2012, 134, 3615. doi: 10.1021/ja2102407
(a) Covell, D. J.; White, M. C. Angew. Chem., Int. Ed. 2008, 47, 6448.
(b) Liu, W.; Ali, S. Z.; Ammann, S. E.; White, M. C. J. Am. Chem. Soc. 2018, 140, 10658.
(b) Fraunhoffer, K. J.; White, M. C. J. Am. Chem. Soc. 2007, 129, 7274.
(c) Ma, R.; Young, J.; Promontorio, R.; Dannheim, F. M.; Pattillo, C. C.; White, M. C. J. Am. Chem. Soc. 2019, 141, 9468.
Li, J.; Ren, Y.; Yue, C.; Fan, Y.; Qi, C.; Jiang, H. ACS Appl. Mater. Interfaces 2018, 10, 36047. doi: 10.1021/acsami.8b14118
Li, J.; Zhang, Z.; Wu, L.; Zhang, W.; Chen, P.; Lin, Z.; Liu, G. Nature 2019, 574, 516. doi: 10.1038/s41586-019-1655-8
Posevins, D.; Qiu, Y.; Bäckvall, J.-E. J. Am. Chem. Soc. 2018, 140, 3210. doi: 10.1021/jacs.7b13563
Zhou, L.; Liu, X.; Ji, J.; Zhang, Y.; Wu, W.; Liu, Y.; Lin, L.; Feng, X. Org. Lett. 2014, 16, 3938. doi: 10.1021/ol501737a
Bolm, C.; Schlingloff, G.; Weickhardt, K. Angew. Chem., Int. Ed. 1994, 33, 1848. doi: 10.1002/anie.199418481
Lopp, M.; Paju, A.; Kanger, T.; Pehk, T. Tetrahedron Lett. 1996, 37, 7583. doi: 10.1016/0040-4039(96)01666-8
Bianchini, G.; Cavarzan, A.; Scarso, A.; Strukul, G. Green Chem. 2009, 11, 1517. doi: 10.1039/b916262n
Wu, W.; Cao, W.; Hu, L.; Su, Z.; Liu, X.; Feng, X. Chem. Sci. 2019, 10, 7003. doi: 10.1039/C9SC01563A
Wang, L.; Chen, M.; Zhang, P.; Li, W.; Zhang J. J. Am. Chem. Soc. 2018, 140, 3467. doi: 10.1021/jacs.8b00178
Barman, S.; Patil, S.; Levy, C. J. Chem. Lett. 2012, 41, 974. doi: 10.1246/cl.2012.974
Zong, L.; Wang, C.; Putra Moeljadi, A. M.; Ye, X.; Ganguly, R.; Li, Y.; Hirao, H.; Tan, C.-H. Nat. Commun. 2016, 7, 13455. doi: 10.1038/ncomms13455
(a) Dai, W.; Li, G.; Wang, L.; Chen, B.; Shang, S.; Lv, Y.; Gao, S. RSC Adv. 2014, 4, 46545.
(b) Dai, W.; Shang, S.; Lv, Y.; Li, G.; Li, C.; Gao, S. ACS Catal. 2017, 7, 4890.
(a) Wu, W.; Jiang, H. Acc. Chem. Res. 2012, 45, 1736.
(b) Huang, H.; Ji, X.; Wu, W.; Jiang, H. Chem. Soc. Rev. 2015, 44, 1155.
(c) Liang, Y.; Jiao, N. Acc. Chem. Res. 2017, 50, 1640.
(d) Tang, X.; Wu, W.; Zeng, W.; Jiang, H. Acc. Chem. Res. 2018, 51, 1092.
(e) Li, J.; Liao, J.; Ren, Y.; Liu, C.; Yue, C.; Lu, J.; Jiang, H. Angew. Chem., Int. Ed. 2019, 58, 17148.

图式 3 Ti催化的烯基醇类化合物的不对称氧化反应
Scheme 3 Ti-catalyzed asymmetric oxidation of enol compounds

图式 4 Sc催化的α, β-不饱和羰基化合物不对称环氧化反应
Scheme 4 Sc-catalyzed asymmetric epoxidation of α, β-unsaturated carbonyl compounds

图式 11 Pd催化的烯烃的不对称氧化环化反应
Scheme 11 Pd-catalyzed asymmetric oxidation cyclization of olefins

图式 13 Cu催化的不对称膦氰基化反应
Scheme 13 Cu-catalyzed asymmetric reaction of phosphine cyanation

图式 15 吲哚分子内不对称氧化Heck反应
Scheme 15 Asymmetric oxidation of Heck reaction in the indole molecule

图式 20 二芳基乙酸与端烯的不对称氧化反应
Scheme 20 Asymmetric oxidation of diarylacetic acid with terminal olefins

图式 24 烯胺分子内不对称氧化胺化反应
Scheme 24 Intramolecular asymmetric oxidative amination of enamine

图式 26 烯肼类化合物的不对称Wacker反应
Scheme 26 Asymmetric Wacker reaction of enhydrazine compounds

图式 33 二茂铁与芳基杂环的不对称偶联反应
Scheme 33 Asymmetric coupling reaction between ferrocene and aryl heterocycle

图式 34 Rh催化的烯酰胺与联烯的不对称氧化反应
Scheme 34 Rh-catalyzed asymmetric oxidation of enamide and allene

图式 41 苄基碳-氢键的不对称氧化偶联
Scheme 41 Asymmetric oxidative coupling of carbon-hydrogen bonds in benzyl

图式 42 分子内苄基C—H的不对称氧化偶联
Scheme 42 Asymmetric oxidative coupling of intramolecular benzyl C—H

图式 48 Sc催化的环己酮不对称BV氧化反应
Scheme 48 Sc-catalyzed asymmetric BV oxidation of cyclohexanone

图式 49 钼酸盐催化的硫醚类化合物的不对称氧化反应
Scheme 49 Molybdate-catalyzed asymmetric oxidation of sulfide compounds

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:


































