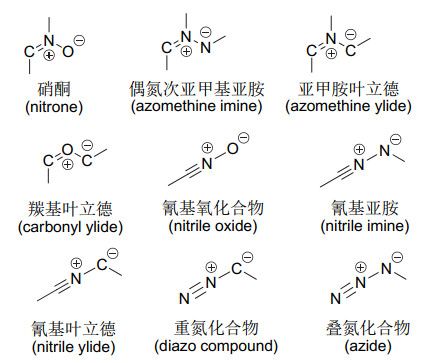
图 1.
常见的偶极子
Figure 1.
Common 1, 3-diploes

1, 3-偶极环加成反应从20世纪60年代由Huisgen[1]发现发展至今, 已有近60年的历史[2].在这半个多世纪里, 1, 3-偶极环加成反应取得了蓬勃的发展, 成为构建杂环骨架最为有效的方法之一.研究内容最初主要集中在1, 3-偶极子与烯烃、炔烃等亲偶极子之间发生的[3+2]环加成, 形成五元杂环化合物, 包括吡咯环、吡唑环及(异)噁唑环等.随后逐渐发展了1, 3-偶极子的[3+3]、[3+4]、[3+5]及[3+6]等环加成反应, 构建六元、七元、八元及桥杂环的化合物.相比之下, 这类环加成研究相对较少, 有待进一步被挖掘.目前, 1, 3-偶极环加成反应的综述较多[3-57], 但它们综述的对象主要是[3+2]的环加成反应, 只有少量文献提及其它类型的环加成.迄今为止, 还没有关于各种1, 3-偶极子的非[3+2]环加成反应的专门综述报道.鉴于该类环加成反应的重要性, 有必要对1, 3-偶极子的[3+n] (n≥3)环加成反应进行系统性综述. 1, 3-偶极子主要包括:硝酮(nitrone)、偶氮次甲基亚胺或亚甲胺亚胺(azomethine imine)、亚甲胺叶立德(azomethine ylide)、氰基氧化物(nitrile oxide)、氰基亚胺(nitrile imine)、氰基叶立德(nitrile ylide)、羰基叶立德(carbonyl ylide)、重氮化合物(diazo compound)、叠氮化合物(azide)(图 1).笔者按照以上的顺序分别对它们进行综述.
硝酮是能稳定存在的1, 3-偶极子, 它的制备方法简单, 早期主要用于与烯烃或炔烃进行[3+2]环加成反应来合成异噁唑类化合物, 现在逐渐应用到其它的环加成反应中.
早在1992年, McCullough课题组[58]报道了取代的烯基醚作为亲硝酮偶极子的前体, 在臭氧氧化下, 产生羰基氧化物活泼中间体I1, 随后被硝酮捕获, 发生[3+3]的环加成反应, 从而获得1, 2, 4, 5-三氧唑六环化合物.当硝酮的R3及R4都为苯基时, 反应产率大部分超过80%;而当R3为氢, R4为脂肪链时, 反应产率普遍较低.大多数产物的顺反异构无法通过核磁氢谱确认, 而在少数几个能指认的产物中, 非对映选择性也较低(≈2:1 dr).对于反应机理, 作者认为是硝酮的氧负离子进攻缺电性的羰基碳, 经过椅式过渡态, 氧负离子对亚胺的碳进攻后环化完成反应(Eq. 1).
|
|
(1) |
近年来, 供-受体环丙烷(DACs)作为高价值的三碳合成子, 被大量应用于各种环加成及重排等反应中, 从而构建碳环及杂环等环状化合物[59-62].施敏课题组[63]报道了Yb(OTf)3(三三氟甲磺酸镱)催化的DACs衍生丙二烯作为亲偶极体, 与硝酮进行快速地环加成, 获得哌啶酮类化合物, 而非正常的产物噁嗪.可能的机理是:环加成形成的噁嗪作为中间体I2, 进一步发生氮氧键的断裂, 重排形成最终的哌啶酮(Eq. 2).
|
|
(2) |
2013年, Hashmi等[64]报道了在氯化铂催化下, 邻炔基苯胺发生分子内的环化形成α, β-不饱和金属卡宾I3, 接着与硝酮进行环加成反应获得吲哚噁嗪烷.该反应体系适用于缺电子芳基的硝酮(如硝基、三氟甲基等)或富电子的炔基苯胺(如甲基), 反应均能获得较高的收率, 且反应不需要严格的无水无氧条件(Eq. 3).
|
|
(3) |
α-溴代酰胺作为重要的砌块被频繁用于各种环加成及串联反应中, 获得各种氮杂化合物[65]. 2016年, 夏宏光及吴劼团队[66]报道了α-溴代酰胺作为亲偶极子, 与异喹啉-N-氧化物的偶极环加成反应.在碳酸钠的作用下α-溴代酰胺首先脱除溴化氢, 形成氮氧烯丙基阳离子I4, 随后与异喹啉偶极子发生环加成获得噁二嗪产物(Eq. 4).偶极子的取代基R为甲氧基或R1为对硝基苯时, 形成的环加成产物会进一步重排形成酰胺类化合物.当换成碱性更强的碳酸铯时, 反应也均得到重排后的酰胺化合物.随后, 赵洪武与赵丽娇小组[67], 以及汪舰小组[68]等也分别报道了醛衍生的硝酮与α-溴代酰胺的类似反应.最近, 莫冬亮等[69]又报道了咔唑衍生的硝酮与α-溴代酰胺的环加成反应.
|
|
(4) |
2005年, Sibi小组[70]利用镍催化剂实现DACs与硝酮的不对称环加成反应, 获得噁嗪烷衍生物.该反应具有高的收率(普遍在95%~99%)及优异的对映选择性(89%~99% ee).但反应体系不适用于以下底物:环丙烷的R为叔丁基时, 几乎不反应; 当R1及R2都有取代基时, 产物的非对映选择性一般.另外硝酮的取代基对反应也有较大的影响, 当R3为呋喃基及肉桂基时, 反应的对映选择性会下降, ee值仅为70%左右(Eq. 5).
|
|
(5) |
随后, Kerr小组[71]也报道了手性DACs的类似反应. DACs在Yb(OTf)3的作用下开环, 并与硝酮进行[3+3]环加成, 获得手性基本保持的顺式噁嗪衍生物.该反应的对映选择性对温度较为敏感.当反应温度上升时, 部分环丙烷在开环后发生消旋化, 从而显著降低环丙烷自身及产物的ee值, 这也证实了反应是分步进行的(Eq. 6).
|
|
(6) |
Doyle小组一直致力于金属铑卡宾参与的有机反应, 并开发了多个手性铑催化剂. 2011年他们[72]报道了β-TBSO(叔丁基二甲硅氧)的烯基重氮乙酸甲酯作为卡宾前体, 在手性铑催化剂下形成的金属卡宾互变异构化为中间体I5, 捕获硝酮偶极子, 接着发生环化、消除RhH, 最后形成噁嗪衍生物.该反应体系尤其适用于R为富电子苯基的硝酮, 不太适用于R为缺电子苯基及环己基的, 因其对映选择性会有显著降低(R为对氟苯基或环己基, ee值都为77%); 而且当拓展至环状的硝酮时, 也仅获得54%~80%的ee值(Eq. 7).
|
|
(7) |
随后, 他们[73]又报道了类似的环加成反应, 利用烯基重氮化合物在AgSbF6(六氟锑酸银)作用下原位形成的环丙烯作为亲偶极子, 三步“一锅煮”获得顺式噁嗪产物(>20:1 dr), 而催化剂Rh2(OAc)4几乎得不到环加成的产物.作者认为: Ag(I)作为路易斯酸活化硝酮, 随后被重氮甲烷的烯醇醚亲核进攻.手性噁唑啉配体L2能有效地实现手性诱导, 获得高的收率及优异的对映选择性(普遍具有≥90% ee); 但当硝酮的Ar、Ar1分别为对溴苯基及4-氯苯基时, 产物的ee值降低至82%, 但重结晶后可以获得大于99.5%的ee值; 另外当Ar1为呋喃基时, 反应的ee值也有显著下降(79% ee) (Eq. 8).
|
|
(8) |
2007年, Hayashi团队[74]报道了在钯催化剂及手性亚磷酰胺配体L3的条件下, 三亚甲基甲烷(TMM)前体形成不稳定的I7作为亲偶极子, 与硝酮发生不对称[3+3]环加成反应.该催化体系具有优异的对映选择性(88%~93% ee), 但非对映选择性略显不足(3.2:1~8.1:1 dr).当亲偶极子的Ar换成氢时, 该催化体系无手性诱导, 另外需要在底物的普适性方面进一步提升, 如杂芳基及脂肪烃基等(Eq. 9).
|
|
(9) |
2010年, 张俊良小组[75]报道了金诱导炔酮的环异构化/[3+3]环加成的串联反应, 发现手性双膦配体L4及L5均给出优秀的立体选择性(绝大多数>20:1 dr及90%~98% ee).该催化体系尤其适用于R为缺或富电子苯基的硝酮(Eq. 10).随后, 作者发现手性亚砜膦配体Ming-Phos也能很好地催化该反应, 通过对配体构型的调控, 可以分别获得顺式产物的两个异构体[76].
|
|
(10) |
2017年, 周剑小组[77]报道了在手性镍催化下的硝酮与消旋环丙基吲哚酮的[3+3]环加成反应, 获得高的收率及优异的立体选择性, 而且还能实现该消旋体的拆分, 获得高ee值的吲哚酮.该反应体系官能团兼容性好, 硝酮的R无论是芳基还是烷基, 都能很顺利地进行反应.另外作者也扩展到了酮衍生的偶极子, 反应在0 ℃下即可顺利进行, 获得令人满意的结果(Eq. 11).
|
|
(11) |
王忠文小组[78]报道了炔基环丙酮作为亲偶极体, 用于硝酮的环异构化/环加成, 获得呋喃并氮氧杂环庚烷的串联反应.环丙基酮在Cu(Ⅱ)或Au(Ⅲ)催化下, 首先发生分子内的环异构化形成类似I8呋喃中间体(Eq. 10), 随后被硝酮的氧负离子进攻开环, 反过来碳金属键对亚胺部分的亲核进攻, 金属解离, 最后形成顺式氮氧杂䓬.取代基的电子效应对反应影响不明显, 硝酮的取代基R为芳基及烷基, 都能有效地进行.作者还考察了环丙烷的取代基效应, 当R1为氢时, 金催化的效率相比铜的更高(Eq. 12).
|
|
(12) |
2011年, Pagenkopf课题组[79]在Kerr小组的基础上(Eq. 6), 在相同的催化条件下, 利用供-受体环丁烷作为亲偶极子与硝酮进行形式上的[3+4]环加成反应, 获得氮氧杂䓬.两个非对映异构体产物可以相互转化:在0 ℃下, 最初会形成顺反异构体的混合物, 15 min内就能完成反应; 当升至室温并延长时间时, 反式异构体全部转化为热力学更稳定的顺式并环产物, 但产率要低近20%.该催化体系也适用于吡喃并环丁烷及乙氧基环丁烷, 获得顺反异构体的混合物(Eq. 13).
|
|
(13) |
2012年, 张俊良小组[80]在前期工作基础上(Eq. 10), 又报道了以消旋炔基环丙烷作为亲偶极子与硝酮进行[3+4]环加成反应, 在Au(I)/L5催化条件下, 实现炔基环丙烷的动力学拆分, 获得有用的手性炔基环丙烷砌块; 而该手性砌块目前无法通过手性诱导直接实现烯炔的不对称环丙烷化而获得(Eq. 14).
|
|
(14) |
2015年, 唐勇课题组[81]在Pagenkopf小组的基础上(Eq. 13), 报道供-受体环丁烷环加成的不对称反应, 利用手性噁唑啉配体L7、高氯酸酮作为催化剂, 实现了环丁烷与硝酮的不对称环加成, 获得高的立体选择性(大部分产物均有大于99:1 dr及90% ee).该催化剂尤其适用于R1及R2都为芳基的硝酮, 反应均能获得高的收率及立体选择性(Eq. 15).
|
|
(15) |
Hayashi小组[82-83]在前期的基础上(Eq. 9), 又发展了γ-亚甲基-δ-戊内酯作为亲偶极体与硝酮的环加成反应.在钯催化下, 戊内酯脱羧形成活性中间体I9, 随后与硝酮反应, 获得氮氧杂䓬.最初作者利用非手性亚磷酰胺配体实现该反应的非对映选择性合成, 产率绝大多数大于90%, 但非对映选择性只有中等偏上(≤15.7:1 dr).随后又利用手性亚磷酰胺类配体, 实现高对映选择性的反应, 但非对映选择性不理想(2.3:1~6.1:1 dr).最后作者将该反应扩展至偶氮次甲基亚胺偶极子以及丙烯酸酯, 同样也能获得相似的反应结果.但在底物的普适性方面还需进一步提升(Eq. 16).
|
|
(16) |
2012年, 刘瑞雄团队[84]报道了金催化下的1, 6-烯炔环异构化形成5/3并环的金卡宾中间体I10, 接着与硝酮发生[3+2+2]环加成的串联反应.该反应同时实现了构建5/7并环骨架, 获得单一非对映异构体, 并且该反应体系适用于各种底物, R为苯基及脂环基的烯炔以及富电子或缺电子(杂)芳基的硝酮, 均能以高收率得产物.最后, 作者利用手性双膦配体(R)-L5, 实现了该反应的不对称合成, 同样获得高的收率及优异的对映选择性(Eq. 17).
|
|
(17) |
偶氮次甲基亚胺(又称亚甲胺亚胺), 通常由吡唑烷酮与醛或酮在醇中回流脱水形成.这类偶极子是能稳定存在的固体, 但在碱性条件下会缓慢分解.最近它被广泛应用于[3+n] (≥3), 特别是[3+3]的环加成反应中.
2006年, Hayashi课题组[85]在前期硝酮环加成的研究基础上(Eqs. 9, 16), 又报道了三亚甲基甲烷(TMMs)与偶氮次甲基亚胺的[3+3]环加成反应.该反应利用三甲硅基烯丙基醚作为前体, 在Pd(PPh3)4作用下产生与I7相似的TMMs, 随后与偶极子进行环加成.该反应能兼容R为各种取代基的偶极子, 包括(杂)芳基, 叔丁基及环己基(Eq. 18).
|
|
(18) |
那日松小组[86]利用不稳定的亚甲胺叶立德在质子酸或路易斯酸的催化下, 与1, 3-偶极子实现[3+3]的环加成反应.作者利用亚甲胺叶立德前体, 在酸的作用下形成不稳定的亚甲胺叶立德I11, 随后与偶氮次甲基亚胺发生交叉环加成, 反应需要加入过量的叶立德前体抑制自身的二聚(Eq. 19).
|
|
(19) |
2018年, 王凯凯课题组[87]也实现了亚甲胺叶立德I11与喹啉衍生的1, 3-偶极子的[3+3]环加成反应.该反应体系的产率相比Eq. 19中的更高, 取代基对反应性至关重要, 偶极子氮上的取代苯甲酰基(Bz)换成对甲苯磺基(p-Ts)时, 反应则无法进行(Eq. 20).
|
|
(20) |
2008年, Charette小组[88]报道了Ni(ClO4)2催化的DACs作为亲偶极子与偶氮次甲基亚胺的反应, 获得哒嗪类化合物. R为富电子芳基的环丙烷适用于该反应体系, 而R为氢或乙烯基时则不太适用.总体而言, 该反应的产率及非对映选择性一般, 有提高的空间, 不过异喹啉衍生的1, 3-偶极子也能进行该反应, 高非对映选择性地生成产物(Eq. 21).
|
|
(21) |
Truskhov等[89]发现二氮环甲烷作为前体, 在Ni(Cl- O4)2•6H2O(六水合高氯酸镍)作用下形成的1, 3-偶极子DACs进行[3+3]环加成, 以中高的非对映选择性及中等产率获得哒嗪类产物.作者发现利用手性环丙烷获得手性保持的产物, 因此认为其机理是二氮环甲烷作为亲核试剂对环丙烷的类似SN2反应, 然后形成丙二酸酯的负离子对亚胺加成.当二氮环甲烷的环为四五元环时, 主要获得反式产物; 六元环时主要获得顺式产物; 非环时产率则大大降低.另外二氮环甲烷在反应条件下形成的偶极子能发生环二聚, 形成反式产物(Eq. 22).另外, 作者[90]还发现, 若在反应体系中有水存在, 形成的中间体发生水解反应, 而非环化反应, 最后得到氮烷基化的产物.
|
|
(22) |
2011年, 郭红超团队[91]报道了以有机膦为催化剂, 联烯为亲偶极子的环加成反应, 发现可以实现[3+2]、[3+3]、[3+4]及[3+2+3]等环化.其中[3+3]环加成形成1, 2-二氮杂环己烷的产率较低, 主要原因是伴随[3+2]及未知的副产物.可能的机理为叔丁基膦首先对联烯共轭加成, 形成的酯α-碳负离子I12接着进攻偶极子的亲电性碳, 反过来, 偶极子的氮负离子对烯烃加成、氢迁移、解离催化剂, 最后生成产物(Eq. 23).
|
|
(23) |
2012年, 黄有小组[92]报道了三苯基膦催化炔酮与1, 2-偶极子的环加成反应.由三苯基膦对炔酮的共轭加成启动反应, 随后发生质子交换形成中间体I13, 再进行后续的环加成.作者发现使用混合溶剂能显著提高反应的收率及缩短反应的时间, 可能的原因是外加的正丁醇能有效降低质子转移步骤的活化能[93].该反应体系整体上表现良好, 产率普遍在中等以上; Ar无论为富电子还是缺电子的苯基, 都能很好地适用于该反应体系(Eq. 24).
|
|
(24) |
2012年, 郭红超课题组[94]又发现丙炔酸甲酯在三叔丁基膦的催化下, 与偶氮次甲基亚胺偶极子能进行[3+2]及[3+3]的环加成反应.随后他们对反应条件进行优化, 发现三苯基膦可以催化炔酮与异喹啉衍生的1, 3-偶极子选择性地进行[3+3]环加成[95], 苯酚作为添加剂有助于产率的提升(Eq. 25).
Doyle课题组[96]在完成硝酮的环加成反应后(Eqs. 7, 8), 又开展了重氮化合物与偶氮次甲基亚胺的环加成研究.该反应体系官能团兼容性优秀, 产物的产率及非对映选择性都较高.反应的机理与硝酮的环加成类似(Eq. 7), 形成铑卡宾中间I5.如果利用Cu(I)或Au(I)作催化剂, 反应的结果与Eq. 8不一样, 主要形成[3+2]环加成产物.重氮化合物的R2为氢、甲基或苯环时, 反应得到混合物; 该催化体系也不太适用于R1为甲基、苄基、氢等的1, 3-偶极子, 因为主要发生氮氮键的裂解形成开链产物(Eq. 26).
|
|
(25) |
|
|
(26) |
2018年, 宣俊小组[97]报道了氮氧烯丙基正离子作为亲偶极子, 与异喹啉衍生的偶氮次甲基亚胺进行[3+3]环加成反应.作者以α-溴代酰胺作为亲偶极子的前体, 在碳酸钾作用下原位形成氮氧烯丙基正离子I4, 而1, 3-偶极子则由邻炔基苯甲醛腙在三氟甲磺酸银催化下原位得到.该反应能实现两步“一锅”反应, 炔基相连的R2可以是芳基、脂肪及脂环链等(Eq. 27).
|
|
(27) |
Sawant等[98]发展了邻叠氮基苯甲醛、异腈及芳基磺酰肼在醋酸钯的催化下制备杂环偶氮次甲基亚胺的反应.在此基础上, 利用该产物与各种亲偶极子, 如硝基烯烃、丁二烯酸酯、环酮及α-溴代酰胺等进行多样性导向合成(DOS)的1, 3-偶极环加成反应, 两步“一锅煮”的方法也能顺利地被实现.其中与α-溴代酰胺的[3+3]环加成, 获得5个三氮六元环产物.该反应体系的效率不高, “一锅煮”的产率比分步反应低4%~14%, 且底物扩展较少, 但一锅反应减少后处理及纯化步骤(Eq. 28).
|
|
(28) |
王锐小组[99]实现了在三乙胺催化下硫代异氰酸吲哚酮与偶氮次甲基亚胺快速进行[3+3]环加成的反应, 以75%~94%的收率及6:1~>20:1 dr获得1, 2, 4-三嗪吲哚酮类化合物.该反应的可能机理为:在碱的作用下, 形成的吲哚酮烯醇负离子进攻偶极子的亲电性碳, 随后亚胺相邻的氮负离子反过来进攻硫代羰基, 最后从体系中获得氢, 异构化生成最终产物.该反应时间短(5 min), 尤其适用于富电子或缺电子芳基的偶极子, 其产率及非对映选择性普遍较高, 而且碱的用量可以降低到1 mol% (Eq. 29).
|
|
(29) |
王春江课题组[100]以巯基乙醛的二聚体1, 4-二噻烷-2, 5-二醇作为亲偶极子, 发现在1 mol%的1, 4-二氮杂双环[2.2.2]辛烷(DABCO)的催化下, 即可与偶极子实现[3+3]环化反应, 获得噻哒嗪化合物.该反应实现了硫杂二氮环己烷的合成, 且非对映选择性高, 由于异头碳效应的影响, 产物都为顺式构型(Eq. 30).
|
|
(30) |
2014年, 徐显秀及刘群[101]利用异腈作为三元合成子, 分别与醛或喹啉形成的偶氮次甲基亚胺进行[3+3]环加成, 最后获得两类1, 2, 4-三嗪化合物.该反应体系条件温和, 底物普适性较好, 有高的区域及非对映选择性(Eqs. 31, 32).
|
|
(31) |
|
|
(32) |
2009年, Toste小组[102]利用金催化剂实现苯甲酸-2-甲基丁炔酯与偶极子非对映选择性的[3+3]环加成反应.该催化体系尤其适用于取代基R为缺电子芳基的偶极子, 产物有不错的收率及非对映选择性, 但当其为邻取代芳环时, 可能因为位阻的影响, 非对映选择性下降; 同样当其为环丙基时, 该反应体系会使产物的收率及非对映选择性都会下降.另外R1及R2的体积越大, 反应的非对映选择性越高, 最高超过20:1 dr.该反应机理是首先炔丙醇酯在金的作用下发生异构化, 形成烯醇酯金卡宾I14, 接着偶极子氮负离子进攻, 随后烯醇酯反过来对偶极子亲电性碳进攻, 最后释放金催化剂, 获得5/6并环产物.该反应还需要在扩展炔丙醇酯上进一步提升(Eq. 33).
|
|
(33) |
随后, Scheidt小组[103]报道了氮杂环卡宾(NHC)C2催化的偶氮次甲基亚胺与肉桂醛的[3+3]环加成反应.该反应首先由氮杂环卡宾与醛加成形成类似Breslow中间体I15, 接着I15对1, 3-偶极子的亲核加成, 异构化形成咪唑盐, 最后酰胺化释放氮杂环卡宾, 获得1, 2-哒嗪酮产物.反应具有高度的顺式选择性, 可能是由于1, 3-偶极子与中间体烯醇之间氢键影响的缘故.总体而言, 该反应的产率及非对映选择性都优异, 但不足之处是, 肉桂醛的苯环有吸电子基, 偶极子的Ar为邻取代苯基或环丙基时, 反应均不能进行(Eq. 34).
|
|
(34) |
Molchanov等[104]发现在银盐的作用下, 乙烯基吡咯作为三碳合成子与1, 3-偶极子发生[3+3]而非[3+2]的环加成.但反应的收率及选择性都一般, 而且反应的温度也较高.可能的原因是富电子的吡咯易发生自身聚合(Eq. 35).作者也把1, 3-偶极子扩展到了硝酮, 高氯酸镍作为催化剂得到相似的结果.
|
|
(35) |
2018年, Ramachary等[105]实现了以对苯二酚类作为亲偶极体, 在叔丁醇钾的作用下与1, 3-偶极子发生[3+3]环化反应, 获得一类新的噁哒嗪.该反应体系的特点是条件温和, 底物普适性广, 大多数产物的非对映选择性优异.但对苯二酚的取代基对反应影响较大, 当R5为甲基或R2为大位阻的仲丁基时, 反应均无法进行(Eq. 36).
|
|
(36) |
2019年, 赵立明小组[106]实现了α-氯代芳香甲醛肟的[3+3]环加成反应.甲醛肟在碱的作用下产生氰基氧化合物偶极子I16, 随后与偶氮次甲基亚胺发生交叉环加成反应.该反应体系适用于缺电子或者富电子(杂)芳基的偶氮次甲基亚胺, 以及缺电子或富电子苯基的甲醛肟.由于α-氯代脂肪族醛肟有部分分解, 导致产率普遍在中等水平(Eq. 37).
|
|
(37) |
2013年, 李兴伟课题组[107]报道了银催化的分子内加成/环化反应, 获得[3+3]的环加成产物.炔基偶极子在银盐的作用下发生6-exo-trig型环化, 接着亲核试剂(如甲基酮、炔烃或水)进攻1, 3-偶极子的亲电性碳, 形成三环哒嗪化合物, 脯氨酸作为添加剂能加速反应.当用甲基酮作亲核试剂时, 反应室温即可进行; 而用非甲基酮时, 需要提高反应温度到80 ℃.当用炔烃作亲核试剂时, 也需要在60 ℃下才能实现反应, 但不需要添加剂.该反应如果用含水溶剂作为反应媒介, 水作为亲核试剂主要形成羟基取代的产物.另外, 该反应体系也适用于硝基烷烃(Eqs. 38, 39).
|
|
(38) |
|
|
(39) |
Matloubi-Mogaddam等[108]利用诺文格尔中间体作为亲偶极子, 与靛红偶氮次甲基亚胺进行反应, 在DA- BCO的催化下进行[3+3]环加成, 获得1, 2-二氮杂环己烷产物.该反应区别于之前的报道, 没有按预想的[3+2]环加成进行(Eq. 40).靛红芳基及氮上的取代基扩展较少, 特别是氮上为氢时, 反应的结果不得而知.因为在类似用靛红衍生的偶极子的反应中, 氮上没有取代基时, 反应几乎得不到产物[109-110].
|
|
(40) |
2013年, 李玉学、唐勇及孙秀丽[111]联合报道了Ni(ClO4)2/手性双噁唑啉配体L9的催化体系, 能有效地促进1, 3-偶极子与DACs的不对称环加成.作者对该反应的机理进行了详细的理论及实验研究, 发现配体茚环部分与环丙烷芳基存在的π-π相互作用对反应的手性诱导关键作用, 因为环丙烷的芳基换成氢等不会产生手性诱导, 而且改变配体上侧链的茚环部分的芳基, 也会显著降低对映选择性.该催化剂体系适用于各种取代的环丙烷, 并且该项研究为设计新的手性催化剂提供了有力的参考价值(Eq. 41).
|
|
(41) |
同年, 郭红超与胡向平[112]合作, 利用手性二茂铁基氮膦配体L10, 实现了铜催化偶氮次甲基亚胺与甲亚胺叶立德的高立体选择性的不对称[3+3]环加成反应.该催化体系适用于大多数的反应底物, 其产物拥有>20:1 dr及≈90% ee (Eq. 42).
|
|
(42) |
2015年, 郭红超小组[113]又报道了Morita-Baylis- Hillman (MBH)碳酸酯作为亲偶极子与二氢异喹啉衍生的1, 3-偶极子不对称[3+3]环化反应.该催化体系适用于各种富电子或缺电子苯基、噻吩及萘环的碳酸酯, 所有产物都有非常优异立体选择性(>20:1 dr; ≥98% ee) (Eq. 43).
|
|
(43) |
2018年, Schmalz小组[114]报道了共轭烯炔酮作为亲偶极子, 在手性金催化剂的作用下, 实现与烯炔酮的环化成类似I8的中间体, 接着与偶极子形式[3+3]环加成获得5/6/5三环哒嗪.利用他们自己发展的手性双膦配体L11, 该反应的产率及立体选择性都优异.当Ar为4-甲基呋喃基时, 反应获得最高的ee值; 该催化体系不太适用于脂肪烃基的烯炔, 如正丙基及正丁基时, 其产物是不可分离的混合物, 且对映选择性大大下降(Eq. 44).
|
|
(44) |
Doyle小组[115]报道了烯酮重氮化合物与偶氮次甲基亚胺的[3+2+1]偶极环加成反应.作者利用自己开发的手性噁唑烷酮催化剂Rh2(4S-MPPIM)4, 反应具有非常优异的非对映选择性, 但遗憾的是无对映选择性.作者通过实验证明, 催化剂首先是作为路易斯酸活化1, 3-偶极子, 而非与重氮形成铑卡宾, 随后偶极子与α-重氮酮发生亲电加成, 释放N2, 第二分子重氮酮再加成、释放N2, 最后环化得到产物(Eq. 45).
郭红超等[91]以有机膦为催化剂, 实现联烯与偶极子的多种环加成反应, 包括[3+2]、[3+3]、[3+4]及[3+2+3]等(Eq. 23).但[3+4]环化反应的结果也不太理想, 仅给出一个反应结果, 以63%总收率获得顺式及反式的1, 2-二氮䓬衍生物, 伴随[3+2]及[3+3]环加成的副产物(Eq. 46).
|
|
(45) |
|
|
(46) |
随后郭红超等[116]将1, 3-偶极子换成异喹啉衍生的偶极子, 发现三叔丁基膦为催化剂, 与联烯反应获得以七元杂环为主的产物; 而当利用三甲基膦时, 主要是五元环为主的产物.还发现如果联烯的芳基换成烷基时, 无论是三苯基膦还是三丁基膦, 都获得[3+2]产物(Eq. 47).
|
|
(47) |
他们[117]在2015年对该催化体系又做了底物的扩展研究, 从原来的喹啉扩展到了喹啉、异喹啉及菲啶等衍生的1, 3-偶极子.在相同反应条件下发生[3+4]环加成反应, 获得1, 2-二氮䓬化合物, 进一步拓展了该反应的应用范围(Eq. 48).
2013年, 肖文精等[118]报道了以α-卤代腙为共轭杂二烯的前体, 在碳酸钾的作用下形成杂二烯I17, 进而与二氢异喹啉衍生的偶氮次甲基亚胺发生[3+4]的环加成反应, 获得1, 2, 4, 5-四氮䓬衍生物.该反应体系的优点是条件温和, 适用于各种取代基的α-卤代腙, 当R为富电子或缺电子芳基、叔丁基时, 都能获得优异的收率, 而且也适用于环状的α-卤代腙(Eq. 49).
|
|
(48) |
|
|
(49) |
2020年, 侯云雷及宫平团队[119]也报道了α-卤代腙作为亲偶极子的前体, 与邻炔基苯甲醛腙的环加成.炔基苯甲醛腙首先在银离子的作用下原位形成1, 3-偶极子, α-卤代腙在碱的作用下消除溴化氢形成氮杂二烯(类似I17), 随后与偶极子进行[4+3]环加成获得1, 2, 4, 5-四氮䓬衍生物.该催化体系适用于Ar为富电子或缺电子的单或二取代苯基的α-卤代腙(Eq. 50).
|
|
(50) |
邻醌亚甲基化合物(o-QMs)作为四元合成子, 已被广泛应用于各种环加成反应来构建氧杂环状化合物[120-122]. 2017年, 石枫小组[123]又发现在消旋联萘膦酸的作用下, o-QMs的前体邻羟基苯甲醇与偶极子能有效地进行[3+4]环加成反应.该反应体系能使各种取代的底物都能顺利进行, 产物具有优异的非对映选择性(大多数>19:1 dr) (Eq. 51).
|
|
(51) |
2018年, 杜志云课题组[124]报道了以氮邻醌亚甲基化合物(aza-oQMs)为亲偶极体, 与二氢异喹啉偶氮次甲基亚胺偶极子进行[3+4]的环加成.作者以邻氯亚甲基芳香胺为亲偶极子前体, 在碳酸钠的作用下消除一分子氯化氢原位形成I19.该反应在室温下即可顺利进行, 亲偶极子上的R2为强吸电子基如硝基时产率相对较低, 取代基的位置对反应几乎不影响; 另外该反应体系也适用于R1为氢、溴或甲基以及R为苯基、对氯苯基、乙酰基的偶极子, 产率都在80%以上(Eq. 52).
|
|
(52) |
随后, 金巧梅等[125]也报道了aza-oQMs与靛红偶极子进行[3+4]的并环反应.该反应体系尤其适用于R1为甲氧基的偶极子, 产率最高为96%, 但R1为甲基或氯原子时产率下降严重.该反应体系不适用于R为氢的偶极子以及R2为对甲苯磺酰基或乙酰基的苯胺.作者对化合物做了进一步的体外抗癌活性筛选, 发现在微摩尔浓度下对人乳腺癌细胞MIF-7及MDA-MB-231具有较好的抑制活性(Eq. 53).
2019年, 石枫团队[126]发现二苯基次膦酸的作用下, 靛红酐在与偶氮次甲基亚胺能有效地进行脱羧/[3+4]环加成反应, 但获得的产物是经过重排之后的1, 2, 4-三氮䓬衍生物.该反应体系适用于R为各种取代的苯基、杂芳基、苄基及环己基的偶极子, 尤其适用于氮上供电子基的靛红酐(Eq. 54).
|
|
(53) |
|
|
(54) |
2020年, 柳红小组[127]报道了钯催化的1, 3-偶极子环加成, 利用BocOCH2-烯丙醇作为亲偶极子的前体.在钯的作用下, 该前体先脱除二氧化碳及叔丁醇形成烯丙基钯的1, 4-两性离子I21, 再与1, 3-偶极子进行分步环加成.该反应体系适合于喹啉及异喹啉衍生的偶极子, 当偶极子的R为芳基磺酰基时, 反应均获得中上等产率.总体而言, 喹啉及异喹啉环上没有取代基比有取代基的产率更高(Eq. 55).
|
|
(55) |
2016年, 王春江小组[128]报道了类似反应的不对称合成, 采用Cu(OTf)2/(S, S)-双噁唑啉L13为催化体系, 能有效地实现简单醛衍生的1, 3-偶极子与α-卤代腙的不对称[3+3]环加成.利用消旋的偶极子作为反应物, 通过动态动力学转化, 最终形成顺式环加成产物, 并且实现了对消旋偶极子的动力学拆分, 该反应体系能适用于各种取代的偶极子及腙(Eq. 56).
|
|
(56) |
郭红超等[91]发现以有机膦为催化剂联烯为亲偶极子的环加成反应, 实现了[3+2]、[3+3]、[3+4]及[3+ 2+3]等环化.其中, 利用三环己基膦催化联烯酯与偶氮次甲基亚胺发生的[3+2+3]环加成反应, 形成了1, 2-二氮杂环辛烷.该反应可能的机理为, 联烯先在三环己基膦的作用下形成活性中间体I12, 再对第二分子的联烯进行迈克尔加成, 形成的1, 5-离子中间体被1, 3-偶极子所捕获, 最后环化、消除三环己基膦形成产物.该反应体系适用于各种缺或富电子的苯基, 尤其适用于2-卤苯基, 有最高的收率(Eq. 57).
|
|
郭红超小组在前面工作的基础上[54, 91, 95, 111-112, 115-116], 于2018年报道了亲偶极子乙烯基碳酸酯(VEC), 在钯催化剂下脱羧, 再与异喹啉或喹喔啉的偶极子发生[3+5]环加成, 获得1, 2-二氮-4-氧杂环辛烯化合物[129].该反应受配体影响很大, 利用双膦配体L14能有效地抑制[3+3]环化.反应体系适用于R1为芳基的亲偶极子, 获得中等到高的收率以及优异的化学选择性.除了乙烯基碳酸酯, 该反应体系还能应用于乙烯基环氧乙烷.
该反应的可能机理为:碳酸酯或环氧乙烷在钯的作用下, 脱羧或开环形成烯丙基钯的1, 5-偶极子I22, 随后其氧负离子亲核进攻亚胺部分, 最后烯丙基部分与氮负离子的关环完成反应(Eq. 58).
|
|
(58) |
陈应春小组[130]报道了在金鸡纳碱的作用下环状MBH醇与偶极子的不对称[3+5]的反应, 以优异的立体选择性构建了5/5/5三并环化合物.硫醇作为活化剂, 它首先对环状MBH醇共轭加成, 脱水形成β-硫代环外不饱和烯酮, 随后与金鸡纳碱反应获得构型匹配的活性烯胺I23, 该中间体对偶极子亲核加成, 随后经过巯基消除、异构化等, 重新形成活性共轭的烯亚胺, 接着1, 3-偶极子部分亲核性氮对其迈克尔加成, 最后脱去催化剂形成[3+5]环加成产物.作者利用控制实验及高分辨质谱检测到关键活性中间体来证实上述机理的合理性.该反应首次利用双催化剂体系, 实现新颖的活化模式及反应途径(Eq. 59).
|
|
(59) |
亚甲胺叶立德是最早被应用于1, 3-偶极环加成反应的偶极子, 它的[3+2]环加成反应研究发展相当成熟, 最近逐渐将研究热点转移至它的[3+3]、[3+4]、[3+5]及[3+6]等环加成上.
对1, 3-偶极子环加成反应的持续研究, 郭红超小组[131]于2015年报道了苯醌单亚胺作为亲偶极子的[3+3]环化反应, 以膦酸C4作为催化剂, 偶极子对单亚胺进行迈克尔加成, 随后羰基异构化成烯醇, 羟基对亚胺部分加成形成苯并噁嗪衍生物.芳基上的取代基为供电子基, 如位阻小的烷基及烷氧基时, 反应获得高的产率(≥82%) (Eq. 60).
|
|
(60) |
2014年, 石枫与屠树江[132]联合报道了3-吲哚基甲醇作为亲偶极子, 与亚甲胺叶立德进行不对称的[3+3]环加成.利用手性磷酸C6作为催化剂, 该反应有优异的对映选择性, 大部分的ee值>90%, 而非对映选择性一般, 大部分的dr值在1:1~3:1之间.该催化体系尤其适合于R1为苄基、取代苄基及甲基的底物, 主产物的ee值均保持在95%以上.作者还考察了芳基醛的取代基效应, 发现吸电子基团, 如硝基、氰基及卤素等具有优异的对映选择性, 而供电子基团, 如甲氧基的非对映选择性大幅提高, 超过19:1 dr.吲哚氮上活泼氢与手性磷酸的氢键作用, 能有效地实现手性诱导, 因为当活泼氢被取代后, 无产物生成(Eq. 61).
|
|
(61) |
2015年, 郭红超小组[133]报道了Cu(I)及手性二茂铁基氮膦配体L15作为催化剂, 实现在温和条件下二氮杂萘的亚甲胺叶立德与亚胺酯偶极子的交叉[3+3]环加成反应.该反应具有优异的立体选择性(≥90% ee, 绝大部分产物具有>20:1 dr).该反应体系官能团兼容性优秀, 亚甲胺叶立德的R1取代基为缺电子或富电子(杂)芳基时, 均获得优异的收率及立体选择性; 氮杂萘上的R基团为强供电子的甲氧基时, 其ee值基本保持(Eq. 62).
|
|
(62) |
2016年, 邓卫平课题组[134]也利用L15作为配体, 以吲哚基硝基乙烯作为亲偶极子, 实现它与偶极子的高立体选择性(≥97% ee及11.5:1~>49:1 dr)环加成反应.作者通过调控反应的温度, 可以显著降低[3+2]环加成产物的比例.该催化体系尤其适合于富电子苯基的叶立德, 具有优异的非对映选择性(>49:1 dr) (Eq. 63).
|
|
(63) |
2019年, Kim等[135]利用Donor-Acceptor氮丙啶作为亚甲胺叶立德前体, 在手性催化剂的存在下与邻烯基二烷基苯胺发生环加成反应.首先苯胺与亚甲胺叶立德进行类似的傅克反应形成中间体I24, 随后它的酯基α-碳负离子与烯酮发生迈克尔加成, 再去氢获得产物.该反应的非对映选择性普遍在中等(2:1~9:1 dr), 当氮丙啶的Ar为对氟苯基或苯胺的R1为杂芳环(2-呋喃或噻吩)时, 其dr值超过30:1, 同时ee值也普遍在60%~70%之间(Eq. 64).
|
|
(64) |
中国药科大学的周庆发等[136]发展了三苯基膦催化的邻羟基苯甲醛亚甲胺叶立德与联烯酯的[3+4]环加成反应, 获得吖庚因类化合物, 而通过在反应中加入碳酸铯和提高反应温度, 可以获得重排的三环吖庚因.该反应首先由三苯基膦对联烯共轭加成, 消除乙酰氧基形成中间体I25, 随后与偶极子环加成获得产物; 若有碱的存在, 该产物进一步发生分子内酯基跨环迁移, 最后形成内酯(Eqs. 65, 66).
|
|
(65) |
|
|
(66) |
2019年, 赵洪武等[137]也利用VEC (Eq. 58)作为亲偶极体, 在钯催化下形成钯的1, 5-偶极子I22与靛红衍生的亚甲胺叶立德进行反应, 获得[3+5]的环加成产物.该反应体系获得优异的产率(普遍大于80%)及非对映选择性(>20:1 dr) (Eq. 67).
|
|
(67) |
与此同时, 施敏小组[138]也报道了同样的反应, 反应条件略有不同, 需要两步进行, 即先在钯催化剂的作用下, VEC脱CO2形成中间体I22, 接着亚甲胺叶立德对它亲核进攻得到烯丙醇, 随后醇在酸C4的作用下, 与亚胺加成形成最后的产物, 该反应的产率略低于赵洪武所报道的(Eq. 68).
|
|
(68) |
郭红超小组[139]发展了高丝氨酸内酯为1, 3-偶极子前体, 利用醋酸银及三苯基膦为催化剂, 实现三环哌啶化合物的高非对映选择性合成(>20:1 dr), 但反应的产率普遍在50%~70%之间, 除了模型底物(90%)外.底物芳基上的取代基电子效应对反应影响不明显(Eq. 69).
|
|
(69) |
2012年, Waldmann小组[140]报道了富烯作为亲偶极体, 在Cu(I)及手性硫膦配体L17的催化下与亚甲胺叶立德、N-甲基马来酰亚胺进行[3+6]/[4+2]的“一锅”反应, 获得8个手性中心的桥环哌啶化合物.该反应体系两步一锅煮, 简化反应步骤, 绝大部分的产物ee值≥87%, 尤其适用于R基团为芳基的富烯; 但为异丁基或甲基时, 产物的ee值下降至最低36%;叶立德的Ar换成烷基也几乎得不到产物.另外反应的非对映选择性绝大部分只有中等水平, 只有当R及R1为甲基时, 才有高的非对映选择性(>19:1 dr), 但其ee值下降至60%.作者认为配体上大位阻的叔丁基巯基对[3+6]环加成的立体选择性起了决定作用(Eq. 70).
|
|
(70) |
2013年, 王春江团队[141]也利用富烯作为亲偶极子进行不对称的[3+6]环加成, 获得具有3~4个手性碳的环戊二烯基哌啶化合物.该催化体系可以获得优异的产率及对映选择性(55%~92%, ≥92% ee), 尤其适合于R为芳基、脂肪及脂环烃的偶极子以及R1及R2为烷基或脂环烃的富烯.作者也展示该类产物作为合成子的应用价值, 可能的反应机理为:铜离子、手性配体与偶极子先形成复合物, 由于富烯的芳基与配体上磷原子苯基的相互排斥, 偶极子从富烯的位阻小的一面(Re面)进攻, 从而获得相应构型的产物(Eq. 71).
|
|
(71) |
紧接着他们[142]又报道了环庚三烯酮与亚甲胺偶极子的[3+6]环加成, 利用CuBF4/手性氮膦配体作为催化剂, 高立体选择性地实现不对称诱导.该反应体系官能团兼容性优秀, R为富电子或缺电子芳基的偶极子, 都能获得满意的结果, 其中R为对氟苯基给出最好的结果(90%的产率、20:1 dr及98% ee).可能的反应机理为:原位形成的偶极子与铜配位, 其亲核性的碳对环庚三烯酮进行1, 8-共轭加成, 由于配体上磷原子苯基的位阻, 只能从相反一面反应, 随后烯醇负离子对亚胺进行反应, 获得产物(Eq. 72).
|
|
(72) |
随后, 他们[143]又报道了环庚三烯环外酮作为亲偶极子的环加成反应, 配体L18给出高的立体选择性, 获得内型产物.该催化体系尤其适用于R1为富电子或缺电子苯基及脂基的酮, 可以获得优异立体选择性.另外R1为酯基时反应生成[3+2]的产物.可能原因在于酮羰基稳定碳负离子的能力更强, 更能促进第一步的共轭加成(Eq. 73).
|
|
(73) |
最后, 他们[144]还发现在CuBF4/L19的催化体系下, 环庚三烯环外酮反应的结果与Eq. 72不同, 得到外型产物, 由产物的单晶结构显示, 在哌啶环的椅式构象中, 4个取代基都处在直立键上(Eq. 74).
|
|
(74) |
2014年, 郭红超小组[145]以醋酸银及三苯基膦作为催化剂, 成功实现了亚甲亚胺偶极子与环庚三烯酮的非对映选择性[3+6]环加成(3:1~>20::1 dr), 得到内型且顺式的哌啶桥环化合物.同时作者利用手性催化剂Cu(I)/L20实现了该反应的不对称合成.该反应体系适用于芳基取代的偶极子.富或缺电子芳基都能获得不错的对映选择性(≥87% ee), 其中dr值最高大于20:1 (Eq. 75).
|
|
(75) |
羰基叶立德由于活性较高, 很不稳定, 只能在反应体系中原位产生, 因此其应用有一定的局限性.
Werz课题组[146]最近报道了羰基叶立德与DACs的[3+3]的环加成, 形成氧桥环辛或壬烷化合物, 其中环辛烷骨架广泛存在天然产物中.作者利用Rh2(OAc)4及路易斯酸Sc(OTf)3和Yb(OTf)3作为协同催化剂, 该反应首先是重氮化合物与铑作用形成金属卡宾, 接着被酯羰基进攻形成不稳定的环状羰基叶立德I26.随后环丙烷在路易斯酸的活化下, 与该叶立德发生[3+3]环加成, 最后形成桥环化合物.当n=0时, 形成的产物为氧桥环辛烷, 但这类产物的非对映选择性一般, dr值仅在2.0:1~4.6:1之间.该催化体系尤其适合于Ar1为富电子芳环的环丙烷, 可以获得总体不错的产率; 但不适用于缺电子芳基的环丙烷, 产率有较大的降低, 可能是由于重氮化合物在反应条件下容易发生自身[3+2]环加成形成二聚产物的缘故[147].当用另一个重氮化合物(n=1)时, 该反应的产率及非对映选择性都有较大的提升, 有趣的是, 产物的单晶显示所有主要产物结构都是芳基处于直立键, 这与熟知的一般规律有所不同(Eq. 76).
|
|
(76) |
Schneider小组[148]以o-QMs作为羰基叶立德的亲偶极体, 在路易斯酸及手性布朗斯特酸的共催化下, 实现高立体选择性获得三个手性碳的氧桥八元环化合物.首先Rh2(OAc)4与重氮化合物形成铑卡宾, 随即被邻位的羰基进攻卡宾获得高活性的羰基叶立德I27, 在手性磷酸的氢键作用下o-QMs前体邻羟基苯甲醇与上述叶立德进行环加成完成反应.该催化体系尤其适用于R为氢及甲氧基的o-QMs前体, 获得最优异的立体选择性; 而不太适合于R为卤素、叔丁基的底物, 立体选择性都会有明显的降低.重氮酮的R2为2-取代(杂)芳基或缺电子苯基时, 可能由于位阻的影响, 使得反应的非对映选择性会显著下降; 而当为富电子苯或者杂环时, 反应整体的立体选择性都最优秀, 但为叔丁基时, 却几乎不能发生(Eq. 77).
|
|
(77) |
Jørgensen等[149]报道了手性伯胺C8作为催化剂, 硫脲及三氟苯甲酸作为添加剂, 吡喃酮与富烯进行[3+6]的环加成反应.首先是吡喃酮在反应条件脱除离去基团三氟苯甲酰基, 接着羰基的烯醇化, 形成吡喃吡喃鎓盐I28, 随后与富烯对映选择性地加成, 获得二环[6.3.0]十一碳烯酮的骨架结构.该反应体系获得对映选择性不错(>82% ee), 但产率处于中等水平, 可能原因是羰基叶立德的活性太高, 从而引发许多的副反应.富烯端烯的取代基主要是苯基、甲基、乙基以及螺环(4~6元环), 该反应的溶剂及离去基团以及添加剂的选择也很关键, 离去基团比一般的苯甲酰基及乙酰基离去能力更强(Eq. 78).
|
|
(78) |
Werz小组[150]在前期的基础上(Eq. 76), 又利用DACs作为氰基亚胺偶极子I29的反应试剂, 在TiCl4的催化下实现它们的[3+3]环加成, 获得哒嗪衍生物.该反应体系适合于富电子或缺电子苯基的环丙烷, 可以获得高的收率, 但强吸电子基(硝基、三氟甲基)取代环丙烷的产率稍微下降.而对于Ar1为缺电子苯基的α-氯代腙, 该反应体系却不太适合, 产率会急剧下降至11%.作者也尝试将其换成苯甲酰基时, 但遗憾的是反应不能发生, 可能是因为吸电子效应降低了相邻氮原子的亲核性; 而R1可以为芳基及环己甚至环丙基, 反应结果还可以接受, 但当为对氟苯基时, 产率也相当低.因此该反应体系还需进一步的优化, 以提升其应用价值(Eq. 79).
|
|
(79) |
赵洪武课题组[151]也报道了α-卤代酰胺作为亲偶极子与α-氯代苯腙形成噁三嗪衍生物的反应.首先是在碱性条件下α-氯代苯腙形成相同的偶极子I29, 同时α-卤代酰胺形成氮氧烯丙基阳离子I4, 但与I29加成的是中间体I4的碳氧两端, 而非碳氮两端, 这与其它的α-卤代酰胺作为亲偶极子的环加成不同.该反应体系官能团兼容性好, 通常获得优异的收率, 最高为96%.同时作者对α-卤代酰胺也做了扩展, 主要改变R1及R2.当同时为甲基时, 产率都为中上等; 而换成单取代基的甲基、苯基或环己基时, 结果却不如人意, 特别是为氯原子时, 没有反应活性(Eq. 80).
|
|
(80) |
叠氮化合物早已被广泛应用于与各种烯烃或炔烃的[3+2]环加成反应(又被称为“Click”反应).但其它的环加成反应很少被研究.
1994年, Kampf等[152]发展了叠氮化合物分子内的1, 3-偶极环加成反应.通过不同的酸可以控制环加成的选择性: SnCl4作为路易斯酸时, 吲哚衍生的叠氮化合物主要发生分子内的[3+2]环加成; 若是在BF3•OEt2(三氟化硼乙醚复合物)的条件下, 底物中的叔羟基脱除形成烯丙基碳正离子, 随后与另一端的叠氮环加成形成吲哚三嗪产物.但BF3•OEt2/NaBH4的反应产率较低, 并伴随有消除的副反应; 若用NaHCO3代替NaBH4, 反应的产率有较大的提升.作者还将底物扩展到了其它烯烃化合物, 但产率较低(Eqs. 81~83).
|
|
(81) |
|
|
(82) |
|
|
(83) |
West等[153]利用Nazarov中间体作为亲偶极子, 在TfOH的作用下与叠氮化合物进行环加成反应.二烯酮发生Nazarov电环化, 生成的环戊烯醇碳正离子被叠氮捕获, 形成[3+3]的环加成产物, 即三氮桥环化合物.该反应获得单一构型的非对映构体, 但少量产物会有区域异构体, 产物可进一步脱出氮气形成开环结构的化合物(Eq. 84).
|
|
(84) |
徐鹏飞小组[154]报道了DACs作为亲偶极子在TiCl4作用下开环, 与叠氮化合物发生[3+3]环加成反应, 得到三嗪化合物.在化学计量的四氯化钛的条件下, 该反应总体收率比较好(75%~91%), 环丙烷的R1为邻位取代的苯基时, 产率相对略低; 而用催化量(20mol%)时, 经延长反应时间, 得到总体相似的反应结果(Eq. 85).
|
|
(85) |
2018年, 姚和权小组[155]报道了以α-溴代酰胺作为亲偶极子, 与叠氮化合物进行区域选择性的环加成反应.该反应体系能兼容各种官能团, 底物扩展的也比较广泛, 叠氮的取代基主要为苄基、烷基, 以及复杂的氨基酸残基、香豆素、甾体、维生素及药物塞来昔布及齐多夫定等.溴代酰胺主要是氮原子上可以是苄氧基、甲氧基、乙氧基或复杂的烷氧基等(Eq. 86).
|
|
(86) |
2011年, Ryan小组[156]报道了靛红酐作为亲偶极子, 与不稳定的偶极子进行[3+2]/脱羰基的反应, 形成二氮杂䓬.首先原位形成的亚甲胺叶立德与靛红酐的羰基发生[3+2]环加成, 接着开环、脱羰基、再关环形成产物, 作者利用核磁实验及红外证实了中间体I31的存在.当靛红酐的R1为氢时, 反应不能得到产物而是未知的副产物, 而当把条件换成是三氟乙酸催化时, 能以42%的收率获得相应的产物.总体而言, 该反应体系能适合于R1为其它取代基, 如甲基、苯基、苄基及烯丙基的靛红酐, 都获得优异的收率.当靛红酐的芳基上R为吸电子基时, 也能获得不错的收率, 而当R为供电子基时, 取代基的位子对反应影响较大:当R为7-甲基、7-甲氧基或6, 7-二甲氧基时, 反应几乎不发生; 当R为6-甲基或8-甲氧基时, 却能与中高等的收率得到产物(Eq. 87).作者还发现当R为6-或7-硝基时, 反应得到的产物也有所不同, 当6-硝基的靛红酐给出三个产物, 其中两个为苯环发生偶极环加成的[157].
|
|
(87) |
2012年, Ukaji小组[158]利用异腈作为亲偶极子, 与喹啉衍生的偶氮次甲基亚胺偶极子进行反应, 发现该反应具有很高的活性, 在室温下即可快速进行环加成获得噁哒嗪产物.该反应是异腈首先进攻偶极子的缺电性碳, 随后发生烯醇异构化形成的1, 5-两性离子反过来进攻异腈形成[5+1]的产物.该反应体系能获得大部分底物超80%的产率, 尤其适用于叔丁基、环己基取代的异腈, 而当为缺电子的苯基时产率会略微下降; 另外当偶极子上的R为6-或8-甲基时, 产率也会有严重的降低(34%~56%) (Eq. 88).
|
|
(88) |
Hashmi小组[159]又利用氮杂丙烯啶作为亲偶极子, 与二氢异喹啉衍生的偶极子进行反应, 发现在室温下即可进行[3+2]反应, 随后三元环开环、消除一分子亚磺酸进而形成1, 2, 4-三嗪化合物.当氮上的取代基对甲苯磺酰基换成苯甲酰基时, 该反应不发生后续的开环反应, 只获得[3+2]的产物.当R1为供电子基时会提高产率, 而氮杂丙烯啶取代基对产率的影响不大(Eq. 89).
|
|
(89) |
邵志会及邓玉华团队[160]报道了亚胺衍生的TMMs与呋喃基氮杂二烯构建氮杂䓬的[3+4]环加成反应.亚胺叔丁氧羰基酯在钯的作用下, 释放二氧化碳及氢质子, 形成烯丙基钯物种TMMs, 该中间体也可以看做是亚甲胺叶立德, 但反应通过TMMs与氮杂二烯完成[3+4]的环加成, 没有获得叶立德的环加成产物.该反应体系可以获得高的产率及优异的立体选择性.当Ar为取代苯环时, 取代基的电子效应及位子对反应的选择性有一定的影响, 但没有明显的规律(Eq. 90).随后Trost小组[161]也报道了几乎一样的不对称[3+4]环加成反应, 仅配体有点区别, 为联萘基亚磷酰胺.
|
|
(90) |
池永贵课题组[162]最近报道了肉桂醛在氮杂环卡宾的活化下与吡啶叶立德的环加成反应, 获得2-吡喃酮.在该环加成反应中, 吡啶作为离去基团并没有保留在产物中.当叶立德的Ar1为间位及对位取代的苯环、萘环或者乙基时, 该反应也能顺利进行(Eq. 91).
|
|
(91) |
总结了迄今为止1, 3-偶极子的[3+n] (n≥3)环加成反应, 其中包括[3+3]、[3+2+2]、[3+4]、[3+2+3]、[3+5]及[3+6]等.由于可以形成多官能团化的5~8元杂环化合物, 1, 3-偶极环加成反应已经成为被研究与应用最为广泛的反应之一.目前关于1, 3-偶极子的[3+n]环加成反应, 发现具有以下特点: (1)大多数的研究主要集中在常见的三种偶极子, 包括硝酮、偶氮次甲基亚胺及亚甲胺叶立德; (2)利用已经被广泛应用于各种有机反应的亲偶极子, 如D-A环丙烷(DACs)或环丁烷、三亚甲基甲烷(TMMs)、α-卤代酰胺、(氮杂)邻醌亚甲基化合物(o-QMs)、联烯、炔酮、富烯及环庚酮等; (3)在所有的研究中, 超过一半的研究论文由中国学者所贡献; (4)不对称合成, 尤其是不对称催化的环加成也已被逐渐开发, 其中中国学者, 以郭红超教授以及王春江教授为代表, 而国外则是以Doyle教授及Hayashi教授为代表.
虽然1, 3-偶极环加成反应从20世纪60年代开始由Huisgen发现并发展至今, 取得了令人瞩目的成就, 但对于[3+n] (n≥3)环加成, 目前仍存在亟需解决的问题及挑战, 包括以下几点: (1)偶极子, 如羰基叶立德、氰基叶立德及亚胺等的相关研究还比较少; (2)不对称催化的环加成反应, 特别是高立体选择性的研究占比还是太少; (3) [3+2]环加成反应已有比较多的已应用于药物及天然产物的合成中, 但[3+n] (n≥3)环加成目前还没应用于复杂分子的合成; (4)更绿色环保的合成方法, 如光电催化方法也未被用于该类反应中.近年来, 伴随着“经济化学、绿色化学”的提出, 可持续的化学合成已经成为合成化学家们追求的目标, 相信随着时间的推移、研究的深入, 以上问题将逐一被解决.
Seeman, J. I.; Restrepo, G. Angew. Chem. Int. Ed. 2020, 59, 12250. doi: 10.1002/anie.202003034
Huisgen, P. D. R. Angew. Chem. Int. Ed. 1963, 2, 565. doi: 10.1002/anie.196305651
Delpierre, G.; Lamchen, M. Quart. Rev. Chem. Soc. 1965, 19, 329. doi: 10.1039/qr9651900329
Stuckwisch, C. G. Synthesis 1973, 469.
Timpe, H.-J. Heteroaromatic N-Imines. In Advances in Heterocyclic Chemistry, Eds:Katritzky, A. R.; Boulton, A. J., Academic Press, Pittsburgh, 1974; Vol. 17, p. 213.
Black, D. S. C.; Crozier, R. F.; Davis, V. C. Synthesis 1975, 205.
Tamura, Y.; Ikeda, M. Advances in the Chemistry of Heteroaromatic N-Imines and N-Aminoazonium Salts. In Advances in Heterocyclic Chemistry, Eds.:Katritzky, A. R.; Boulton, A. J., Academic Press, Pittsburgh, 1981; Vol. 29, p. 71.
Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863. doi: 10.1021/cr970324e
Rodina, L. L.; Kolberg, A.; Schulze, B. Heterocycles 1998, 49, 587. doi: 10.3987/REV-98-SR4
赵宝祥, 化学进展, 2000, 12, 77.Zhao B.-X. Prog. Chem. 2000, 12, 77(in Chinese).
许家喜, 焦鹏, 化学进展, 2000, 12, 131.Xu J.-X.; Jiao P. Prog. Chem. 2000, 12, 131(in Chinese).
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem. Int. Ed. 2001, 40, 2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5
Kotyatkina, A. I.; Zhabinsky, V N.; Khripach, V. A. Russ. Chem. Rev. 2001, 70, 641. doi: 10.1070/RC2001v070n08ABEH000630
Osborn, H. M. I.; Gemmell, N.; Harwood, L. M. J. Chem. Soc., Perkin Trans. 12002, 2419.
Padwa A.; Pearson W. H. The Chemistry of Heterocyclic Compounds: Synthetic Applications of 1, 3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, Vol. 59, John Wiley & Sons, New York, 2002.
Schantl, J. G. Sci. Synth. 2004, 27, 731.
Harju, K.; Yli-Kauhaluoma, J. Mol. Div. 2005, 9, 187. doi: 10.1007/s11030-005-1339-1
Rück-Braun, K.; Freysoldt, T. H. E.; Wierschem, F. Chem. Soc. Rev. 2005, 34, 507. doi: 10.1039/b311200b
Coldham, I.; Hufton, R. Chem. Rev. 2005, 105, 2765. doi: 10.1021/cr040004c
胡晓芬, 冯亚青, 李筱芳, 有机化学, 2005, 25, 1. http://sioc-journal.cn/Jwk_yjhx/CN/Y2005/V25/I01/1Hu, X.-F.; Feng, Y.-Q.; Li, X.-F. Chin. J. Org. Chem. 2005, 25, 1(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y2005/V25/I01/1
Pinho e Melo, T. M. V. D. Eur. J. Org. Chem. 2006, 2006, 2873. doi: 10.1002/ejoc.200500892
Pandey, G.; Banerjee, P.; Gadre, S. R. Chem. Rev. 2006, 106, 4484. doi: 10.1021/cr050011g
王峰, 白东鲁, 有机化学, 2006, 26, 9. http://sioc-journal.cn/Jwk_yjhx/CN/Y2006/V26/I01/9Wang, F.; Bai D.-L. Chin. J. Org. Chem. 2006, 26, 9(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y2006/V26/I01/9
Moses, J. E.; Moorhouse, A. D. Chem. Soc. Rev. 2007, 36, 1249. doi: 10.1039/B613014N
Nair, V.; Suja, T. D. Tetrahedron 2007, 63, 12247. doi: 10.1016/j.tet.2007.09.065
Stanley, L. M.; Sibi, M. P. Chem. Rev. 2008, 108, 2887. doi: 10.1021/cr078371m
Jewett, J. C.; Bertozzi, C. R. Chem. Soc. Rev. 2010, 39, 1272. doi: 10.1039/b901970g
Adrio, J.; Carretero, J. C. Chem. Commun. 2011, 47, 6784. doi: 10.1039/c1cc10779h
Yang, J. Synlett 2012, 2293.
Lashgari, N.; Ziarani, G. M. ARKIVOC 2012, i, 277.
Tanimoto, H.; Kakiuchi, K. Nat. Prod. Commun. 2013, 8, 1021.
Xu, X.; Doyle, M. P. Acc. Chem. Res. 2014, 47, 1396. doi: 10.1021/ar5000055
韩骞, 易超; 熊兴泉, 有机化学, 2014, 34, 1092. doi: 10.6023/cjoc201311044Han, Q.; Yi, C.; Xiong, X., Chin. J. Org. Chem. 2014, 34, 1092(in Chinese). doi: 10.6023/cjoc201311044
Hashimoto, T.; Maruoka, K. Chem. Rev. 2015, 115, 5366. doi: 10.1021/cr5007182
Najera, C.; Sansano, J. M.; Yus, M. Org. Biomol. Chem. 2015, 13, 8596. doi: 10.1039/C5OB01086A
Padwa, A.; Bur, S. Recent Advances of 1, 3-Dipolar Cycloaddition Chemistry for Alkaloid Synthesis. In Advances in Heterocyclic Chemistry, Eds.:Scriven, E. F. V.; Ramsden C. A., Academic Press, Pittsburgh, 2016, Vol. 119, p. 241.
Sears, J. E.; Boger, D. L. Acc. Chem. Res. 2016, 49, 241. doi: 10.1021/acs.accounts.5b00510
Meyer, A. G.; Ryan, J. H. Molecules 2016, 21, 935. doi: 10.3390/molecules21080935
Belskaya, N. P.; Bakulev, V. A.; Fan, Z. Chem. Heterocycl. Compd. 2016, 52, 627. doi: 10.1007/s10593-016-1943-2
袁贝贝, 李雅宁, 郭娇美, 王琪琳, 卜站伟, 化学研究, 2017, 28, 135.Yuan, B.-B.; Li, Y.-M.; Wang, Q.-L.; Bu, Z.-W. Chem. Res. 2017, 28, 135(in Chinese).
Pozgan, F.; Al Mamari, H.; Groselj, U.; Svete, J.; Stefane, B. Molecules 2017, 23, 3. doi: 10.3390/molecules23010003
Xuan, J.; Cao, X.; Cheng, X. Chem. Commun. 2018, 54, 5154. doi: 10.1039/C8CC00787J
Li, J.; Ye, Y.; Zhang, Y. Org. Chem. Front. 2018, 5, 864. doi: 10.1039/C7QO01077J
Arrastia, I.; Arrieta, A.; Cossío, F. P. Eur. J. Org. Chem. 2018, 5889.
Grošelj, U.; Svete, J.; Mamari, H. H. A.; Požgan, F.; Štefane, B. Chem. Heterocycl. Compd. 2018, 54, 214. doi: 10.1007/s10593-018-2258-2
Grošelj, U.; Požgan, F.; Štefane, B.; Svete, J. Synthesis 2018, 4501.
Frankowski, S.; Romaniszyn, M.; Skrzynska, A.; Albrecht, L. Chemistry 2019.
Gulevskaya, A. V.; Nelina-Nemtseva, J. I. Chem. Heterocycl. Compd. 2019, 54, 1084.
Gao, K.; Zhang, Y. G.; Wang, Z.; Ding, H. Chem. Commun. 2019, 55, 1859. doi: 10.1039/C8CC09077G
Sukhorukov, A. Adv. Synth. Catal. 2019, 362, 724.
Nebra, N.; García-Álvarez, J. Molecules 2020, 25, 2015. doi: 10.3390/molecules25092015
Zhu, Y.; Huang, Y. Synthesis 2020, 1181.
Brunel, D.; Dumur, F. New J. Chem. 2020, 44, 3546. doi: 10.1039/C9NJ06330G
Gui, H.-Z.; Wei, Y.; Shi, M. Chem. Asian J. 2020, 15, 1225. doi: 10.1002/asia.202000054
Dhameja, M.; Kumar, H.; Gupta, P. Asian J. Org. Chem. 2020, in press.
Breugest, M.; Reissig, H. U. Angew. Chem. Int. Ed. 2020, 59, 12293. doi: 10.1002/anie.202003115
Wei, L.; Chang, X.; Wang, C.-J. Acc. Chem. Res. 2020, 53, 1084. doi: 10.1021/acs.accounts.0c00113
Mori, M.; Sugiyama, T.; Nojima, M.; Kusabayashi, S.; Mc- Cullough, K. J. J. Org. Chem. 1992, 57, 2285. doi: 10.1021/jo00034a018
Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem. Int. Ed. 2014, 53, 5504. doi: 10.1002/anie.201309886
Singh, P.; Varshnaya, R. K.; Dey, R.; Banerjee, P. Adv. Syn. Catal. 2020, 362, 1447. doi: 10.1002/adsc.201901332
Werz, D. B.; Biju, A. T. Angew. Chem. Int. Ed. 2020, 59, 3385. doi: 10.1002/anie.201909213
Ivanova, O. A.; Trushkov, I. V. Chem. Rec. 2020, 19, 2189.
Wu, L.; Shi, M. Chem. Eur. J. 2010, 16, 1149. doi: 10.1002/chem.200902510
Yang, W.; Wang, T.; Yu, Y.; Shi, S.; Zhang, T.; Hashmi, A. S. K. Adv. Syn. Catal. 2013, 355, 1523. doi: 10.1002/adsc.201300338
El Bouakher, A.; Martel, A.; Comesse, S. Org. Biomol. Chem. 2019, 17, 8467. doi: 10.1039/C9OB01683J
An, Y.; Xia, H.; Wu, J. Chem. Commun. 2016, 52, 10415. doi: 10.1039/C6CC03650C
Zhao, H.-W.; Zhao, Y.-D.; Liu, Y.-Y.; Zhao, L.-J.; Song, X.-Q.; Chen, X.-Q.; Pang, H.-L.; Du, J.; Feng, N.-N. RSC Adv. 2017, 7, 55106. doi: 10.1039/C7RA09766B
Jia, Q.; Li, D.; Lang, M.; Zhang, K.; Wang, J. Adv. Syn. Catal. 2017, 359, 3837. doi: 10.1002/adsc.201700415
Luo, Y.; Chen, C.-H.; Zhang, J.-Q.; Liang, C.; Mo, D.-L. Synthesis 2019, 52, 424.
Sibi, M. P.; Ma, Z.; Jasperse, C. P. J. Am. Chem. Soc. 2005, 127, 5764. doi: 10.1021/ja0421497
Sapeta, K.; Kerr, M. A. J. Org. Chem. 2007, 72, 8597. doi: 10.1021/jo701606u
Wang, X.; Xu, X.; Zavalij, P. Y.; Doyle, M. P. J. Am. Chem. Soc. 2011, 133, 16402. doi: 10.1021/ja207664r
Xu, X.; Zavalij, P. J.; Doyle, M. P. Chem. Commun. 2013, 49, 10287. doi: 10.1039/c3cc46415f
Shintani, R.; Park, S.; Duan, W. L.; Hayashi, T. Angew. Chem. Int. Ed. 2007, 46, 5901. doi: 10.1002/anie.200701529
Liu, F.; Qian, D.; Li, L.; Zhao, X.; Zhang, J. Angew. Chem. Int. Ed. 2010, 49, 6669. doi: 10.1002/anie.201003136
Zhou, L.; Xu, B.; Ji, D.; Zhang, Z. M.; Zhang, J. Chin. J. Chem. 2020, in press.
Xu, P. W.; Liu, J. K.; Shen, L.; Cao, Z. Y.; Zhao, X. L.; Yan, J.; Zhou, J. Nat. Commun. 2017, 8, 1619. doi: 10.1038/s41467-017-01451-1
Bai, Y.; Fang, J.; Ren, J.; Wang, Z. Chem. Eur. J. 2009, 15, 8975. doi: 10.1002/chem.200901133
Stevens, A. C.; Palmer, C.; Pagenkopf, B. L. Org. Lett. 2011, 13, 1528. doi: 10.1021/ol200220d
Zhang, Y.; Zhang, J. Chem. Commun. 2012, 48, 4710. doi: 10.1039/c2cc30309d
Hu, J.-L.; Wang, L.; Xu, H.; Xie, Z.; Tang, Y. Org. Lett. 2015, 17, 2680. doi: 10.1021/acs.orglett.5b01077
Shintani, R.; Murakami, M.; Hayashi, T. J. Am. Chem. Soc. 2007, 129, 12356. doi: 10.1021/ja073997f
Shintani, R.; Murakami, M.; Hayashi, T. Pure Appl. Chem. 2008, 80, 1135. doi: 10.1351/pac200880051135
Gawade, S. A.; Bhunia, S.; Liu, R. S. Angew. Chem. Int. Ed. 2012, 51, 7835. doi: 10.1002/anie.201203507
Shintani, R.; Hayashi, T. J. Am. Chem. Soc. 2006, 128, 6330. doi: 10.1021/ja061662c
Li, S.-N.; Yu, B.; Liu, J.; Li, H.-L.; Na, R. Synlett 2016, 27, 282.
Wang, K.-K.; Li, Y.-L.; Wang, Z.-Y.; Hu, M.-W.; Qiu, T.-T.; Zhu, B.-K. Org. Biomol. Chem. 2018, 17, 244.
Perreault, C.; Goudreau, S. R.; Zimmer, L. E.; Charette, A. B. Org. Lett. 2008, 10, 689. doi: 10.1021/ol702414e
Chagarovskiy, A. O.; Vasin, V. S.; Kuznetsov, V. V.; Ivanova, O. A.; Rybakov, V. B.; Shumsky, A. N.; Makhova, N. N.; Trushkov, I. V. Angew. Chem. Int. Ed. 2018, 57, 10338. doi: 10.1002/anie.201805258
Chagarovskiy, A. O.; Kuznetsov, V. V.; Ivanova, O. A.; Goloveshkin, A. S.; Levina, I. I.; Makhova, N. N.; Trushkov, I. V. Eur. J. Org. Chem. 2019, 2019, 5475. doi: 10.1002/ejoc.201900579
Na, R.; Jing, C.; Xu, Q.; Jiang, H.; Wu, X.; Shi, J.; Zhong, J.; Wang, M.; Benitez, D.; Tkatchouk, E.; Goddard, W. A.; Guo, H.; Kwon, O. J. Am. Chem. Soc. 2011, 133, 13337. doi: 10.1021/ja200231v
Liang, L.; Huang, Y. Org. Lett. 2016, 18, 2604. doi: 10.1021/acs.orglett.6b00988
Li, Y.; Zhang, Z. New J. Chem. 2019, 43, 13600. doi: 10.1039/C9NJ01943J
Liu, J.; Liu, H.; Na, R.; Wang, G.; Li, Z.; Yu, H.; Wang, M.; Zhong, J.; Guo, H. Chem. Lett. 2012, 41, 218. doi: 10.1246/cl.2012.218
Li, Z.; Yu, H.; Liu, Y.; Zhou, L.; Sun, Z.; Guo, H. Adv. Synth. Catal. 2016, 358, 1880. doi: 10.1002/adsc.201600223
Qian, Y.; Zavalij, P. J.; Hu, W.; Doyle, M. P. Org. Lett. 2013, 15, 1564. doi: 10.1021/ol400339c
Cheng, X.; Cao, X.; Xuan, J.; Xiao, W.-J. Org. Lett. 2018, 20, 52. doi: 10.1021/acs.orglett.7b03344
Ansari, A. J.; Pathare, R. S.; Kumawat, A.; Maurya, A. K.; Verma, S.; Agnihotri, V. K.; Joshi, R.; Metre, R. K.; Sharon, A.; Pardasani, R. T.; Sawant, D. M. New J. Chem. 2019, 43, 13721. doi: 10.1039/C9NJ02874A
Zhu, G.; Sun, W.; Wu, C.; Li, G.; Hong, L.; Wang, R. Org. Lett. 2013, 15, 4988. doi: 10.1021/ol402295m
Fang, X.; Li, J.; Tao, H.-Y.; Wang, C.-J. Org. Lett. 2013, 15, 5554. doi: 10.1021/ol402724h
Du, J.; Xu, X.; Li, Y.; Pan, L.; Liu, Q. Org. Lett. 2014, 16, 4004. doi: 10.1021/ol501829k
Shapiro, N. D.; Shi, Y.; Toste, F. D. J. Am. Chem. Soc. 2009, 131, 11654. doi: 10.1021/ja903863b
Chan, A.; Scheidt, K. A. J. Am. Chem. Soc. 2007, 129, 5334. doi: 10.1021/ja0709167
Efremova, M. M.; Kostikov, R. R.; Stepakov, A. V.; Panikorovsky, T. L.; Shcherbakova, V. S.; Ivanov, A. V.; Molchanov, A. P. Tetrahedron 2017, 73, 671. doi: 10.1016/j.tet.2016.12.034
T. P.; Krishna, A. V.; Ramachary, D. B. Org. Lett. 2018, 20, 6979. doi: 10.1021/acs.orglett.8b02719
Fang, Q-Y.; Jin, H-S.; Wang, R-B.; Zhao, L-M. Chem. Commun. 2019, 55, 10587. doi: 10.1039/C9CC05367K
Liu, Y.; Zhen, W.; Dai, W.; Wang, F.; Li, X. Org. Lett. 2013, 15, 874. doi: 10.1021/ol4000108
Moghaddam, F. M.; Eslami, M.; Siahpoosh, A.; Golfam, H. New J. Chem. 2019, 42, 10318.
Wang, X.; Yang, P.; Zhang, Y.; Tang, C.-Z.; Tian, F.; Peng, L.; Wang, L.-X. Org. Lett. 2017, 19, 646. doi: 10.1021/acs.orglett.6b03815
Hu, S.; Zhang, J.; Jin, Q. New J. Chem. 2018, 42, 7025. doi: 10.1039/C8NJ00234G
Zhou, Y. Y.; Li, J.; Ling, L.; Liao, S. H.; Sun, X. L.; Li, Y. X.; Wang, L. J.; Tang, Y. Angew. Chem. Int. Ed. 2013, 52, 1452. doi: 10.1002/anie.201207576
Guo, H.; Liu, H.; Zhu, F.-L.; Na, R.; Jiang, H.; Wu, Y.; Zhang, L.; Li, Z.; Yu, H.; Wang, B.; Xiao, Y.; Hu, X.-P.; Wang, M. Angew. Chem. Int. Ed. 2013, 52, 12641. doi: 10.1002/anie.201307317
Zhang, L.; Liu, H.; Qiao, G.; Hou, Z.; Liu, Y.; Xiao, Y.; Guo, H. J. Am. Chem. Soc. 2015, 137, 4316. doi: 10.1021/jacs.5b01138
Du, Q.; Neudoerfl, J.-M.; Schmalz, H.-G. Chem. Eur. J. 2018, 24, 2379. doi: 10.1002/chem.201800042
Xu, X.; Xu, X.; Zavalij, P. Y.; Doyle, M. P. Chem. Commun. 2013, 49, 2762. doi: 10.1039/c3cc41119b
Jing, C.; Na, R.; Wang, B.; Liu, H.; Zhang, L.; Liu, J.; Wang, M.; Zhong, J.; Kwon, O.; Guo, H. Adv. Synth. Catal. 2012, 354, 1023. doi: 10.1002/adsc.201100831
Li, Z.; Yu, H.; Feng, Y.; Hou, Z.; Zhang, L.; Yang, W.; Wu, Y.; Xiao, Y.; Guo, H. RSC Adv. 2015, 5, 34481. doi: 10.1039/C5RA04374C
Hu, X.-Q.; Chen, J.-R.; Gao, S.; Feng, B.; Lu, L.-Q.; Xiao, W.-J. Chem. Commun. 2013, 49, 7905. doi: 10.1039/c3cc43888k
Li, Z.; Li, S.; Kan, T.; Wang, X.; Xin, X.; Hou, Y.; Gong, P. Adv. Synth. Catal. 2020, 362, 2626. doi: 10.1002/adsc.202000398
Water, R. W. V. D.; Pettus, T. R. R. Tetrahedron 2002, 58, 5367. doi: 10.1016/S0040-4020(02)00496-9
Grotenhuis, C. T.; Bruin, B. D. Synlett 2018, 29, 2238. doi: 10.1055/s-0037-1610204
Yang, B.; Gao, S. Chem. Soc. Rev. 2018, 47, 7926. doi: 10.1039/C8CS00274F
Mei, G.-J.; Zhu, Z.-Q.; Zhao, J.-J.; Bian, C.-Y.; Chen, J.; Chen, R.-W.; Shi, F. Chem. Commun. 2017, 53, 2768. doi: 10.1039/C6CC09775H
Chen, L.; Yang, G.; Wang, J.; Jia, Q.; Wei, J.; Du, Z. RSC Adv. 2015, 5, 76696. doi: 10.1039/C5RA15903B
Jin, Q.; Zhang, J.; Jiang, C.; Zhang, D.; Gao, M.; Hu, S. J. Org. Chem. 2018, 83, 8410. doi: 10.1021/acs.joc.8b01055
Li, C.; Wang, C. S.; Li, T. Z.; Mei, G. J.; Shi, F. Org. Lett. 2019, 21, 598. doi: 10.1021/acs.orglett.8b03604
Dai, W.; Li, C.; Liu, Y.; Han, X.; Li, X.; Chen, K.; Liu, H. Org. Chem. Front. 2020, 7, 2612.. doi: 10.1039/D0QO00320D
Wei, L.; Wang, Z. F.; Yao, L.; Qiu, G.; Tao, H.; Li, H.; Wang, C. J. Adv. Syn. Catal. 2016, 358, 3955. doi: 10.1002/adsc.201600961
Yuan, C.; Wu, Y.; Wang, D.; Zhang, Z.; Wang, C.; Zhou, L.; Zhang, C.; Song, B.; Guo, H. Adv. Syn. Catal. 2018, 360, 652. doi: 10.1002/adsc.201701247
Yang, Q.-Q.; Yin, X.; He, X.-L.; Du, W.; Chen, Y.-C. ACS Catal. 2019, 9, 1258. doi: 10.1021/acscatal.8b04942
Wu, Y.; Qiao, G.; Liu, H.; Zhang, L.; Sun, Z.; Xiao, Y.; Guo, H. RSC Adv. 2015, 5, 84290. doi: 10.1039/C5RA12401H
Shi, F.; Zhu, R. Y.; Dai, W.; Wang, C. S.; Tu, S. J. Chem.-Eur. J. 2014, 20, 2597. doi: 10.1002/chem.201304187
Yuan, C.; Liu, H.; Gao, Z.; Zhou, L.; Feng, Y.; Xiao, Y.; Guo, H., Org. Lett. 2015, 17, 26. doi: 10.1021/ol503169d
Yang, W.-L.; Li, C.-Y.; Qin, W.-J.; Tang, F.-F.; Yu, X.; Deng, W.-P. ACS Catal. 2016, 6, 5685. doi: 10.1021/acscatal.6b01596
Kim, S.; Kim, S.-G. Asian J. Org. Chem. 2019, 8, 1621. doi: 10.1002/ajoc.201900073
Dai, Z.; Zhu, J.; Wang, J.; Su, W.; Yang, F.; Zhou, Q. Adv. Synth. Catal. 2019, 362, 545.
Zhao, H.-W.; Wang, L.-R.; Guo, J.-M.; Ding, W.-Q.; Song, X.-Q.; Wu, H.-H.; Tang, Z.; Fan, X.-Z.; Bi, X.-F. Adv. Synth. Catal. 2019, 361, 4761. doi: 10.1002/adsc.201900651
Niu, B.; Wu, X. Y.; Wei, Y.; Shi, M. Org. Lett. 2019, 21, 4859. doi: 10.1021/acs.orglett.9b01748
Wu, Y.; Liu, H.; Zhang, L.; Sun, Z.; Xiao, Y.; Huang, J.; Wang, M.; Guo, H. RSC Adv. 2016, 6, 73547. doi: 10.1039/C6RA14018A
Potowski, M.; Bauer, J. O.; Strohmann, C.; Antonchick, A. P.; Waldmann, H. Angew. Chem. Int. Ed. 2012, 51, 9512. doi: 10.1002/anie.201204394
He, Z.-L.; Teng, H.-L.; Wang, C.-J. Angew. Chem. Int. Ed. 2013, 52, 2934. doi: 10.1002/anie.201208799
Teng, H. L.; Yao, L.; Wang, C. J. J. Am. Chem. Soc. 2014, 136, 4075. doi: 10.1021/ja500878c
Li, Q. H.; Wei, L.; Wang, C. J. J. Am. Chem. Soc. 2014, 136, 8685. doi: 10.1021/ja503309u
He, Z. L.; Sheong, F. K.; Li, Q. H.; Lin, Z.; Wang, C. J. Org. Lett. 2015, 17, 1365. doi: 10.1021/acs.orglett.5b00011
Liu, H.; Wu, Y.; Zhao, Y.; Li, Z.; Zhang, L.; Yang, W.; Jiang, H.; Jing, C.; Yu, H.; Wang, B.; Xiao, Y.; Guo, H. J. Am. Chem. Soc. 2014, 136, 2625. doi: 10.1021/ja4122268
Petzold, M.; Jones, P. G.; Werz, D. B. Angew. Chem. Int. Ed. 2019, 58, 6225. doi: 10.1002/anie.201814409
Petzold, M.; Günther, A.; Jones, P. G., Werz, D. B. Chem.-Eur. J. 2020, in press.
Suneja, A.; Loui, H. J.; Schneider, C. Angew. Chem. Int. Ed. 2020, 59, 5536. doi: 10.1002/anie.201913603
McLeod, D.; Cherubini-Celli, A.; Sivasothirajah, N.; McCulley, C. H.; Christensen, M. L.; Jørgensen, K. A. Chem.-Eur. J. 2020, 26, 11417. doi: 10.1002/chem.202001369
Garve, L. K.; Petzold, M.; Jones, P. G.; Werz, D. B. Org. Lett. 2016, 18, 564. doi: 10.1021/acs.orglett.5b03598
Zhao, H.-W.; Zhao, Y.-D.; Liu, Y.-Y.; Zhao, L.-J.; Song, X.-Q.; Chen, X.-Q.; Pang, H.-L.; Du, J.; Feng, N.-N. RSC Adv. 2017, 7, 55106. doi: 10.1039/C7RA09766B
Pearson, W. H.; Fang, W.; Kampf, J. W. J. Org. Chem. 1994, 59, 2682. doi: 10.1021/jo00089a007
Scadeng, O.; Ferguson, M. J.; West, F. G. Org. Lett. 2011, 13, 114. doi: 10.1021/ol102651k
Zhang, H.-H.; Luo, Y.-C.; Wang, H.-P.; Chen, W.; Xu, P.-F. Org. Lett. 2014, 16, 4896. doi: 10.1021/ol5024079
Xu, X.; Zhang, K.; Li, P.; Yao, H.; Lin, A. Org. Lett. 2018, 20, 1781. doi: 10.1021/acs.orglett.8b00280
D'Souza, A. M.; Spiccia, N.; Basutto, J.; Jokisz, P.; Wong, L. S.-M.; Meyer, A. G.; Holmes, A. B.; White, J. M.; Ryan, J. H. Org. Lett. 2011, 13, 486. doi: 10.1021/ol102824k
D'Souza, A. M.; Rivinoja, D. J.; Mulder, R. J.; White, J. M.; Meyer, A. G.; Hyland, C. J. T.; Ryan, J. H. Aust. J. Chem. 2018, 71, 690. doi: 10.1071/CH18196
Soeta, T.; Tamura, K.; Ukaji, Y. Org. Lett. 2012, 14, 1226. doi: 10.1021/ol2034542
Wu, Y.; Tian, B.; Hu, C.; Sekine, K.; Rudolph, M.; Rominger, F.; Hashmi, S. Org. Biomol. Chem. 2019, 17, 5505. doi: 10.1039/C9OB00740G
Kumari, P.; Liu, W.; Wang, C. J.; Dai, J.; Wang, M. X.; Yang, Q. Q.; Deng, Y. H.; Shao, Z. Chin. J. Chem. 2019, 38, 151.
Trost, B. M.; Zuo, Z. Angew. Chem. Int. Ed. 2020, 59, 1243. doi: 10.1002/anie.201911537
Zheng, P.; Li, C.; Mou, C.; Pan, D.; Wu, S.; Xue, W.; Jin, Z.; Chi, Y. R. Asian J. Org. Chem. 2019, 8, 1067. doi: 10.1002/ajoc.201900153

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




























































































 下载:
下载: