Figure Chart 1.
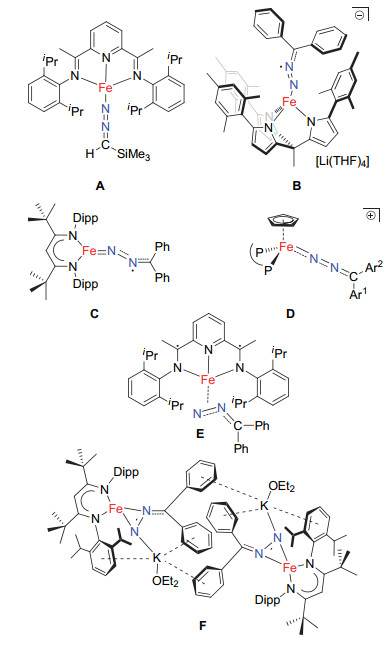
Examples of isolable iron diazoalkane complexes
Reactions of Iron(II) Complexes Supported by Tripodal Amido-Phosphine-Amido Ligands with Diazo Compounds
Jian Liu , Jie Xiao , Sudipta Mondal , Xuebing Leng , Liang Deng

In the reactions of metal species with diazoalkanes, diazoalkane complexes[1-5] are recognized as intermediates en route to the products, alkylidene complexes, that could facilitate useful transformations such as alkene cyclopropa- nation, C—H alkylation, alkene metathesis, and so on.[6-10] Consequently, there are great interests on the sythesis, structural feature, and reactivity of transition-metal diazoalkane complexes. The known transition-metal diazo- alkane complexes are dominated by the complexes of cobalt, nickel, and ruthenium, [11-16] whereas the diazoalkane complexes of iron are scarce, which form a sharp contrast to the soaring research interest on iron-catalyzed carbene- transfter reactions.[17]
The limited examples of iron diazoalkane complexes include the bis(imino)pyridine complex [(PDI)Fe(η1-N2CH-SiMe3)] (A in Chart 1), [18] the tris(pyrrolido)ethane iron complex [(tpe)Fe(η1-N2CPh2)][Li(THF)4] (tpe=tris(5-me- sitylpyrrolyl)ethane, B), [19] the β-diketiminate iron complex [LBu-tFe(η1-N2CPh2)] (LBu-t=[(DippNCBut)2CH]-, C)[20], the cyclopentadienyliron complexes [(η5-C5H5) Fe(η1-N2C-Ar1Ar2)(P-P)]BPh4 (P-P=1, 2-bis(diphenylphosphine)etha- ne, 1, 3-bis(diphenylphosphine)propane; Ar1=Ar2=Ph; Ar1=Ph, Ar2=p-tolyl; Ar1Ar2=C12H8, D), [21] as well as the side-on type diazo complexes [(PDI)Fe(η2-N, N-N2C-Ph2)] (E)[22] and [LBu-tFe(η2-N, N-N2CPh2)K(OEt2)]2 (F)[20]. Interestingly, spectroscopic and theoretical studies revealed that the diazo ligands in these complexes could be present in different electronic structures. For example, the diazoalkane ligands in B and C have been assigned as radical anions. The side-on diazoalkane ligand in E is thought to receive little backdonation from its iron center and can be viewed as merely a σ-donor. The side-on diazoalkane ligand in F on the other hand is a radical anion. In addition to the aforementioned studies, herein, we wish to report the reactions of high-spin iron(II) complexes featuring tripodal amido-phosphine-amido ligands, from which two new iron diazoalkane complexes [(κ3-N, N, P-MesN2PBu-t)Fe(η2-N, N-(p-tol-yl)2CN2)] and [(κ3-N, N, P-MesN2PBu-t)Fe(κ2-N, O-PhC(N2)-CO2Et)] have been isolated and structurally characterized.
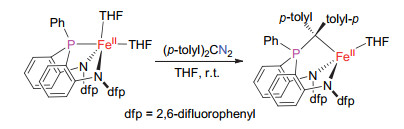
Previously, we found that the reaction of an (amido-phosphine-amido)iron(II) complex [(κ3-N, N, P-dfpN2PPh)Fe-(THF)2] with (p-tolyl)2CN2 furnished a diamido-phosphine ylide iron(II) complex [(κ3-N, N, C-dfpN2PPhC(tolyl-p)2)-Fe(THF)] (Scheme 1).[23] The ylide complex was proposed to be formed from the migratory insertion reaction of an iron(II) alkylidene intermediate. Aiming to attenuate the ease of the migratory insertion reaction of the proposed iron(II) alkylidene intermediates, new amido-phosphine- amido ligands having tert-butyl on phosphorus atom were then developed.
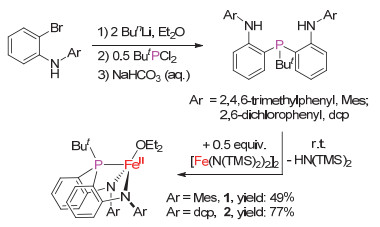
The synthetic procedures of the new ligands (o-(N-(2, 4, 6-Me3C6H2)NH)C6H4)2PBut and (o-(N-(2, 6-Cl2C6H3)NH)-C6H4)2PBut (denoted as H2(MesN2PBu-t) and H2(dcpN2-PBu-t)) follow that used for H2(dfpN2PPh).[23] Treatment of ButPCl2 with 2 equiv. of (2, 4, 6-Me3C6H2)(2-LiC6H4)NLi and (2, 6- Cl2C6H3)(2-LiC6H4)NLi, which were generated in situ by the interaction of (2, 4, 6-Me3C6H2)(2-BrC6H4)NH and (2, 6- Cl2C6H3)(2-BrC6H4)NH with 2 equiv. of BunLi, followed by quenching with water, afforded the amine-phosphine-amine compounds H2(MesN2PBu-t) and H2(dcpN2PBu-t) in 50% and 53% isolated yields, respectively (Scheme 2). H2(MesN2-PBu-t) and H2(dcpN2PBu-t) have been characterized by NMR, FT-IR and HRMS.
The new amine-phosphine-amine ligands H2(MesN2PBu-t) and H2(dcpN2PBu-t) readily undergo amine-elimination reaction with [Fe(N(SiMe3)2)2]2 (0.5 euiqv.) at room temperature to afford (amido-phosphine-amido)iron(II) complexes [(κ3-N, N, P-MesN2PBu-t)Fe(OEt2)] (1) and [(κ3-N, N, P-dcpN2-PBu-t)Fe(OEt2)] (2) in 49% and 77% isolated yields, respectively (Scheme 2). Complexes 1 and 2 are sensitive to air and moisture. They are soluble in diethyl ether, toluene and tetrahydrofuran, and have been characterized by 1H NMR, solution magnetic moment measurement, 57Fe Mössbauer spectroscopy, as well as elemental analysis. Their measured solution magnetic moments (μeff=4.7(1)μB and 5.0(1)μB in C6D6 at room temperature for 1 and 2, respectively) and 57Fe Mössbauer spectroscopic data (isomer shift δ=0.75 mm/s, quadrupole splitting |ΔEQ|=1.19 mm/s for 1; δ=0.82 mm/s, |ΔEQ|=2.40 mm/s for 2) are indicative of their high-spin iron(II) nature (S=2). The difference in the 57Fe Mössbauer data of 1 and 2 should be caused by their different N-aryl substituents.
The molecular structure of 2 has been confirmed by single-crystal X-ray diffraction study. As shown in Figure 1, the amido-phosphine-amido ligand in 2 is coordinating with the iron center in a tripodal conformation, being similar to the reported (amido-phosphine-amido)iron(II) complex [(κ3-N, N, P-MesN2PPh)Fe(OEt2)].[24] Notably, while the Fe—N distances in 2 are comparable to those of their congeners in [(κ3-N, N, P-MesN2PPh)Fe(OEt2)][24], its Fe—P bond with a distance of 0.24116(5) nm is longer than that in [(κ3-N, N, P-MesN2PPh)Fe(OEt2)][24] [0.2367(1) nm]. The long Fe—P distance in 2 reflects the influence of the steric demanding nature of tert-butyl versus phenyl group. Another worth mentioning structural feature of 2 is its Fe—Cl(3) distance of 0.2845 nm, which lies between the sum of the two van der Waals radii (0.307 nm) and the Fe(II)—Cl distance in the five-coordinate (bis(imino)pyridine)iron(II) chloride complex [(PDI)FeCl2][25] (0.2301 nm), indicating weaker interaction between the two atoms.
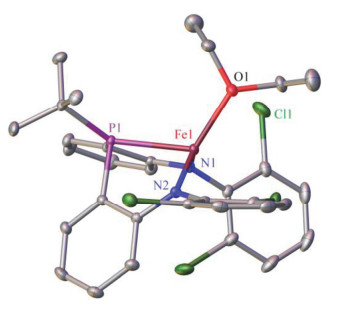
Selected distances (nm) and angles (°): Fe1—N1, 0.19877(14); Fe1—N2, 0.19926(14); Fe1—P1, 0.24116(5); Fe1—Cl1, 0.2845; Fe1—O1, 0.20626(13); N1—Fe1—N2, 123.31(6); N1—Fe1—P1, 80.99(4); N1—Fe1—O1, 113.78(6); N2—Fe1—P1, 84.25(4); N2—Fe1—O1, 121.62(6); P1— Fe1—O1, 117.17(4); P1—Fe1—Cl1, 153.00
With the new (amido-phosphine-amido)iron (II) com- pounds 1 and 2 in hand, we further studied their reactions with (p-tolyl)2CN2, PhC(N2)CO2Et, and DmpC(N2)H (Dmp=2, 6-dimesitylphenyl), which were found to give different outcomes.
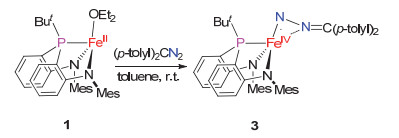
The interaction of 1 with (p-tolyl)2CN2 (1 equiv.) in toluene or THF at room temperature produced a brown solution without N2-evolution. Further workup and recrys-tallization led to the isolation of a diazoalkane complex [(κ3-N, N, P-MesN2PBu-t)Fe(η2-N, N-(p-tolyl)2CN2)] (3) in 56% yield (Scheme 3). The attainment of 3 differs from the formation of ylide complex in the reaction of [(κ3-N, N, P-dfpN2PPh)Fe(THF)2] with (p-tolyl)2CN2, which demonstrates the effect of the ligand subsituents on the stability of the iron diazoalkane complexes. The attempts to access alkylidene iron species by thermolysis of 3 in benzene gave an intractable mixture.
Complex 3 has been characterized by 1H NMR, 13C NMR, and 31P NMR spectroscopies, 57Fe Mössbauer spectroscopy, infrared spectroscopy, single-crystal X-ray diffraction, as well as elemental analysis. The molecular structure of 3 established by X-ray diffraction study revealed a side-on coordination mode of the diazoalkane ligand that has the Fe—N2 and Fe—N1 distances of 0.17524(11) and 0.19601(11) nm, respectively (Figure 2). The Fe1—N2 distance is obviously shorter than its congener in the side-on diazoalkane complexes [(PDI)Fe(η2-N, N-N2CPh2)][22] (0.1903 nm) and [LBu-tFe(η2-N, N-N2CPh2)-K(OEt2)]2[20] [0.1839(2) nm], and is more close to the Fe—N distances of the end-on diazoalkane complexes [(IPr)Fe(NArTrip)(η1-N2C(tolyl-p)2)][26] [0.1727(2) nm], [(tpe)Fe(η1-N2CPh2)][Li(THF)4][19] [0.1781(9) nm], and [LBu-tFe(η1-N2CPh2)][0.1752(1) nm], reaching the long end of the Fe—N distances of terminal iron imido complexes.[26] The N—N and N—C bonds of the side-on diazoalkane ligand with the distances of 0.13292(16) and 0.13054(17) nm, respectively, are comparable to those of the middle transition-metal complexes that feature dianionic diazo ligands, e.g. [Mo(OBut)4(NNPh2)][27] and [Cr(OSi-But3)2(NNCPh2)2].[28] As compared to the diazo radical ligands, the N—N and N—C bonds in 3 are longer and shorter, respectively, than their counterparts in [(tpe)Fe(η1-N2CPh2)][Li(THF)4][19] and [LBu-tCo(η1-N2CPh2)].[20] The characteristic bond distances of the diazo ligand in 3, in addition with the short Fe—N (amido) [0.18571(11) and 0.18559(11) nm] and Fe—P [0.21628(4) nm] distances seem to agree with the assignment of 3 as an iron(IV) complex having a dianionic diazo ligand [η2-N, N-(p-tolyl)2-CN2)]2-.
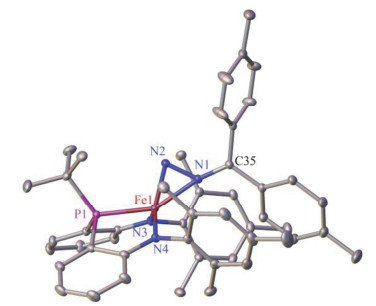
Selected distances (nm) and angles (°): Fe1—P1, 0.21628(4); Fe1—N3, 0.18571(11); Fe1—N4, 0.18559(11); Fe1—N1, 0.19601(11); Fe1—N2, 0.17524(11); N3—Fe1—P1, 85.38(3); N4—Fe1—P1, 85.93(4); N4— Fe1—N3, 128.91(5)
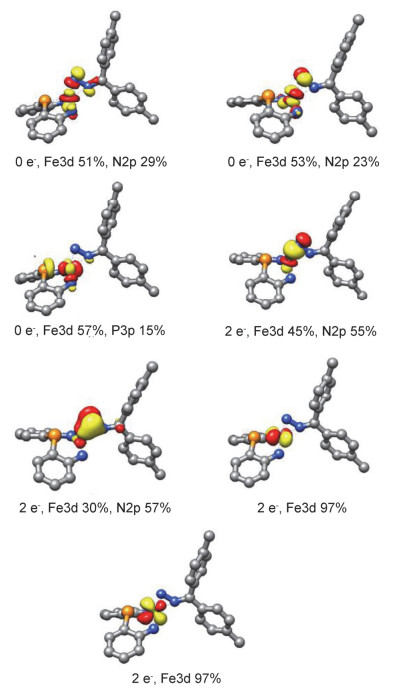
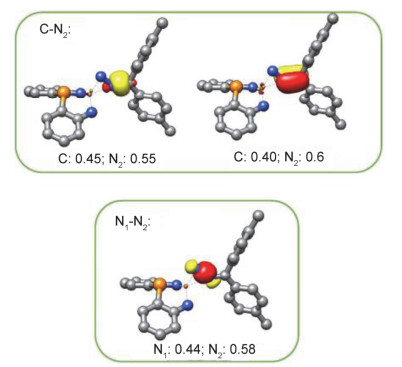
Theoretical studies on 3 have further corroborated the assignment of 3 as an iron(IV) complex having a dianionic diazo ligand. Single point energy calculations at the B3- LYP/TZVP/SVP level of theory based on the molecular structure of 3 established by X-ray diffraction study indicate that the complex at its S=0 spin state is 55.76 and 128.41 kJ/mol, respectively, lower in energy than those of the S=1 and S=2 spin states. Localized orbital analysis on the S=0 state revealed the presence of both σ- and π-type interactions between the iron center and the terminal nitrogen atom of the diazo ligand (Figure 3). Meanwhile, the N—N and N—C bonds of the diazo ligand are found single- and double-bond in character, respectively (Figure 4). The composition of the frontier orbitals (Figure 3) points to an electronic configuration of (dxy)2(dxz)2(dx-y22)0(dz2)0(dyz)0 for the covalent iron(IV) complex 3. Being supportive to this assignment, complex 3 is diamagnetic and its zero-field 57Fe Mössbauer data measured at 80 K (δ=0.02 mm/s and |ΔEQ|=2.98 mm/s) is in reasonable agreement with those of the calculated ones (δ=-0.08 mm/s and ΔEQ=3.61 mm/s).
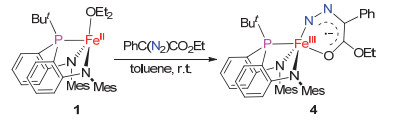
Complex 1 can also readily react with the diazo compound PhC(N2)CO2Et at room temperature, producing the diazo iron complex [(κ3-N, N, P-MesN2PBu-t)Fe(κ2-N, O-PhC(N2)CO2Et)] (4) in 60% isolated yield (Scheme 4). As isolated, complex 4 presents a brown-green crystalline solid, and is sensitive to air and water. Its solution magnetic moment (μeff=3.2(1)μB in C6D6) indicates an S=1 ground state. The zero-field 57Fe Mössbauer spectrum of 4 measured at 80 K features one quadrupole doublet with δ=0.25 mm/s and |ΔEQ|=2.55 mm/s. The Mössbauer data are apparently different from those of 1 or 3, hinting at the distinct electronic nature of their iron centers (Figure 5). The attempts to access alkylidene iron complexes by thermolysis of 4 in benzene were unsuccessful.
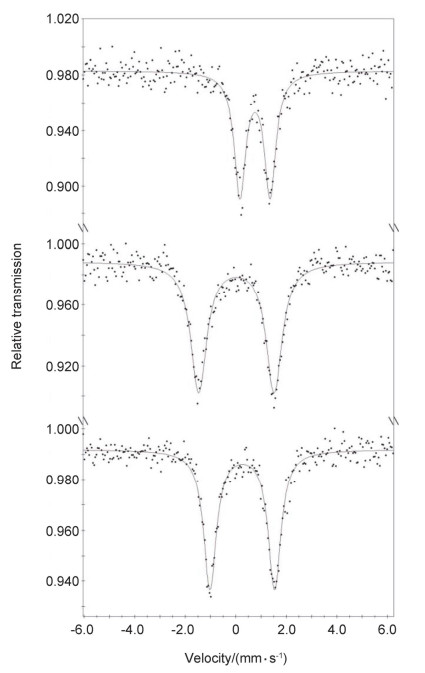
The data (dots) and best fits (solid line) are shown. The fitting data are compiled in Table 1
 下载:
导出CSV
下载:
导出CSV
| Complex | δ/(mm•s-1) | |ΔEQ|/(mm•s-1) |
| 1 | 0.75 | 1.19 |
| 2 | 0.82 | 2.40 |
| 3 | 0.02 | 2.98 |
| 4 | 0.25 | 2.55 |
| 5 | 0.70 | 1.24 |
The solid-state structure of 4, determined by X-ray crystallography, revealed that the diazo ligand is coordinating to the iron center via its terminal nitrogen atom and carbonyl oxygen atom, forming a planar six-membered chelating ring (Figure 6). Thus, complex 4 is among the rare examples of metal complexes featuring chelating diazo-ketone/ester ligands after [RhCl(PPri3)2(κ2-N, O-Me2-CHCH2C(O)C(Ph)N2)][15] and [(But2P(NSiMe3)2)Cu(κ2-N, O-O(N2)C2(C12H8))].[29] The distances of Fe1—N3 and Fe1—O1 bonds in 4 [0.1873(2) and 0.19696(17) nm, respectively] are typical as single bonds. The N3—N4, N4—C36, C36—C35, C35—O1 distances [0.1212(3), 0.1353(3), 0.1414 and 0.1248 nm, respectively] deviate from those of the single and double bonds, indicative of electron-delo-calization within the chelating ring.
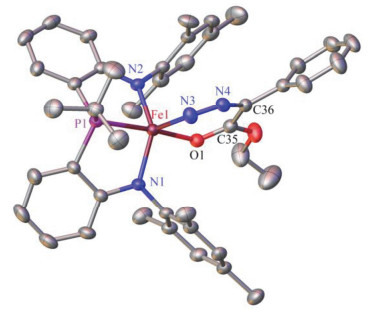
Selected distances (nm) and angles (°): Fe1—P1, 0.22431(8); Fe1—O1, 0.19696(17); Fe1—N1, 0.19258(19); Fe1—N2, 0.19382(19); Fe1—N3, 0.1874(2); O1—Fe1—P1, 169.88(5); N1—Fe1—P1, 83.23(6); N1— Fe1—O1, 93.65(7); N1—Fe1—N2, 138.58(9); N2—Fe1—P1, 83.55(6); N2—Fe1—O1, 92.61(8)
Density functional theory (DFT) calculations have been used to further probe the electronic structure of 4. The calculation study at the B3LYP/TZVP/SVP level of theory indicates that the complex has an S=1 ground spin-state that lies lower in energy than the S=0 and S=2 spin states by 70.27 and 57.23 kJ/mol, respectively. The calculated S=1 ground spin-state is consistent with that referring from the solution magnetic susceptibility of 4. At the ground spin state, maximal alignment of the α- and β-spins of the molecular orbital diagram indicated significant spin polarization, where one electron pair has an orbital overlap of 76% between an iron 3d orbital and the π-orbital of the N—N moiety of the diazo ligand (Figure 7), indicating an electronic structure of an intermediate spin iron(III) (SFe=3/2) antiferromagnetically coupled with a ligand-based radical (Sdiazo=1/2) for 4. In line with this assignment, Fe—N [0.19258(19) and 0.19382(19) nm] and Fe—P [0.22431(8) nm] distances between the amido-phosphine-amido ligand and the iron center are found to be locating between those of 1 and 3 that have high-spin iron(II) and low-spin iron(IV) centers, respectively.
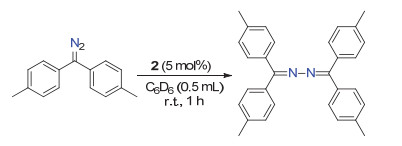
The amido-posphine-amido ligand in [(κ3-N, N, P-dcpN2-PBu-t)Fe(OEt2)] (2) has 2, 6-dichlorophenyl as its N-substituents that should have different steric and electronic character when compared to the mesityl groups in 1. Examining the reaction of 2 with (p-tolyl)2CN2 revealed the capability of the iron complex in catalzying the conversion of (p-tolyl)2CN2 to (p-tolyl)2C=NN=C(tolyl-p)2, wherein the reaction in an 1:1 ratio produced the azine along with the majority of 2 remained unreactive according to 1H NMR analysis. With a catalyst loading of 5 mol% 2, the reaction at room temperature in C6D6 for 1 h produced (p-tolyl)2C=NN=C(tolyl-p)2 in 73% NMR yield (Scheme 5). The result hints that the iron(IV) alkylidene intermediate (κ3-N, N, P-dcpN2PBu-t)Fe(C(tolyl-p)2) might be formed during the reaction course. This intermediate, however, might be highly active and susceptible to the attack by (p-tolyl)2CN2, leading to the formation of the azine (p-tolyl)2C=NN=C(tolyl-p)2.
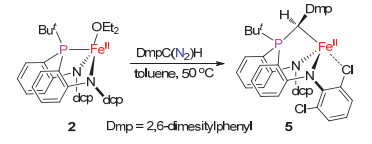
The other diazo compound was examined in the reactions with 2 is DmpC(N2)H (Dmp=2, 6-dimesityphenyl) that bears a sterically hindered Dmp group. No reaction took place when 2 was treated with DmpC(N2)H in toluene at room temperature. Elevating temperature to 50 ℃ could lead to the occurence of the reaction. Further workup on the reaction mxiture led to the isolation of the diamido-phos- phine ylide iron(II) complex [(κ3-N, N, C-dcpN2-PBu-tCH, Dmp)-Fe] (5) in 70% yield (Scheme 6). Complex 5 has been fully characterized by various spectrsocopic methods, and its moelcular structure was confirmed by single-crystal X-ray diffraction study (Figure 8). The formation of 5 implies the readiness of the mono-substitued alkylidenemetal species (κ3-N, N, P-dcpN2PBu-t)Fe[CH(Dmp)] to undergo migratory insertion reaction in spite of the presence of the bulky substituents on the phosphine and alkylidene moieties.
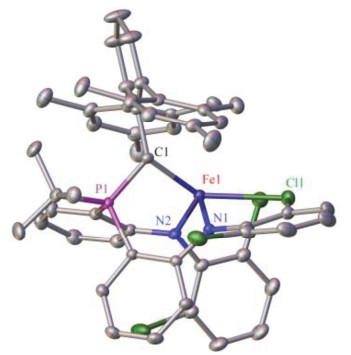
Selected distances (nm) and angles (°): Fe1—Cl1, 0.26554(6); Fe1—P1, 0.28358(6); Fe(1)—N(1), 0.20286(17); Fe1—N2, 0.19863(17); Fe1—C1 0.2146(2); P(1)—C(1), 0.1786(2); Cl1—Fe1—P1, 157.063(19); N1— Fe1—Cl1, 75.25(5); N1—Fe1—P1, 82.53(5); N1—Fe1—C1, 88.10(7); N2—Fe1—Cl1, 115.76(5) N2—Fe1—P1, 77.93(5); N2—Fe1—N1, 112.567; N2—Fe1—C1, 111.30(7); C1—Fe1—Cl1, 132.93(5); C1— Fe1—P1, 39.01(5)
Two new high-spin iron(II) complexes supported by tripodal amido-phosphine-amido ligands [(κ3-N, N, P-MesN2PBu-t)Fe(OEt2)] (1) and [(κ3-N, N, P-dcpN2PBu-t)-Fe(OEt2)] (2) were synthesized, and their reactions with diazoalkane compounds were examined. Complex 1 can react with the diazo compounds (p-tolyl)2CN2 and PhC-(N2)CO2Et to form the iron diazo complexes [(κ3-N, N, P-MesN2PBu-t)Fe(η2-N, N-(p-tolyl)2CN2)] (3) and [(κ3-N, N, P-MesN2PBu-t)Fe(κ2-N, O-PhC(N2)CO2Et)] (4). Spectroscopic characterization in combination with DFT studies suggest that 3 is a low-spin iron(IV) complex featuring a dianionic diazo ligand [η2-(p-tolyl)2CN2)]2-, whereas 4 can be viewed as an intermediate iron(III) complex bearing an antiferromagnetically coupled diazo radical anion [κ2-N, O-PhC(N2)CO2Et)]•1-. Reactivity studies of 2 revealed its ability in catalyzing the conversion of (p-tolyl)2CN2 to its azine, and its reaction with DmpC(N2)H was found to produce the high-spin iron(II) ylide complex [(κ3-N, N, C-dcpN2PBu-tCH, Dmp)Fe] (5). These results demonstrated that the diversified reactivity of iron(II) species supported by tripodal amido-phosphine-amido scaffolds toward diazoalkenes.
All experiments were performed either under an atmosphere of dry dinitrogen with the rigid exclusion of air and moisture using standard Schlenk techniques or in a glovebox. Organic solvents were dried with a solvent purification system (Innovative Technology) and bubbled with dry N2 gas prior to use. [Fe(N(TMS)2)2]2, [30] (p-tolyl)2CN2, [31] DmpC(N2)H[32] were synthesized according to literature procedures. All other chemicals were purchased from either Strem or J & K Chemical Co. and used as received unless otherwise noted. 1H NMR, 13C NMR and 31P NMR spectra were recorded on an Agilent 400 MHz spectrometers. Chemical shifts were reported with references to the residue of the deuterated solvents for proton and carbon chemical shifts, and to external 85% phosphoric acid solution for phosphorous chemical shifts. For the 1H NMR data, the widths of peaks at their half heights are denoted as v1/2. Elemental analyses were performed by the Analytical Laboratory of Shanghai Institute of Organic Chemistry (CAS). Magnetic moments were measured by the method originally described by Evans with stock and experimental solutions containing a known amount of (CH3)3SiOSi(CH3)3 standard.[33] IR spectra were recorded with a NICOLET AVATAR 330 FT-IR spectrophotometer.
Crystals were coated with Paratone-N oil and mounted on a Bruker APEX CCD-based diffractometer equipped with an Oxford low-temperature apparatus. Cell parameters were retrieved with SMART software and refined using SAINT software on all reflections. Data integration was performed with SAINT, which corrected for Lorentz polarization and decay. Absorption corrections were applied using SADABS.[34] Space groups were assigned unambiguously by analysis of symmetry and systematic absences determined by XPREP. All structures were solved and refined using SHELXTL SADABS.[34]Metal and first coordination sphere atoms were located from direct-methods E-maps. Non-hydrogen atoms were found in alternating difference Fourier synthesis and least-squares refinement cycles, and were refined anisotropically during final cycles. The crystal data, and summary of data collection and refinement for the complexes were summarized in Tables S1. CCDC 2006056~2006059 contain the supplementary crystallographic data for complexes 2~4, and 5•Et2O. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_ request/cif.
All solid samples for 57Fe Mössbauer spectroscopy were run on non-enriched samples of the as-isolated complexes. Each sample was loaded into a Delrin Mössbauer sample cup for measurements and loaded under liquid nitrogen. Low temperature 57Fe Mössbauer measurements were performed using a SeeCo. MS4 Mössbauer spectrometer integrated with a Janis SVT-400T He/N2 cryostat for measurements at 80 K. Isomer shifts were determined relative to α-Fe at 298 K. All Mössbauer spectra were fitted using the program WMoss (SeeCo).
To an Et2O (15 mL) solution of (2, 4, 6-Me3C6H2)(2-Br- C6H4)NH (1.45 g, 5 mmol) was added nBuLi (2.5 mol•L-1 in n-hexane, 4 mL, 10 mmol) slowly at -78 ℃. The resulting mixture was allowed to warm to room temperature and further stirred for 5 h. To this solution, an Et2O solution (10 mL) of tBuPCl2 (0.40 g, 2.5 mmol) was added at -78 ℃. The resulting mixture was allowed to warm to room temperature and further stirred for 48 h. The mixture was then quenched with a saturated aqueous solution of NaHCO3, and extracted with CH2Cl2 (50 mL×3). The combined organic phases were washed with a saturated NaHCO3 aqueous solution, brine, and dried over Na2SO4. After filtration and removal of volatiles, the crude product was purifiedby flash column chromatography [silica gel, n-hexanes/CH2Cl2 (V:V=10:1) as elute] to give H2(MesN2PBu-t) as a white solid (0.64 g, 50% yield). m.p. 165~168 ℃; 1H NMR (400 MHz, CDCl3, 293 K) δ: 7.63~7.55 (m, 2H), 7.10 (dd, J=11.3, 4.0 Hz, 2H), 6.95 (s, 2H), 6.83 (s, 2H), 6.77 (t, J=7.4 Hz, 2H), 6.27 (d, J=7.6 Hz, 2H), 6.19~6.16 (m, 2H), 2.31 (s, 6H), 2.21 (s, 6H), 1.64 (s, 6H), 1.53 (d, J=13.7 Hz, 9H); 13C NMR (101 MHz, CDCl3, 293 K) δ: 149.87 (d, JC—P=17.8 Hz), 136.23, 135.81 (d, JC—P=2.0 Hz), 135.53, 135.21, 134.21, 130.04, 129.09, 118.39 (d, JC—P=11.3 Hz), 117.58, 111.80 (d, JC—P=2.8 Hz), 31.69 (d, JC—P=9.1 Hz), 28.80 (d, JC—P=13.8 Hz), 21.04, 18.40, 17.50; 31P NMR (162 MHz, CDCl3, 293 K) δ: -28.42; IR (film) v: 3360 (w), 2944 (w), 2917 (w), 2859 (w), 1585 (m), 1487 (s), 1438 (s), 1275 (m), 1158 (w), 1033 (w), 1011 (w), 854 (m), 809 (m), 746 (s), 683 (w), 478 (m) cm-1. HRMS (ESI) calcd for C34H42N2P [M+H]+: 509.3080; found 509.3081
To an Et2O (40 mL) solution of (2, 6-Cl2C6H3)(2-Br-C6H4)NH (3.17 g, 10.0 mmol) was added nBuLi (2.5 mol/L in n-hexane, 8 mL, 20 mmol) slowly at -78 ℃. The resulting mixture was allowed to warm to room temperature and further stirred for 5 h. To this solution, an Et2O solution (10 mL) of tBuPCl2 (0.80 g, 5 mmol) was added at -78 ℃. The resulting mixture was allowed to warm to room temperature and further stirred for 48 h. The mixture was then quenched with a saturated aqueous solution of NaHCO3, and extracted with CH2Cl2 (50 mL×3). The combined organic phases were washed with a saturated NaHCO3 aqueous solution, brine, and dried over Na2SO4. After filtration and removal of volatiles, the crude product was purifiedby flash column chromatography [silica gel, n-hexanes/CH2Cl2 (V:V=10:1) as elute] to give H2(dcpN2PBu-t) as a white solid (1.50 g, 53% yield). m.p. 220~222 ℃; 1H NMR (400 MHz, CDCl3, 293 K) δ: 7.59~7.49 (m, 2H), 7.39~7.19 (m, 4H), 7.14 (t, J=7.7 Hz, 2H), 7.00 (t, J=8.1 Hz, 2H), 6.89 (t, J=7.4 Hz, 2H), 6.64 (d, J=5.9 Hz, 2H), 6.33 (dd, J=7.6, 5.5 Hz, 2H), 1.46 (d, J=13.6 Hz, 9H); 13C NMR (101 MHz, CDCl3, 293K) δ: 147.18 (d, JC—P=18.4 Hz), 136.98, 136.95, 134.22, 132.31~131.94 (m), 129.55, 128.70 (d, JC—P=30.6 Hz), 125.55, 121.65 (d, JC—P=13.3 Hz) 120.22, 114.60, 31.77 (d, JC—P=10.3 Hz), 28.71 (d, JC—P=13.8 Hz); 31P NMR (162 MHz, CDCl3, 293K) δ: -24.43; IR (film) v: 3330 (w), 3298 (w), 2957 (w), 2863 (w), 1569 (m), 1493 (m), 1439 (s), 1411 (m), 1296 (m), 1277 (m), 1192 (w), 1156 (w), 1095 (w), 838 (s), 773 (s), 504 (w) cm-1. HRMS (ESI) calcd for C28H26N2PCl4 [M+H]+: 561.0582; found 561.0571.
To a toluene (20 mL) solution of H2(MesN2PBu-t) (125 mg, 0.25 mmol) was added [Fe(N(TMS)2)2]2 (111 mg, 0.15 mmol). The resulting mixture was stirred at 50 ℃ for 2 h to afford a red solution. After removal of the solvent under vacuum, the red residue was washed by n-hexane (10 mL) to produce 1 as an orange solid (77 mg, 49%). m.p. > 163 ℃ (dec.); 1H NMR (400MHz, C6D6, 293 K) δ: 64.08 (very br), 59.64 (v1/2=49 Hz), 47.72 (v1/2=87 Hz), 39.46 (v1/2=75 Hz), 37.57 (v1/2=93 Hz), 29.53 (v1/2=246 Hz), 23.22 (v1/2=51 Hz), 10.12 (very br), 6.21 (very br), 3.93 (v1/2=107 Hz), -37.23 (v1/2=86 Hz); IR (film) v: 2966 (s), 2916 (s), 2864 (m), 1586 (s), 1452 (s), 1431 (s), 1312 (m), 1266 (m), 1201 (m), 1039 (m), 876 (3), 856 (m), 748 (s) cm-1. Magnetic susceptibility (C6D6, 293 K): μeff=4.7(1)μB. Anal. calcd for C38H49FeN2OP: C 71.69, H 7.76, N 4.40; found C 71.55, H 7.52, N 4.38. 57Fe Mössbauer spectrum (Zero-field, 80 K) δ: 0.75 mm/s, |ΔEQ|=1.19 mm/s.
To a tetrahydrofuran (THF) (20 mL) solution of H2(dcpN2- PBu-t) (105 mg, 0.19 mmol) was added [Fe(N(TMS)2)2]2 (106 mg, 0.14 mmol). The resulting mixture was stirred at room temperature for 2 h to afford a orange solution. After removal of the solvent under vacuum, the orange residue was washed by n-hexane (10 mL) to produce 2 as an orange solid (100 mg, 77%). Single-crystals of 2 suitable for X-ray diffraction study were obtained by standing its saturated Et2O solution at room temperature via slow evaporation of Et2O overnight. m.p. > 201 ℃ (dec.); 1H NMR (400 MHz, C6D6, 293 K) δ: 41.55 (v1/2=56 Hz), 36.74 (v1/2=63 Hz), 33.97 (v1/2=95 Hz), 29.80 (v1/2=179 Hz), 16.53 (v1/2=41 Hz), 5.17 (very br), -8.68 (v1/2=128 Hz), -27.67 (v1/2=45 Hz), -40.58 (very br), -42.64 (v1/2=49 Hz); IR (film) v: 1583 (s), 1453 (s), 1450 (s), 1316 (m), 1071 (m), 1032 (m), 863 (m), 780 (s), 766 (s), 748 (m) cm-1. Magnetic susceptibility (C6D6, 293 K): μeff=5.0(1)μB. Anal. calcd for C32H33FeN2OCl4P: C 55.68, H 4.82, N 4.06; found C 55.57, H 4.80, N 4.10. 57Fe Mössbauer spectrum (Zero-field, 80 K) δ: 0.82 mm/s, |ΔEQ|=2.40 mm/s.
To a red solution of 1 (217 mg, 0.34 mmol) in toluene (15 mL) was added (p-tolyl)2CN2 (76 mg, 0.34 mmol) at room temperature for 2 h. The color of the solution changed from red to brown gradually. After removal of the solvent under vacuum, the brown residue was washed by n-pentane (10 mL) to produce 3 as a brown solid (149 mg, 56% yield). Single-crystals of 3 suitable for X-ray diffraction study were obtained by standing its saturated n-pentane solution at room temperature via slow evaporation of n-pentane overnight. m.p. > 167 ℃ (dec.); 1H NMR (400 MHz, C6D6, 293 K) δ: 7.77 (d, J=7.8 Hz, 2H), 7.31 (t, J=7.3 Hz, 2H), 7.02 (d, J=7.8 Hz, 2H), 6.94 (t, J=7.4 Hz, 2H), 6.61 (t, J=7.0 Hz, 4H), 6.43 (s, 4H), 6.42~6.34 (m, 2H), 5.80 (d, J=7.9 Hz, 2H), 2.41 (s, 6H), 2.07 (d, J=20.5 Hz, 6H), 1.94 (s, 6H), 1.62 (d, J=15.4 Hz, 9H), 1.54 (s, 6H); 13C NMR (101 MHz, C6D6, 293 K) δ: 170.99 (d, JC—P=24.5 Hz), 146.12, 138.33, 137.34, 136.18, 134.67, 134.16, 133.74, 133.54, 131.51, 130.84, 129.25, 129.20, 129.04, 128.86, 118.68, 115.53, 115.12, 114.61, 114.50, 33.35 (d, JC—P=29.9 Hz), 27.80, 21.34 (d, JC—P=7.2 Hz), 21.04, 20.07, 18.40; 31P NMR (162 MHz, C6D6, 293 K) δ: 108.22; IR (film) v: 2967 (w), 2918 (m), 2867 (m), 2035 (s), 1593 (m), 1512 (s), 1451 (s), 1432 (s), 1316 (m), 1272 (m), 1064 (m), 1041 (m), 858 (w), 813 (m), 749 (m), 623 (w), 566 (w) cm-1. Anal. calcd for C49H53FeN4P: C 74.99, H 6.81, N 7.14; found C 75.20, H 6.75, N 7.40. 57Fe Mössbauer spectrum (Zero-field, 80 K) δ: 0.02 mm/s, |ΔEQ|=2.98 mm/s.
To a red solution of 1 (151 mg, 0.24 mmol) in toluene (15 mL) was added PhC(N2)CO2Et (45 mg, 0.24 mmol) at room temperature for 2 h. The color of the solution changed from red to green gradually. After removal of the solvent under vacuum, the green residue was washed by n-pentane (10 mL) to produce 4 as a green solid (108 mg, 60% yield). Single-crystals of 4 suitable for X-ray diffraction study were obtained by standing its saturated n-pentane solution at room temperature via slow evaporation of n-pentane overnight. m.p. > 160 ℃ (dec.); 1H NMR (400 MHz, C6D6, 293 K) δ: 34.16 (v1/2=41 Hz), 26.71 (v1/2=40 Hz), 24.59 (v1/2=24 Hz), 22.33 (v1/2=20 Hz), 15.61 (v1/2=31 Hz), -0.83 (v1/2=78 Hz), -31.30 (v1/2=45 Hz), -31.63 (v1/2=25 Hz), -52.10 (v1/2=47 Hz); IR (film) v: 2994 (w), 2963 (w), 2920 (w), 2090 (m), 1709 (m), 1577 (s), 1435 (s), 1371 (m), 1288 (s), 1276 (s), 1255 (s), 1195 (m), 876 (m), 756 (m), 741 (w) cm-1. Magnetic susceptibility (C6D6, 293 K): μeff=3.2(1)μB. Anal. calcd for C44H49FeN4O2P: C 70.21, H 6.56, N 7.44; found C 69.49, H 6.45, N 7.37. 57Fe Mössbauer spectrum (Zero-field, 80 K) δ: 0.25 mm/s, |ΔEQ|=2.55 mm/s.
To a C6D6 solution (0.2 mL) of 2 (5.1 mg, 0.007 mmol) in a J-Young tube was added (p-tolyl)2CN2(33 mg, 0.148 mmol) and C6D6 (0.3 mL) at room temperature. After 1 h, an internal standard 1, 3, 5-trimethoxybenzene (2.1 mg, 0.013 mmol) was then added. 1H NMR analysis on the reaction mixture indicated the formation of the (p-tolyl)2- C=NN=C(p-tolyl)2 in 73% yield.
To an orange solution of 2 (267 mg, 0.47 mmol) in toluene (15 mL) was added DmpC(N2)H (202 mg, 0.57 mmol). The mixture was then heated at 50 ℃ for 2 h. The color of the solution changed from orange to yellow-green gradually. After removal of the solvent under vacuum, the yellow-green residue was washed by n-hexane (10 mL) and dried under vaccum, which leaved 5 as a yellow-green solid (270 mg, 70% yield). Single-crystals of 5 suitable for X-ray diffraction study were obtained by standing its saturated Et2O solution at room temperature via slow evaporation of Et2O overnight. m.p. > 190 ℃ (dec.); 1H NMR (400 MHz, C6D6, 293 K) δ: 1H NMR (400 MHz, C6D6, 293 K) δ: 61.44 (v1/2=42 Hz), 44.44 (v1/2=31 Hz), 39.97 (v1/2=35 Hz), 36.05 (v1/2=32 Hz), 31.52 (v1/2=49 Hz), 30.36 (v1/2=55 Hz), 28.31 (v1/2=49 Hz), 23.74 (v1/2=29 Hz), 19.32 (v1/2=29 Hz), 17.62 (very br), 16.82 (v1/2=36 Hz), 16.19 (v1/2=22 Hz), 15.45 (v1/2=54 Hz), 13.99 (v1/2=14 Hz), 12.73 (v1/2=21 Hz), 9.43 (v1/2=28 Hz), -7.61 (v1/2=203 Hz), -9.10 (very br), -15.32 (v1/2=28 Hz), -22.49 (v1/2=28 Hz), -26.67 (v1/2=28 Hz), -36.60 (very br), -36.68 (very br), -37.36 (v1/2=32 Hz), -74.56 (very br); IR (film) v: 2963 (m), 2916 (m), 2864 (m), 1587 (m), 1553 (w), 1468 (s), 1454 (w), 1443 (s), 1299 (m), 1293 (m), 1093 (w), 1067 (w), 867 (w), 847 (m), 773 (m), 743 (m) cm-1. Magnetic susceptibility (C6D6, 293 K): μeff=4.9(1)μB. Anal. calcd for C52H49Cl4FeN2P: C 67.53, H 5.24, N 2.97; found C 67.37, H 5.34, N 3.01. 57Fe Mössbauer spectrum (Zero-field, 80 K) δ: 0.70 mm/s, |ΔEQ|=1.24 mm/s.
To have a better understanding of the electronic structures of 3 and 4, density functional theory (DFT)[35-36] study has been performed with the ORCA 4.1.0 program[37] using the B3LYP methods.[38-39] The SVP[40-41] basis set was used for the C, N, and H atoms, and the TZVP[42] basis set was used for the other atoms. The RIJCOSX[43] approximation with matching auxiliary basis sets[40-41] was employed to accelerate the calculations. The calculation utilizes the atom-pairwise dispersion correction with the Becke-John- son damping scheme (D3BJ).[44-45] The atomic coordinates of 3 and 4 used for calculation were obtained from X-ray diffraction studies, and only the positions of hydrogen atoms were optimized. The selected localized orbitals for 3 are listed in Figures 3 and 4. The frontier molecular orbital diagram for 4 is listed in Figure 6.
Supporting Information NMR, IR, Mössbauer spectra for the new complexes, Table for crystal data (PDF). The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
Herrmann, W. A. Angew. Chem., Int. Ed. 1978, 17, 800. doi: 10.1002/anie.197808001
Sutton, D. Chem. Rev. 1993, 93, 995. doi: 10.1021/cr00019a008
Mizobe, Y.; Ishii, Y.; Hidai, M. Coord. Chem. Rev. 1995, 139, 281. doi: 10.1016/0010-8545(94)01118-U
Poverenov, E.; Leitus, G.; Shimon, L. J. W.; Milstein, D. Organo- metallics 2005, 24, 5937.
Samant, R. G.; Graham, T. W.; Rowsell, B. D.; McDonald, R.; Cowie, M. Organometallics 2008, 27, 3070. doi: 10.1021/om700756q
Brookhart, M.; Studabaker, W. B. Chem. Rev. 1987, 87, 411. doi: 10.1021/cr00078a008
Chauvin, Y. Angew. Chem., Int. Ed. 2006, 45, 3740. doi: 10.1002/anie.200601234
Schrock, R. R. Angew. Chem., Int. Ed. 2006, 45, 3748. doi: 10.1002/anie.200600085
Grubbs, R. H. Angew. Chem., Int. Ed. 2006, 45, 3760. doi: 10.1002/anie.200600680
Zhu, S.-F.; Zhou, Q.-L. Natl. Sci. Rev. 2014, 1, 580. doi: 10.1093/nsr/nwu019
Albertin, G.; Antoniutti, S.; Forcolin, M.; Gasparato, D. Polyhedron 2016, 104, 46. doi: 10.1016/j.poly.2015.11.021
Albertin, G.; Antoniutti, S.; Botter, A.; Castro, J. Inorg. Chem. 2015, 54, 2091. doi: 10.1021/ic502963n
Albertin, G.; Antoniutti, S.; Castro, J.; Dottorello, G. Dalton Trans 2015, 44, 9289. doi: 10.1039/C5DT00755K
Albertin, G.; Antoniutti, S.; Bortoluzzi, M.; Castro, J.; Marzaro, L. Dalton Trans 2015, 44, 15470. doi: 10.1039/C5DT02113H
Werner, H.; Mahr, N.; Wolf, J.; Fries, A.; Laubender, M.; Bleuel, E.; Garde, R.; Lahuerta, P. Organometallics 2003, 22, 3566. doi: 10.1021/om0302037
Bonyhady, S. J.; Goldberg, J. M.; Wedgwood, N.; Dugan, T. R.; Eklund, A. G.; Brennessel, W. W.; Holland, P. L. Inorg. Chem. 2015, 54, 5148. doi: 10.1021/acs.inorgchem.5b00673
Li, Y.; Huang, J.-S.; Zhou, Z.-Y.; Che, C.-M.; You, X.-Z. J. Am. Chem. Soc. 2002, 124, 13185. doi: 10.1021/ja020391c
Bart, S. C.; Bowman, A. C.; Lobkovsky, E.; Chirik, P. J. J. Am. Chem. Soc. 2007, 129, 7212. doi: 10.1021/ja070056u
Sazama, G. T.; Betley, T. A. Inorg. Chem. 2014, 53, 269. doi: 10.1021/ic402210j
Bonyhady, S. J.; DeRosha, D. E.; Vela, J.; Vinyard, D. J.; Cowley, R. E.; Mercado, B. Q.; Brennessel, W. W.; Holland, P. L. Inorg. Chem. 2018, 57, 5959. doi: 10.1021/acs.inorgchem.8b00468
Albertin, G.; Antoniutti, S.; Bortoluzzi, M.; Botter, A.; Castro, J.; Sibilla, F. RSC Adv. 2016, 6, 97650. doi: 10.1039/C6RA22051G
Russell, S. K.; Hoyt, J. M.; Bart, S. C.; Milsmann, C.; Stieber, S. C. E.; Semproni, S. P.; DeBeer, S.; Chirik, P. J. Chem. Sci. 2014, 5, 1168. doi: 10.1039/C3SC52450G
Liu, J.; Hu, L.; Wang, L.; Chen, H.; Deng, L. J. Am. Chem. Soc. 2017, 139, 3876. doi: 10.1021/jacs.7b00484
Xiao, J.; Deng, L. Dalton Trans. 2013, 42, 5607. doi: 10.1039/c3dt50518a
Smit, T. M.; Tomov, A. K.; Gibson, V. C.; White, A. J. P.; Williams, D. J. Inorg. Chem. 2004, 43, 6511. doi: 10.1021/ic0490775
Cheng, J.; Liu, J.; Leng, X.; Lohmiller, T.; Schnegg, A.; Bill, E.; Ye, S.; Deng, L. Inorg. Chem. 2019, 58, 7634. doi: 10.1021/acs.inorgchem.9b01147
Chisholm, M. H.; Folting, K.; Huffman, J. C.; Ratermann, A. L. Inorg. Chem. 1984, 23, 2303. doi: 10.1021/ic00183a019
Sydora, O. L.; Kuiper, D. S.; Wolczanski, P. T.; Lobkovsky, E. B.; Dinescu, A.; Cundari, T. R. Inorg. Chem. 2006, 45, 2008. doi: 10.1021/ic051481w
Straub, B. F.; Rominger, F.; Hofmann, P. Organometallics 2000, 19, 4305. doi: 10.1021/om000430y
Olmstead, M. M.; Power, P. P.; Shoner, S. C. Inorg. Chem. 1991, 30, 2547. doi: 10.1021/ic00011a017
Javed, M. I.; Brewer, M. Org. Lett. 2007, 9, 1789. doi: 10.1021/ol070515w
Iluc, V. M.; Laskowski, C. A.; Hillhouse, G. L. Organometallics 2009, 28, 6135. doi: 10.1021/om900566k
Evans, D. F. J. Chem. Soc. 1959, 2003.
Sheldrick, G. M. SADABS: Program for Empirical Absorption Correction of Area Detector Data, University of G ttingen, G ttingen, Germany, 1996.
Hohenberg, P.; Kohn, W. Phys. Rev. 1964, 136, B864. doi: 10.1103/PhysRev.136.B864
Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, A1133. doi: 10.1103/PhysRev.140.A1133
Neese F.. ORCA-an ab initio, Density Functional and Semiempirical Program Package, Version 4.1.0[J]. Max-Planck Institute for Bioinorganic Chemistry, Mulheim an der Ruhr, Germany, 2017, : .
Becke, A. D. J. Chem. Phys. 1993, 98, 5648. doi: 10.1063/1.464913
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785. doi: 10.1103/PhysRevB.37.785
Schäfer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys. 1992, 97, 2571. doi: 10.1063/1.463096
Schäfer, A.; Huber, C.; Ahlrichs, R. J. Chem. Phys. 1994, 100, 5829. doi: 10.1063/1.467146
Pantazis, D. A.; Chen, X.-Y.; Landis, C. R.; Neese, F. J. Chem. Theory Comput. 2008, 4, 908. doi: 10.1021/ct800047t
Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Chem. Phys. 2009, 356, 98. doi: 10.1016/j.chemphys.2008.10.036
Grimme, S.; Ehrlich, S.; Goerigk, L. J. Comput. Chem. 2011, 32, 1456. doi: 10.1002/jcc.21759
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi: 10.1063/1.3382344

Figure 1 Molecular structure of 2 (hydrogen atoms were omitted)
Selected distances (nm) and angles (°): Fe1—N1, 0.19877(14); Fe1—N2, 0.19926(14); Fe1—P1, 0.24116(5); Fe1—Cl1, 0.2845; Fe1—O1, 0.20626(13); N1—Fe1—N2, 123.31(6); N1—Fe1—P1, 80.99(4); N1—Fe1—O1, 113.78(6); N2—Fe1—P1, 84.25(4); N2—Fe1—O1, 121.62(6); P1— Fe1—O1, 117.17(4); P1—Fe1—Cl1, 153.00

Figure 2 Molecular structure of complex 3 (hydrogen atoms were omitted)
Selected distances (nm) and angles (°): Fe1—P1, 0.21628(4); Fe1—N3, 0.18571(11); Fe1—N4, 0.18559(11); Fe1—N1, 0.19601(11); Fe1—N2, 0.17524(11); N3—Fe1—P1, 85.38(3); N4—Fe1—P1, 85.93(4); N4— Fe1—N3, 128.91(5)

Figure 3 Selected localized orbitals for 3 (hydrogen atoms and substituents on ligands were omitted)

Figure 4 Selected localized orbitals for the N—N and N—C bonds in 3 (hydrogen atoms and substituents on ligands were omitted)

Figure 5 57Fe Mössbauer spectra (80 K, zero-field) of 1 (top), 3 (middle), and 4 (bottom)
The data (dots) and best fits (solid line) are shown. The fitting data are compiled in Table 1

Figure 6 Molecular structure of 4 (hydrogen atoms were omitted)
Selected distances (nm) and angles (°): Fe1—P1, 0.22431(8); Fe1—O1, 0.19696(17); Fe1—N1, 0.19258(19); Fe1—N2, 0.19382(19); Fe1—N3, 0.1874(2); O1—Fe1—P1, 169.88(5); N1—Fe1—P1, 83.23(6); N1— Fe1—O1, 93.65(7); N1—Fe1—N2, 138.58(9); N2—Fe1—P1, 83.55(6); N2—Fe1—O1, 92.61(8)

Figure 8 Molecular structure of complex 5 (hydrogen atoms were omitted)
Selected distances (nm) and angles (°): Fe1—Cl1, 0.26554(6); Fe1—P1, 0.28358(6); Fe(1)—N(1), 0.20286(17); Fe1—N2, 0.19863(17); Fe1—C1 0.2146(2); P(1)—C(1), 0.1786(2); Cl1—Fe1—P1, 157.063(19); N1— Fe1—Cl1, 75.25(5); N1—Fe1—P1, 82.53(5); N1—Fe1—C1, 88.10(7); N2—Fe1—Cl1, 115.76(5) N2—Fe1—P1, 77.93(5); N2—Fe1—N1, 112.567; N2—Fe1—C1, 111.30(7); C1—Fe1—Cl1, 132.93(5); C1— Fe1—P1, 39.01(5)
Table 1. Zero-Field 57Fe Mössbauer spectroscopic data of 1~5 measured at 80 K
| Complex | δ/(mm•s-1) | |ΔEQ|/(mm•s-1) |
| 1 | 0.75 | 1.19 |
| 2 | 0.82 | 2.40 |
| 3 | 0.02 | 2.98 |
| 4 | 0.25 | 2.55 |
| 5 | 0.70 | 1.24 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们