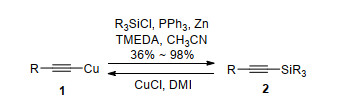
Scheme 1.
Introduction of trialkylsilyl groups to terminal acetylenes

The protection/deprotection of functional group is one of the fundamental technologies in organic synthesis. Other reactive sites must be temporarily blocked if we want to carry out a chemical reaction selectively at one reaction site in a multifunctional compound. Therefore, many protective groups have been, and are being, developed. An ideal protecting group needs to satisfy the following issues: facile introduction to the target functional group, stability during the desired transformation such as C—C bond formation and facile deprotection under mild reaction conditions.[1]
Alkynes, important unsaturated compounds, presenting in many pharmaceuticals, argochemicals, natural products, organic materials and so on, [2] are also key intermediates in organic synthesis.[3] In preparation of alkynes, protected terminal alkynes are usually used as starting materials. In terminal alkyne molecule, hydrogen atom on alkynyl group has strong acidity. And the presence of this hydrogen atom in metal organic synthesis will affect the normal progress of the reaction.[1] Therefore, a number of protecting groups were developed and utilized in synthesis of structurally complicated compounds, and new protecting groups were still being explored.[4] Less polar groups such as trimethylsilyl (TMS), [5] trimethylgermanium group (Me3Ge)[6] and high polar groups like (3-cyanopropyl)dimethylsilyl (CPDMS), [7] (3-cyanopropyl) diisopropylsilyl (CPDIPS)[8] and (Ph2P(O))[9] were developed as suitable protecting groups for terminal alkynes.
Trialkylsilyl groups were widely used as protecting groups for terminal acetylenes for the bulk of silane.[5] Trialkylsilylacetylenes were often used as a convenient reagent to introduce an acetylenic unit because they tend to be easily handled liquids or solids, as opposed to gaseous acetylene.[1] Trialkylsilanes could be formed by reaction of copper acetylide with silyl chloride. In the presence of PPh3, Zn and tetramethylethylenediamine (TMEDA), alkynylcopper reagents 1 reacted with trialkylsilyl chloride to afford the trialkylsilyl protected acetylenes 2 (Scheme 1). It is interesting to note that the reaction can be reversed to give the alkynylcopper(Ⅰ) reagent 1 in the presence of CuCl and 1, 3-dimethyl-2-imidazolidinone (DMI).[10] Other method such as addition of lithium or Grignard reagent to silylchloride also produced trialkylsilanes.[11]
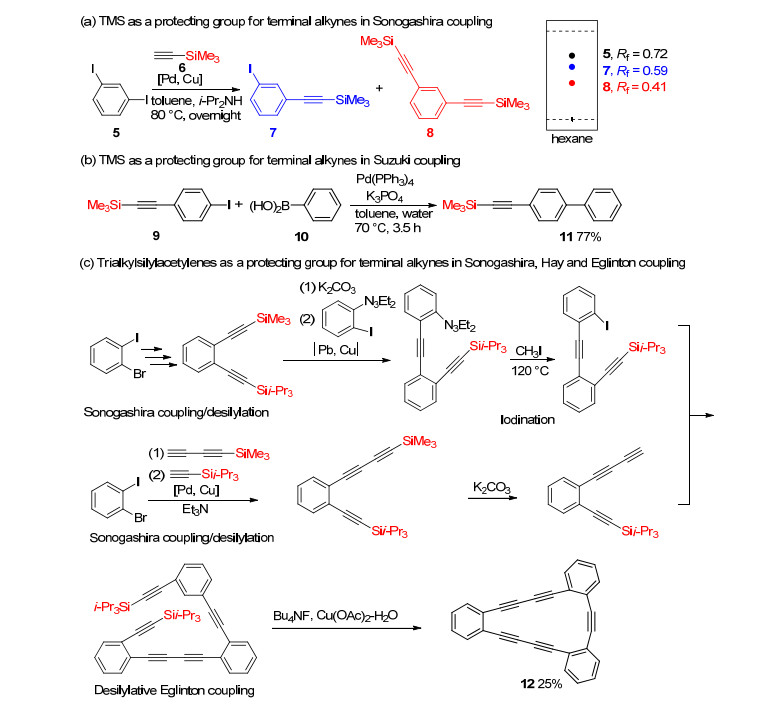
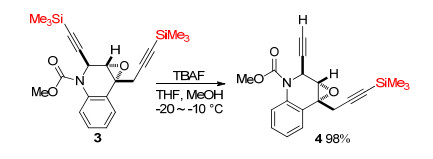
Trialkylsilyl groups were usually deprotected in the presence of suitable bases.[12] As shown in Scheme 2, the desired deprotected compounds 4 was obtained in 98% yield when trialkylsilyl 3 was treated with tetrabutylam-moniumfluoride (TBAF) in tetrahydrofuran (THF) and MeOH at -20~10 ℃.[13] And limited use of TBAF (0.4~0.5 equiv.) enabled regioselective desilylation of 3. Trialkylsilyl groups, stable under carbon-carbon bond formation reaction conditions, are widely used to synthesize acetylene derivatives.[14] As shown in Scheme 3, desired mono-silyl-protected acetylene 7 and di-silyl-protected acetylene 8 were produced when diiodide 5 and TMS-protected acetylene 6 were treated with Pd and Cu catalyst in toluene at 80 ℃. But in this reaction, isolation of 7 and 8 was kind of difficult for the similar Rf values among 5, 7 and 8 (Rf=0.72 for 5, Rf=0.59 for 7, Rf=0.41 for 8 in hexane) (Scheme 3a).[9] In 2001, Suzuki coupling of TMS-protected alkyne was reported by Li.[15] Treatment of TMS-ethyl-substituted iodide 9 and phenylboronic acid 10 with Pd(PPh3)4 and K3PO4 in toluene gave the corresponding compound 11 in 77% yield (Scheme 3b). In 1997, Haley et al.[16] synthesized cyclic pentayne 12 in 25% total yield by repeating Sonogashira coupling/desilyl stepwise reaction, iodination and desilylation/Eglinton coupling stepwise reaction (Scheme 3c).
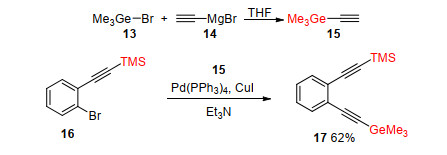
Trimethylgermanium group (Me3Ge) was another less polar protecting group for terminal acetylenes. In 1996, Ernst et al.[6] prepared several TMS- and Me3Ge-protected diynes and achieved mono regioselective deprotection by protodesilylation and protodegermylation. Me3Ge-prote- cted acetylene 15 was prepared from bromotrimethylgermane 13 and ethynylmagnesium bromide 14. Ortho-substituted TMS- and Me3Ge-protected diyne 17 was obtained in 62% yield by Sonogashira coupling reaction of 15 and 16 (Scheme 4).
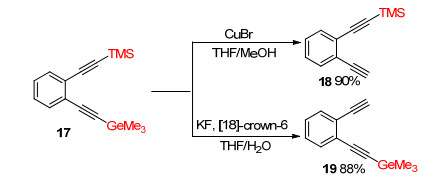
Under suitable reaction conditions, selective deprotection of TMS or Me3Ge proceeded and gave pure mono TMS- or Me3Ge-protected diynes. Treatment of ortho-substituted TMS- and Me3Ge-protected diyne 17 with CuBr gave mono-TMS-protected diyne 18 in 90% yield. Subjection of 17 to KF/[18]-crown-6 catalyzed deprotection produced mono-Me3Ge-protected diyne 19 in 88% yield (Scheme 5).[6]
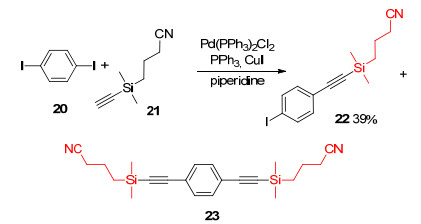
Although trialkylsilyl protecting groups were stable under carbon-carbon bond formation reaction conditions, the major drawback of those groups was the difficult separation of desired coupling compounds from remaining starting materials and byproducts because of their similar polarities. In 2000, Höger and Bonrad et al. reported a polar protecting group (3-cyanopropyl)dimethylsilyl (CPDMS) for terminal alkyne. In the presence of Pd(PPh3)2Cl2, PPh3, CuI and piperidine, diiodide 20 reacted with CPDMS-protected acetylene 21 to produce monoadduct 22 in 39% yield and diadduct 23. 22 and 23 were easy to separate for their different Rf values (Rf=0.50 for 22, Rf=0.20 for 23 in hexane/CH2Cl2, V:V=1:1) (Scheme 6).[7]
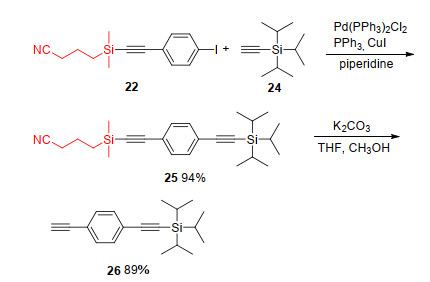
Mono-protected-diyne could be obtained by selective CPDMS-deprotection. Coupling of the mono-CPDMS-adduct 22 with triisopropylsilyl (TIPS)-acetylene 24, purification of the CPDMS- and TIPS-protected-diyne 25 and subsequent treatment with K2CO3 gave the mono-TIPS-protected-diyne 26 in 89% yield (Scheme 7).[7]
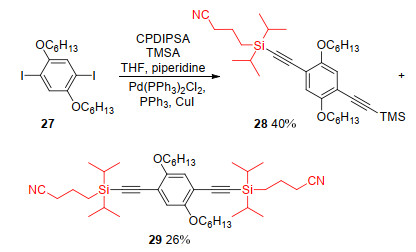
In 2008, Höger and Gaefke reported another polar protecting group (3-cyanopropyl)diisopropylsilyl (CPDIPS) for terminal acetylenes. When Pd(PPh3)2Cl2 and CuI were used as catalysts and PPh3 was employed as a ligand, the reaction of diiodobenzene 27 with (3-cyanopropyl) diisopropylsilyl-acetylene (CPDIPSA) and TMS-acetylene (TMSA) proceeded smoothly and produced TMS- and CPDIPS-protected-diyne 28 in 40% yields and CPDIPS- protected-diyne 29 in 26% yields (Scheme 8).
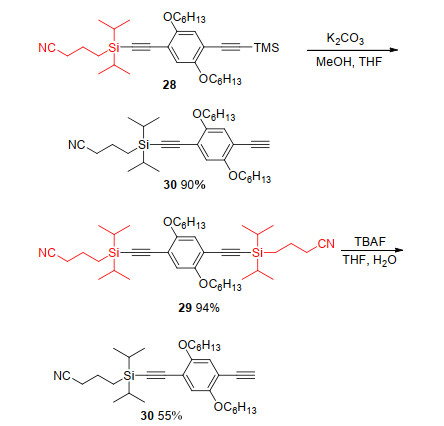
By treatment with K2CO3, TMS group in TMS- and CPDIPS-protected-diyne 28 can be selectively deprotected to give mono-CPDIPS-protected-diyne 30 in 90% yield. CPDIPS-protected-diyne 29 was ideally suited for partial deprotection. Treatment of 29 with TBAF and H2O in THF afforded mono-CPDIPS-protected-diyne 30 in 55% yield (Scheme 9).[8]
Trialkylsilyl groups are often invoked routinely in preparation of aromatic acetylenes having expanded π-systems. But these reactions frequently suffered from a severe drawback about isolation of the products.[17] In 2011, Orita and Otera et al.[9] developed a polar protecting group, diphenylphosphoryl (Ph2P(O)), for terminal ethynes which enabled facile separation of aromatic acetylenes.
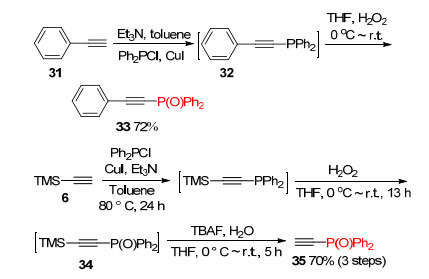
Ph2P(O) group was introduced to terminal ethynes by phosphination with Ph2PCl followed by oxidation with H2O2. Treatment of phenylacetylene 31 with chlorodiphenylphosphine, copper(Ⅰ) iodide and triethylamine afforded diphenyl(phenylethynyl)phosphine 32. Addition of H2O2 to THF solution of 32 gave the desired product 33 in 72% yield. Ph2P(O)-protected acetylene 35 was prepared from TMS-acetylene 6 through phosphination, oxidation and selective deprotection of TMS group using TBAF as a cleaving reagent (Scheme 10).[9, 18]
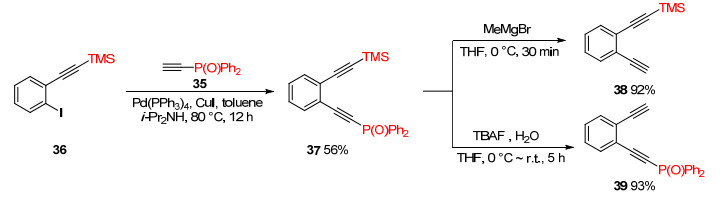
Ph2P(O), stable under acidic conditions, could be deprotected in the presence of suitable bases like t-BuOK and MeMgBr. Facile selective deprotection of TMS and Ph2P(O) group was achieved by changing deprotecting reagent. In the presence of TMS group, selective deprotection of Ph2P(O) was achieved by using MeMgBr as a deprotecting reagent, while TBAF enabled selective deprotection of TMS group. Sonogashira coupling between iodide 36 and phosphorylethyne 35 gave ortho-substituted TMS- and Ph2P(O)-protected diyne 37 in 56% yield.[19] Treatment of 37 with MeMgBr afforded ortho-substituted mono-TMS-protected diyne 38 in 92% yield.[6] Subjection of 37 to TBAF and small amount of water gave ortho-substituted mono-Ph2P(O)-protected diyne 39 in 93% yield (Scheme 11).[20]
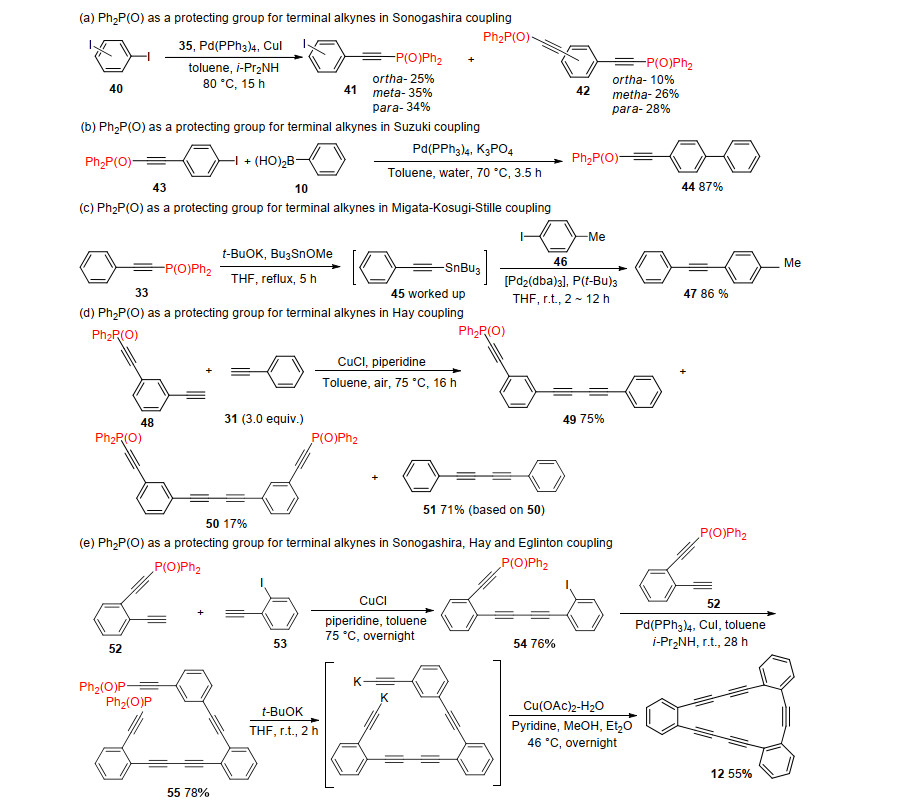
Ph2P(O) was a promising protecting group for terminal acetylene in carbon-carbon bond formation reactions such as Sonogashira coupling, [21] Suzuki coupling, [15] Migita- Kosugi-Stille coupling, [22-23] Hay coupling, [24] and Eglinton couplings.[18] By taking advantage of this highly polar protecting group, a series of ethyne derivatives have been synthesized successfully. Sonogashira coupling between diiodobenzene 40 and phosphorylethyne 35 formed mono-adduct 41 in 25%~35% yield and bis-adduct 42 in 10%~28% yield. And the high polarity of Ph2P(O) enabled facile isolation of 41 and 42 for their large different Rf values (Rf=0.55 for meta-41, Rf=0.23 for meta-42 in ethyl acetate) (Scheme 12a).[9] Ph2P(O)-protected acetylene could be applied in Suzuki coupling as well. Treatment of phosphorylethyl-substituted iodide 43 with phenylboronic acid 40 in Suzuki coupling conditions provided the desired product ethynylphosphine oxide 44 in 87% yield (Scheme 12b).[15] Phosphorylethynes could be applied to Migata- Kosugi-Stille coupling as well. Treatment of phosphorylethyne 33 with t-BuOK and Bu3SnOMe in refluxing temperature obtained the crude product stannylethyne. Treatment of the crude stannylethyne with 1-iodo-4-methyl- benzene 46 under Stille coupling conditions gave the desired coupling product 47 in 86% yield (Scheme 12c).[25] Mono-Ph2P(O)-protected diynes could be applied in Hay coupling to prepare unsymmetrically substituted yne- diynes. When mono-phosphoryl-protected diyne 48 and phenylethyne 31 were heated in presence of CuCl and piperidine, 49 was obtained in 76% yield. And desired product 49 was easy to isolate from by-products 50 and 51 because of their large different Rf values (Rf=0.55 for 49, Rf=0.24 for 50, Rf=0.97 for 51 in ethyl acetate) (Scheme 12d).[18] Hay coupling between monoprotected diyne 52 and iodoethyne 53 gave iodotriyne 54 in 76% yield, and Sonogashira coupling of the resulting iodide 54 with 52 provided bis-Ph2P(O)-protected pentayne 55 in 78% yield. When 55 was subjected to t-BuOK-catalyzed deprotection followed by Cu(OAc)2-catalyzed Eglinton coupling, the expected cyclization occurred to produce cyclic pentayne 12 in 55% yield (Scheme 12e).[16, 18]
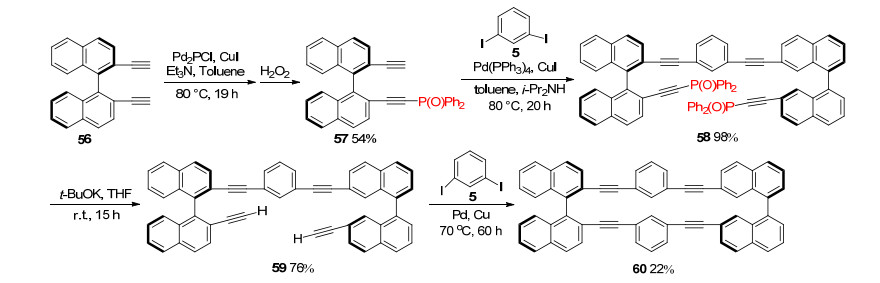
By taking advantage of Ph2P(O) protecting group, enantiopure acetylenic cyclophane could be synthesized successfully. The synthetic route was shown in Scheme 13, diethynyl compound 56 was converted to mono-Ph2P(O)- protected 2, 2'-diethynyl-1, 1'-binaphthyl derivative 57 in 54% yield. Sonogashira coupling of 57 with 1, 3-diiodo- benzene 5 proceeded smoothly to give 58 in 98% yield. Treatment of 58 with t-BuOK afforded 59 in 76% yield. Then, 59 was subjected to Sonogashira coupling with 1, 3-diiodobenzene 5, intermolecular coupling and the subsequent cyclization proceeded successfully to afford cyclophane 60 in 22% yield (Scheme 13).[9]
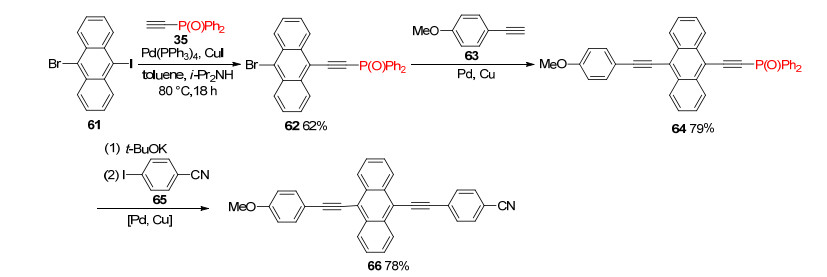
Unsymmetrically substituted anthracene could be prepared efficiently from Ph2P(O)-protected acetylene. Sonogashira coupling of 9-bromo-10-iodoanthracene 61 with phosphorylethyne 35 gave 62 exclusively in 62% yield, and 62 was transformed to 64 in 79% yield by coupling with 4-methoxyphenylethyne 63. Successive subjection of 64 to t-BuOK-promoted dephosphorylation and coupling with 65 provided 9, 10-bis(ethynyl)anthracenes 66 in 78% yield. (Scheme 14).[26]
In summary, less polar protecting groups like trialkylsilyl and Me3Ge, and high polar protecting groups such as CPDMS, CPDIPS and Ph2P(O) for terminal acetylenes were introduced. Trialkylsilyl groups are widely used to synthesize acetylene derivatives, but the major drawback of these protecting groups was the difficult separation of desired coupling compounds. High polarity of CPDMS enabled facile separation. High polar Ph2P(O) was also developed as a protecting group for terminal acetylenes. Ph2P(O), facilely introduced to terminal acetylenes, easily deprotected in bases and stable under acidic conditions, could be applied to transition metal catalyzed carbon-carbon bond formation reactions. High polarity of Ph2P(O) enabled facile isolation of desired coupling products from remaining starting materials and by-products.
Greene, T. W.; Wuts. P. G. M. Protective Groups in Organic Synthesis, 3th ed., John Wiley & Sons, Inc., New York, 1999.
(a) Stang, P. J.; Diederich. F. Modern Acetylene Chemistry, VCH, Weinheim, 1995.
(b) Diederich, F.; Stang, P. J.; Tykwinski, R. R. Acetylene Chemistry, Willey-VCH Verlag GmbH & CO. KgaA, Weinheim, 2005.
(c) Peng, L.-F.; Zhang, S.-W.; Wang, B.-H.; Xun, M.-S.; Tang, Z.-L.; Jiao, Y.-C.; Xu, X.-H. Chin. J. Org. Chem. 2018, 38, 519(in Chinese).
(彭丽芬, 张思维, 王丙昊, 寻梦硕, 唐子龙, 焦银春, 许新华, 有机化学, 2018, 38, 519.)
(d) Peng, L.-F.; Wang, B.-H.; Wang, M.; Tang, Z.-L.; Jiang, Y.-Z.; Jiao, Y.-C.; Xu, X.-H. J. Chem. Res. 2018, 42, 235.
(e) Wang, Z.; Yang, L.; Liu, H.-L.; Bao, W.-H.; Tan, Y.-Z.; Wang, M.; Tang, Z.; He, W.-M. Chin. J. Org. Chem. 2018, 38, 2639(in Chinese).
(王峥, 杨柳, 刘慧兰, 谭英芝, 包文虎, 汪明, 唐子龙, 何卫民, 有机化学, 2018, 38, 2639.)
(f) Peng, L.-F.; Lei, J.-Y.; Wu, L.; Tang, Z.-L.; Luo, Z.-P.; Jiao, Y.-C.; Xu, X.-H. J. Chem. Res. 2018, 42, 271.
(g) Li, W.-Y.; Yin, G.-X.; Huang, L.; Xiao, Y.; Fu, Z.-M.; Xin, X.; Liu, F.; Li, Z.-Z. He, W.-M. Green Chem. 2016, 18, 4879.
(h) Wu, C.; Wang, Z.; Hu, Z.; Zeng, F.; Zhang, X.-Y.; Cao, Z.; Tang, Z.; . He, W.-M.; Xu, X.-H. Org. Biomol. Chem. 2018, 16, 3177.
(i) Peng, L.-F.; Peng, C.; Wang, M.; Tang, Z.-L.; Jiao, Y.-C.; Xu, X.-H. Chin. J. Org. Chem. 2018, 38, 3048(in Chinese).
(彭丽芬, 彭超, 汪明, 唐子龙, 焦银春, 许新华, 有机化学, 2018, 38, 3048.)
(j) Wu, L.; Peng, L.-F.; Hu, Z.-F.; Wang, H.; Tang, Z.-L.; Jiao Y.-C.; Xu, X.-H. J. Chem. Res. 2019, 43, 503.
(a) Mao, G.; Orita, A.; Fenenko, L.; Yahiro, M.; Adachi, C.; Otera, J. Mater. Chem. Phys. 2009, 115, 378.
(b) Fenenko, L.; Shao, G.; Orita, A.; Yahiro, M.; Otera, J.; Svechnikov, S.; Adachi, C. Chem. Commun. 2007, 2278.
(c) Matsuo, D.; Yang, X.; Hamada, A.; Morimoto, K.; Kato, T.; Yahiro, M.; Adachi, C.; Orita, A.; Otera, J. Chem. Lett. 2010, 39, 1300.
(d) Oyamada, T.; Shao, G.; Uchiuzou, H.; Nakanotani, H.; Orita, A.; Otera, J.; Yahiro, M.; Adachi, C. Jpn. J. Appl. Phys., Part 2 2006, 45, 46.
(e) Yang, X.; Kajiyama, S.; Fang, J.-K.; Xu, F.; Uemura, Y.; Koumura, N.; Hara, K.; Orita, A.; Otera, J. Bull. Chem. Soc. Jpn. 2012, 85, 687.
(f) Yang, X.; Fang, J.-K.; Suzuma, Y.; Xu, F.; Orita, A.; Otera, J.; Kajiyama, S.; Koumura, N.; Hara, K. Chem. Lett. 2011, 40, 620.
(g) Peng, L.-F.; Hu, Z.-F.; Wang, H.; Wu, L.; Jiao, Y.-C.; Tang, Z.-L.; Xu, X.-H. RSC. Adv. 2020, 10232.
(h) Peng, L.-F.; Lei, J.-Y.; Wu, L.; Tang, Z.-L.; Luo, Z.-P.; Jiao Y.-C.; Xu, X.-H. J. Chem. Res. 2018, 42, 271.
(i) Peng, L.-F.; Li, R.-Z.; Tang, Z.-L.; Chen, J.-Y.; Yi, R.-N.; Xu, X.-H. Tetrahedron 2017, 73, 3099.
(l) Peng, L.-F.; Jiang, J.; Peng, C.; Dai, N.-N.; Tang, Z.-L.; Jiao Y.-C.; Chen, J.-Y.; Xu, X.-H. Chin. J. Org. Chem. 2017, 37, 3013(in Chinese).
(彭丽芬, 蒋娟, 彭超, 代宁宁, 唐子龙, 焦银春, 陈锦杨, 许新华, 有机化学, 2017, 37, 3013.)
(m) Peng, L.-F.; Hu, Z.-F.; Lu, Q.-C.; Tang, Z.-L.; Jiao Y.-C.; Xu, X.-H. Chin. Chem. Lett. 2019, 30, 2151.
(n) Wu, L.; Peng, L.-F.; Hu, Z.-F.; Jiao, Y.-C.; Tang, Z.-L.; Xu, X.-H. Curr. Org. Synth. 2020, 17, 271.
(a) Rankin, G. M.; Maxwell-Cameron, I.; Painter, G. F.; Larsen, D. S. J. Org. Chem. 2013, 78, 5264.
(b) Urones, B.; Gómez Arrayás, R.; Carretero, J. C. Org. Lett. 2013, 15, 1120.
(c) Balbuena, P.; Gonçalves-Pereira, R.; Jiménez Blanco, J. L.; García-Moreno, M. I.; Lesur, D.; Ortiz Mellet, C.; García Fernández, J. M. J. Org. Chem. 2013, 78, 1390.
(d) Muranaka, K.; Ichikawa, S.; Matsuda, A. J. Org. Chem. 2011, 76, 9278.
(e) Ihara, H.; Koyanagi, M.; Suginome, M. Org. Lett. 2011, 13, 2662.
(f) Liang, H.; Corey, E. J. Org. Lett. 2011, 13, 4120.
(a) Palmer, C. J.; Casida, J. E. Tetrahedron Lett. 1990, 31, 2857.
(b) Andreev, A. A.; Konshin, V. V.; Komarov, N. V.; Rubin, M.; Brouwer, C.; Gevorgyan, V. Org. Lett. 2004, 6, 421.
(c) Jiang, H.; Zhu, S. Tetrahedron Lett. 2005, 46, 517.
Ernst, A.; Gobbi, L.; Vasella, A. Tetrahedron Lett. 1996, 37, 7959. doi: 10.1016/0040-4039(96)01838-2
Höger, S.; Bonrad, K. J. Org. Chem. 2000, 65, 2243. doi: 10.1021/jo991746m
Gaefke, G.; Höger, S. Synthesis 2008, 2155.
Yang, X.; Matsuo, D.; Suzuma, Y.; Fang, J.-K.; Xu, F.; Orita, A.; Otera, J. Synlett 2011, 2402.
(a) Ito, H.; Arimoto, K.; Senusui, H.O.; Hosomi, A. Tetrahedron Lett. 1997, 38, 3977.
(b) Sugita, H.; Hatanaka, Y.; Hiyama, T. Chem. Lett. 1996, 25, 379.
Davidsohn, W. E.; Henry, M. C. Chem. Rev. 1967, 67, 73. doi: 10.1021/cr60245a003
(a) Cai, C.; Vasella, A. Helv. Chim. Acta 1995, 78, 732.
(b) Nishikawa, T.; Ino, A.; Isobe, M.; Tetrahedron 1994, 50, 1449.
(c) Scott, L. T.; Cooney, M. J.; Johnels, D. J. Am. Chem. Soc. 1990, 112, 4054.
(d) Lu, Y.-F.; Fallis, A. G. Tetrahedron Lett. 1993, 34, 3367.
(e) Nielsen, M. B.; Diederich, F. Synlett 2002, 544.
(f) Tobe, Y.; Utsumi, N.; Kawabata, K.; Naemura, K. Tetrahedron Lett. 1996, 37, 9325.
Nishikawa, T.; Ino, A.; Isobe, M. Tetrahedron 1994, 50, 1449. doi: 10.1016/S0040-4020(01)80629-3
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874. doi: 10.1021/cr050992x
Li, H.-B.; Petersen, J. L.; Wang, K.-K. J. Org. Chem. 2001, 66, 7804. doi: 10.1021/jo010687l
(a) Haley, M. M.; Bell, M. L.; English, J. J.; Johnson, C. A.; Weakley, T. J. R. J. Am. Chem. Soc. 1997, 119, 2956.
(b) Bell, M. L.; Chiechi, R. C.; Johnson, C. A.; Kimbal, D. B.; Matzger, A.; Wan, W. B.; Weakley, T. J. R.; Haley, M. M. Tetrahedron 2001, 57, 3507.
(a) Sonogashira, K.; Tohda, Y.; Hagihara, N. Tetrahedron Lett. 1975, 4467.
(b) Tohda, Y.; Sonogashira, K.; Hagihara, N. Synthesis 1977, 777.
(c) Takahashi, S.; Kuroyama, Y.; Sonogashira, K.; Hagihara, N. Synthesis 1980, 627.
Peng, L.-F.; Xu, F.; Suzuma, Y.; Orita, A.; Otera, J. J. Org. Chem. 2013, 78, 12802. doi: 10.1021/jo402176w
Peng, L.-F.; Xu, F.; Shinohara, K.; Orita, A.; Otera, J. Chem. Lett. 2014, 43, 1610. doi: 10.1246/cl.140579
Tahara, K.; Furukawa, S.; Uji-i, H.; Uchino, T.; Ichikawa, T.; Zhang, J.; Mandouh, W.; Sonoda, M.; Schryver, F. C. D.; Feyter, S. D.; Tobe, Y. J. Am. Chem. Soc. 2006, 128, 16613. doi: 10.1021/ja0655441
(a) Peña-López, M.; Ayán-Varela, M.; Sarandeses, L. A.; Sestelo, J. P. Chem. Eur. J. 2010, 16, 9905.
(b) Dogan, J.; Schulte, J. B.; Swiegers, G. F.; Wild, S. B. J. Org. Chem. 2000, 65, 951.
(c) Lu, E.; Chen, Y.; Zhou, J.; Leng, X. Organometallics 2012, 31, 4574.
(a) Kosugi, M.; Fugami, K. Handbook of Organopalladium Chemistry for Organic Synthesis, Ed.: Negishi, E., Wiley, New York, 2002.
(b) Tsuji, J. Palladium Reagents and Catalysts, Wiley, Chichester, U. K. 2004.
(c) Stille, J. K. Angew. Chem., Int. Ed. Engl. 1986, 25, 508.
Warner, B. P.; Buchwald, S. L. J. Org. Chem. 1994, 59, 5822. doi: 10.1021/jo00098a052
(a) Mössinger, D.; Jester, S. S.; Sigmund, E.; Müller, U.; Höger, S. Macromolecules 2009, 42, 7974.
(b) Nicolaou, K. C.; Zipkin, R. E.; Petasis, N. A. J. Am. Chem. Soc. 1982, 104, 5558.
Peña-López, M.; Ayán-Varela, M.; Sarandeses, L. A.; Sestelo, J. P. Chem. Eur. J. 2010, 16, 9905. doi: 10.1002/chem.201000726
(a) Peng, L.-F.; Xu, F.; Shinohara, K.; Orita, A.; Otera, J. Org. Chem. Front. 2015, 2, 248.
(b) Ikegashira, K.; Nishihara, Y.; Hirabayashi, K.; Mori, A.; Hiyama, T. Chem. Commun. 1997, 1039.
(c) Nishihara, Y.; Ikegashira, K.; Mori, A.; Hiyama, T. Chem. Lett. 1997, 26, 1233.
(d) Nishihara, Y.; Ikegashira, K.; Hirabayashi, K.; Ando, J.; Mori, A.; Hiyama, T. J. Org. Chem. 2000, 65, 1780.

Scheme 3 Trialkylsilyl group as a protecting group for terminal acetylene in transition metal-catalyzed coupling reactions

Scheme 12 Application of Ph2P(O) protection group in transition metal-catalyzed coupling reactions

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:










