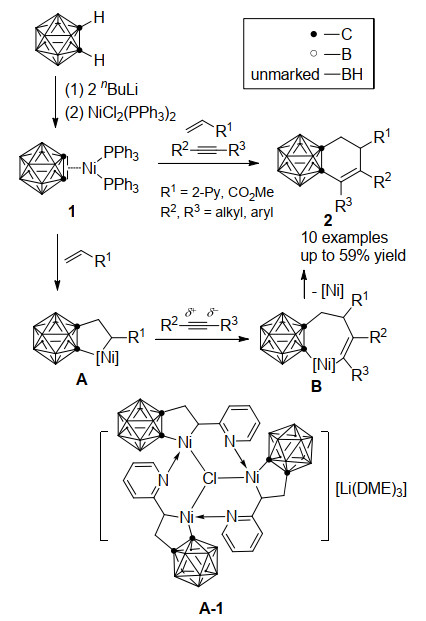
图式 1.
镍-介导碳硼炔、烯烃、炔烃的三组分环加成反应
Scheme 1.
Nickel-mediated three-component cycloaddition reaction of carboryne, alkenes and alkynes

二十面体碳硼烷是一类具有26个离域电子的硼碳簇合物, 它们与苯有许多相似的性质, 例如疏水性、芳香性、优异的热稳定性和化学稳定性.另一方面, 碳硼烷有其独有的性质, 例如几何球面、三维电子离域等[1].这些特质使碳硼烷及其衍生物得到了广泛的应用, 例如:碳硼烷化合物在治疗癌症的硼中子俘获疗法(BNCT)中用作高硼含量药物[2]; 在金属有机化学中用作多变配体[3]; 在超分子及纳米材料中用作功能砌块[4]; 在有机光学材料中用作电子阱等[5].为了拓展其应用, 高效获取碳硼烷官能化衍生物已成为碳硼烷化学领域的重要研究方向.
在碳顶点官能团化方面, 十硼烷与取代炔烃反应可以直接制备取代邻-碳硼烷[6]; 另一方面, 由于碳硼烷上CH具有一定的酸性, 容易被强碱拔除质子, 生成的碳硼烷阴离子与亲电试剂进一步反应, 是制备多种碳顶点取代碳硼烷衍生物的有效方法[7].近年来, 过渡金属介导或催化的碳顶点官能团化方法得到了迅速的发展, 为碳-取代碳硼烷衍生物的合成提供了有效的方法[8].
而相对于已发展较为成熟的碳顶点官能团化, 碳硼烷硼顶点活化及进一步衍生化的研究较为滞后, 目前硼-取代碳硼烷化合物的种类及数量都较为有限, 大大限制了后续的性质研究和应用开发.近年来, 除了直接对硼顶点进行亲核取代反应外[9], 碳硼烷B—H键催化官能团化过程也受到了合成化学家们的广泛关注, 相继报道了过渡金属催化的系列区域选择性B—H键芳基化[10]、烯基化[11]、炔基化[12]、硼基化[13]、氟化[14]、氧化[15]、胺基化[16]、卤化[17]等过程.这些基于直接B—H键活化的合成策略可以简化原料, 缩短反应流程, 从而提升合成效率和原子经济性[18].
多组分反应、串联反应及一锅法反应的系列合成方法都具有高原子及步骤经济性, 原料简单易得, 操作简便等特点, 可以不经中间产物分离, 直接得到结构复杂的最终产物.在以上一系列过渡金属参与的碳硼烷区域选择性官能团化方法学报道中, 除了可以引入单一种类的取代基外, 也能通过多组分反应、串联反应及一锅法反应的策略, 以双取代、多取代等不同形式实现复杂碳硼烷衍生物的高效构筑.本文将围绕C, C, C, B, B, B三种不同取代类型, 综述过渡金属参与的复杂碳硼烷衍生物可控合成的研究进展, 并对该领域所面临的挑战及发展前景做出了总结和展望.
碳硼炔(1, 2-脱氢碳硼烷)可以看作苯炔的三维类似物, 而过渡金属-邻-碳硼炔配合物中金属-碳键具有不同于经典金属-碳键的化学性质, 能与多种不饱和分子发生反应, 其反应模式取决于中心金属离子的电子构型[19]. 2009年, 谢作伟课题组[20]开发了镍介导的碳硼炔与烯烃和炔烃的三组分[2+2+2]环加成反应.在该反应体系中, 邻碳硼烷在丁基锂的作用下得到碳硼烷二锂盐, 其与NiCl2(PPh3)2通过盐消除原位制得镍-碳硼炔配合物1. 1同时与过量的烯烃和炔烃在加热条件下可以得到一系列二氢苯并碳硼烷衍生物2 (Scheme 1).其中, 参与反应的烯烃可以是2-乙烯基吡啶或丙烯酸甲酯(R1=2-Py, CO2Me), 而炔烃则需是烷基或芳基取代的中间炔(R2, R3=alkyl, aryl), 产物分离收率最高可达59%.该反应的路径首先是烯烃插入镍-碳硼炔1的Ni—C键, 得到碳硼烷并镍杂环戊烷中间体A, 随后炔烃插入Ni—C(烷基)键, 形成镍杂七元中间体B, 最后经还原消除得到最终产物2.反应中由于活化烯烃的反应活性高于炔烃, 使得三组分反应可以获得优秀的化学选择性; 而由于烯烃取代基团的配位作用和烷基-芳基取代的非对称炔烃具有一定的极性, 它们的插入步骤也分别具有优秀的区域选择性.由于活化烯烃中配位基团与金属中心的作用, 稳定反应中间体A可以由1与烯烃的单独反应制备, 其中2-乙烯基吡啶的产物A-1的结构经单晶X射线衍射确证. A与炔烃的反应也能以大于90%的收率获得最终产物, 进一步证明了三组分反应获得二氢苯并碳硼烷2的路径.
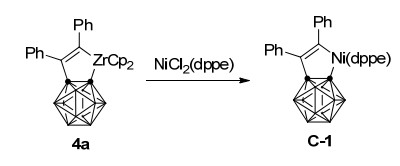
过渡金属-碳硼炔配合物的反应性质取决于金属中心的性质, 其中镍-碳硼炔1与炔烃作用可以获得与两分子相同炔烃的[2+2+2]环加成产物[21], 而锆-碳硼炔前体3只能与一分子炔烃发生Zr—C键插入反应, 得到稳定的碳硼烷并锆杂环戊烯产物4[22].此锆杂五元环化合物并不能进一步与炔烃反应, 考虑到镍杂五元环化合物更高的反应活性, 4在NiCl2(PMe3)2存在下可以与另一分子炔烃反应(Scheme 2)[23]:通过转金属作用先得到镍杂环戊烯中间体C, 炔烃插入Ni—C(烯基)键形成镍杂七元中间体D, 再经还原消除, 最终得到过渡金属锆与镍共同介导下碳硼炔与两分子不同炔烃[2+2+2]环加成产物5.该反应同样具有优秀的炔烃插入区域选择性, 可以以最高85%的分离收率得到不同取代的苯并碳硼烷衍生物.而通过双齿配体dppe的稳定作用, 可以分离到镍转金属中间体C-1, 其结构经由单晶X射线衍射确证(Scheme 3).
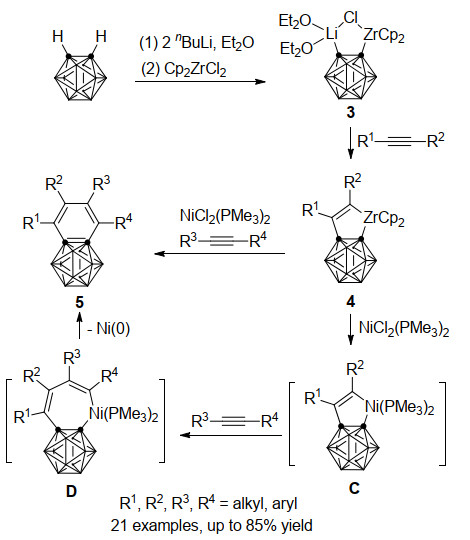
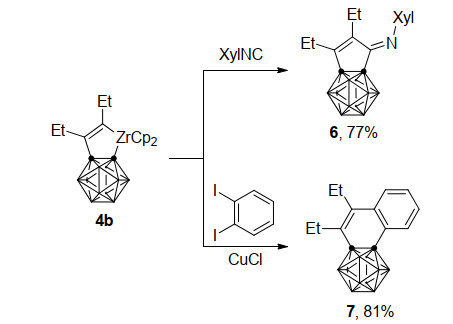
2009年, 谢作伟课题组[24]在先前工作基础上进一步研究了锆环戊烯4b的反应性质(Scheme 4).一方面, 将4b与2, 6-二甲苯基异腈(XylNC)在甲苯中回流, 异腈碳很容易地插入到Zr—C(烯基)键中, 以77%的产率得到碳硼烷并五元环产物6.另一方面, 在CuCl存在下, 锆杂环戊烯4b与邻二碘苯共同加热得到萘并碳硼烷产物7, 产率为81%, 其结构经单晶X射线衍射证实.这个反应过程中铜是必须的, 若没有CuCl的存在, 则反应无法进行.
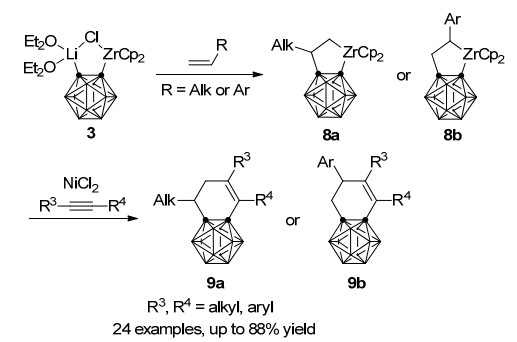
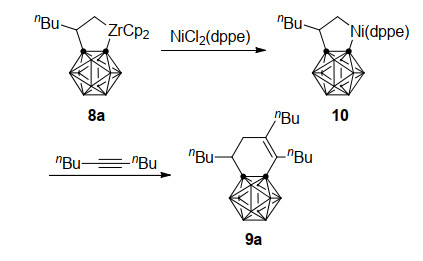
与炔烃的反应类似, 锆-碳硼炔前体3同样能与一分子末端烯烃发生Zr—C键插入反应, 得到稳定的碳硼烷并锆杂环戊烷8[25], 插入反应的区域选择性由烯烃上取代基的电性决定(Scheme 5):给电子烷基位于金属β位(8a)而吸电子芳基位于金属α位(8b).同样8与炔烃在NiCl2存在下进一步反应, 能以最高88%收率合成二氢苯并碳硼烷9[26].该方法既高效又实用, 可以从锆-碳硼炔前体3出发, 与烯烃和炔烃一锅法合成二氢苯并碳硼烷衍生物9a和9b.值得注意的是, 在没有炔烃的情况下, 8与NiCl2直接作用生成含有镍杂环戊烷中间体β-H消除产物在内的复杂混合物.而锆杂环戊烷的镍转金属中间体10同样可经由dppe配体稳定并得到分离鉴定, 它可以在炔烃的作用下高效转化为9a (Scheme 6), 这也进一步证明了此一锅法反应的路径.
当上述反应中使用芳基和三甲硅基取代炔烃时, 在NiCl2(PMe3)2存在下可以最高可达86%的收率获得系列二氢富烯并碳硼烷化合物11 (Scheme 7)[27].在对该特殊[2+2+1]环加成反应的机理研究中, 林振阳小组和谢作伟小组[28]共同通过密度泛函理论(DFT)对过渡态能量进行了计算.结果表明, 亚乙烯基镍过渡态在能量上较为不利, 反应历经炔烃插入、η1-烯基至η2-烯基重排、硅基迁移的系列过程, 而炔烃上芳基取代基的位阻效应决定最终[2+2+1]环加成产物中环外双键的顺反构型.
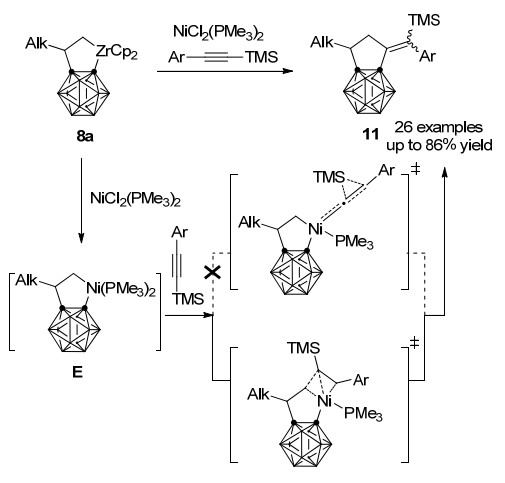
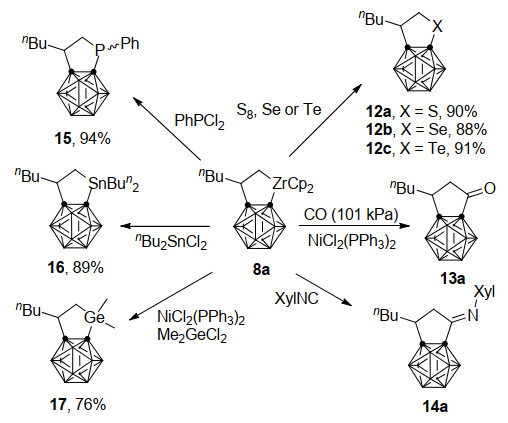
2018年, 谢作伟、邱早早课题组[29]又进一步探索了碳硼烷并锆环戊烷8a对一系列底物的反应性(Scheme 8).例如, 8a与过量硫族元素单质在110 ℃甲苯中反应得到碳硼烷并硫族元素杂环戊烷12a~12c. 8a与一氧化碳在NiCl2(PPh3)2存在条件下得到插羰产物13a, 而具有较强活性的异腈XylNC在室温下插入8a的Zr—C键中, 形成亚胺环戊烷14a.另一方面, 8a还可以与PhPCl2、nBu2SnCl2和Me2GeCl2在不同条件下通过转金属反应分别以94%、89%和76%的收率得到15~17.这些实验结果表明, 碳硼烷并锆杂环戊烷化合物与苯并锆杂环戊烷在反应性上有相似之处, 可以作为合成子用于合成系列碳硼烷并碳环和杂环衍生物.另一方面由于大位阻碳硼烷基团的存在, 8a也表现出一些自己独特的反应性质.
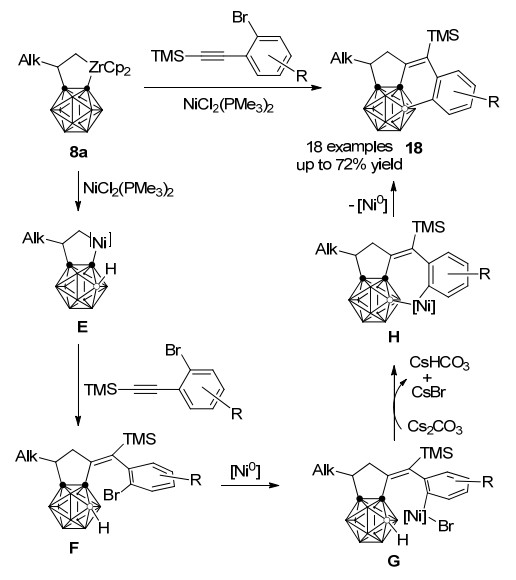
2014年, 谢作伟课题组[30]在之前的锆、镍共同介导碳硼炔与烯烃、三甲硅基芳基乙炔的一锅法[2+2+1]环加成反应基础上, 发现当炔烃的芳基取代基邻位连有溴取代基团时, 最终得到C, C, B-取代的碳硼烷并三环化合物.该反应的机理如Scheme 9所示, 碳硼烷并锆杂环戊烷发生系列金属交换和炔烃插入重排过程后得到[2+2+1]环加成中间体F.该过程中生成的Ni(0)与C(sp2)—Br键发生氧化加成得到中间体G, 并对B(7)—H或B(11)—H进行分子内亲电取代, 在碱的作用下消除一分子HBr得到中间体H, 再经还原消除生成最终产物18.提前淬灭反应并不能观察到体系中F的存在, 这表明整个串联反应过程中B—C(sp2)偶联步骤非常高效.这是首个通过金属催化B—H键活化实现的硼顶点芳基化过程.
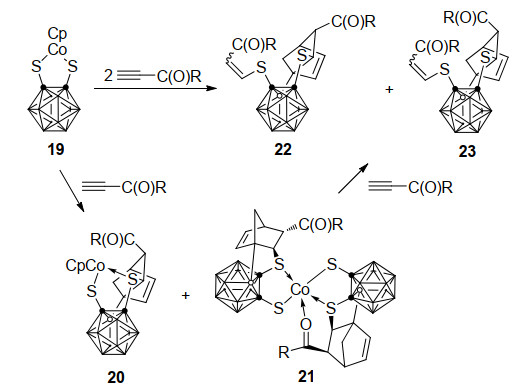
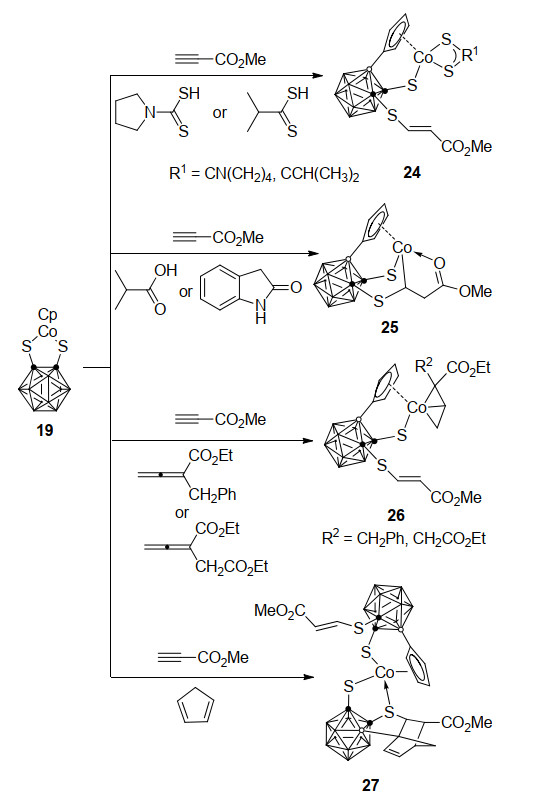
2013年, 燕红课题组[31]报道了一类钴介导的B—H键活化制备硼官能团化碳硼烷衍生物过程.该反应采用16电子的二硫代碳硼烷钴配合物19, 与炔烃反应形成两种B(3)或B(6)连有降冰片烯基的17电子钴配合物20和21 (Scheme 10).经DFT理论计算证明, 反应经过过渡金属引发的B—H键活化、B—Cp偶联、Cp迁移和Diels-Alder加成. 20和21可以与第二分子炔烃进一步发生Co—S键插入反应, 最终得到金属Co消除的烯基硫醚化合物22和23.特别注意的是, 19在温和条件下可以与过量的炔烃作用, 一锅法合成复杂的C, C, B-取代邻碳硼烷衍生物22和23.
16电子的二硫代碳硼烷钴配合物19也能与丙炔酸甲酯和另一3e-配体经三组分反应在室温下制备B-环戊二烯基偶联产物24~27 (Scheme 11)[32].作者通过氘代控制反应提出以下反应机理:活化炔烃首先插入其中一个Co—S键, 并引发B(3)—H键活化和对烯烃的氢转移过程; 在3e-配体的作用下, Cp基团的配位方式由η5转变为η1, 之后再经过α-H消除、H2释放、Co—B和Co—C键断裂、B—C键形成、Cp基团重新回到η5-配位的系列过程, 得到硼顶点被η5-Cp取代的最终产物.本方法为复杂硼官能团化碳硼烷衍生物的合成提供了一条简单、高效的合成路线.
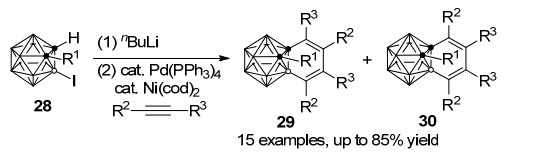
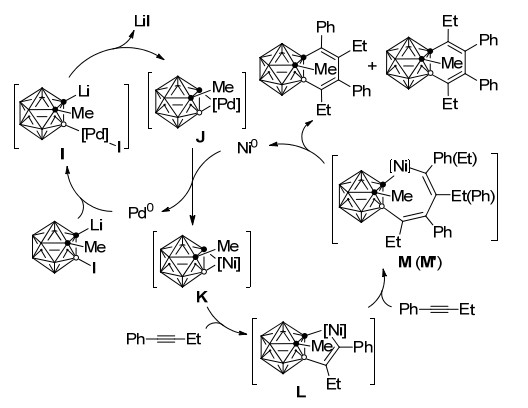
2010年, 谢作伟课题组[33]发展了钯、镍共同催化的1, 3-脱氢-邻-碳硼烷(C, B-碳硼炔)与炔烃的[2+2+2]环加成反应, 以最高可达85%的分离收率制备C, B-取代苯并-邻-碳硼烷衍生物29和30.该方法为碳硼烷的碳硼顶点同时官能团化提供了一种新的方法(Scheme 12).一个碳顶点连有取代基的3-碘代-邻-碳硼烷28在催化剂存在下不能与炔烃发生反应, 需要与正丁基锂作用生成1-锂-3-碘代-邻-碳硼烷后才能发生与炔烃的[2+2+2]环加成反应, 因而本反应是经由金属-C, B-碳硼炔中间体进行的.由于Ni(0)与B—I键很难发生插入反应, 因此1-锂-3-碘代-邻-碳硼烷前体的B—I键与Pd(0)发生氧化加成得到中间体I, 消除LiI形成Pd-1, 3-脱氢-邻-碳硼烷中间体J.由于双组分Pd/Ni催化剂体系在1, 3-脱氢-邻-碳硼烷与炔的反应中比单独的Pd催化剂更有效, 因此反应过程中可能存在Pd与Ni之间的转金属过程得到Ni-1, 3-脱氢-邻-碳硼烷中间体K.由于M—B键较M—C键亲核性更高, 炔烃首先插入M—B键得到镍杂环戊烯中间体L, 第二分子的炔烃以两种形式插入Ni—C键形成M (M'), 最后还原消除得到产物和Ni(0)完成催化循环(Scheme 13).
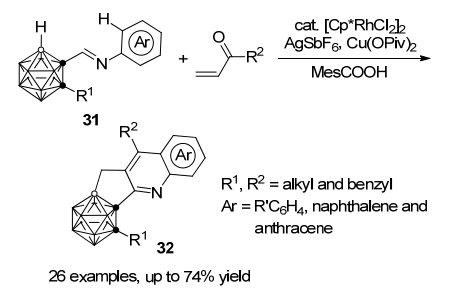
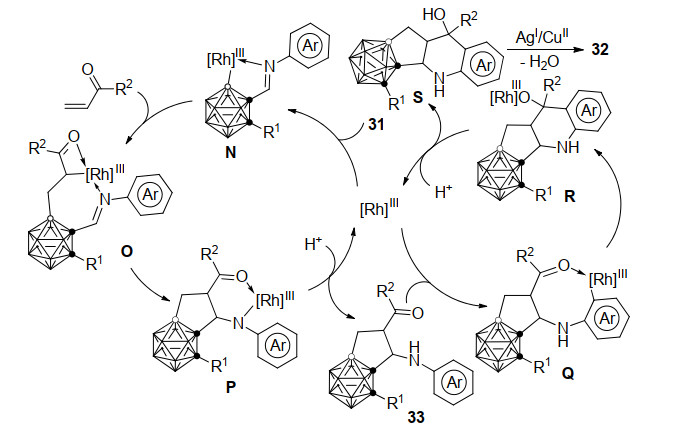
2018年, 谢作伟课题组[34]报道了铑催化的邻-碳硼烷基-N-芳基亚胺31与乙烯基酮的环化反应, 可以通过串联反应高效地合成氮杂多环并碳硼烷的衍生物32 (Scheme 14).该反应采用Cu(OPiv)2和AgSbF6作为添加剂, 同时在MesCOOH的存在下, 利用Rh(Ⅲ)邻碳硼烷B(4)—H键和亚胺的N-芳基C—H键依次活化, 最终以高达74%的收率得到目标产物.提前淬灭本反应可以分离到亚胺导向的烯烃与邻碳硼烷的B—H键活化-环化中间体33, 并经单晶X射线衍射鉴定了其结构.证明了碳硼烷B—H键的活化优先于芳基C—H键, 并通过芳基氘代底物反应的动力学同位素效应(KIE)值(kH/kD=1.7)确认C—H键活化并非本串联反应过程中的决速步.以此为基础, 作者提出了以下可能的反应机理(Scheme 15):底物31首先在亚胺导向基的作用下, Rh(Ⅲ)亲电进攻富电子的B(4/5)—H生成铑杂五元环中间体N, 烯烃插入B—Rh键得到中间体O, 其中C—Rh和C=N间的分子内亲核环化形成P, 随后经由质子化得到33并释放催化剂.接下来Rh(Ⅲ)催化芳基C—H键活化形成铑杂八元环中间体Q, 随后C—Rh和C=O的分子内亲核环化和质子化得到S并释放Rh催化剂, 最后S经脱水及氧化芳构化得到最终产物32.该过程中铑催化剂的价态始终保持不变.
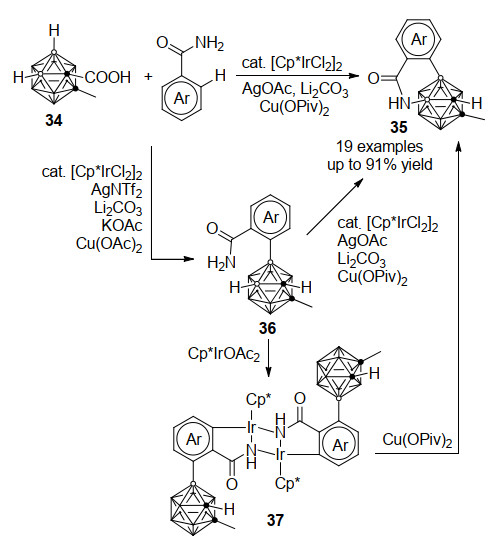
由于碳硼烷笼上十个BH的化学环境非常相似, 因此对两个硼顶点进行连续可控官能团化具有更高的挑战性. 2019年, 谢作伟课题组[35]报道了铱催化羧基碳硼烷34与苯甲酰胺连续脱氢偶联反应合成碳硼烷并异喹啉酮衍生物的方法(Scheme 16).该反应采用[Cp*IrCl2]2为催化剂, 在AgOAc、Li2CO3和Cu(OPiv)2存在下, 经过B—H/C—H和B—H/N—H的两步串联脱氢偶联反应, 高效制备碳硼烷并异喹啉酮衍生物35, 最高收率可达91%.此外在适合的反应条件下, 该连续脱氢偶联反应可以停留在第一个B—H/C—H交叉偶联步骤, 得到产物36.值得注意的是, 该反应中羧基导向基在B—C交叉偶联步骤中起关键作用, 并随后通过原位脱羧去除. 36直接与Cp*Ir(OAc)2作用可得到酰胺基导向的芳基C—H键活化关键中间体37, 其结构经单晶X射线衍射证实. 37可与Cu(Ⅱ)氧化剂反应得到进一步B—H/N—H脱氢偶联产物35, 控制实验和循环伏安实验结果证实, 本步反应中存在Ir(Ⅴ)-氮宾中间体, 通过37中的C—Ir键质子化和氮宾对B—H键的迁移插入最终构筑B—N键.该工作对区域选择性和化学选择性可控的连续脱氢偶联反应具有重要的参考价值.
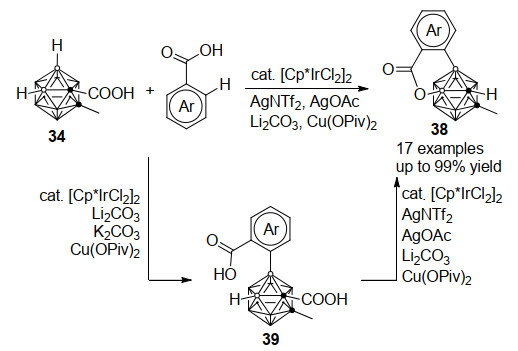
羧基取代碳硼烷34同样也能与苯甲酸经过铱催化B—H/C—H和B—H/O—H的串联脱氢偶联反应, 高效制备碳硼烷并香豆素衍生物38 (Scheme 17)[36].改变反应条件同样可得到单一B—H/C—H交叉偶联产物39.而与Scheme 16中苯甲酰胺反应不同的是, 苯甲酸与碳硼烷的第一步B—H/C—H脱氢偶联完成后, 碳硼烷C(1)位的羧基导向基并不会发生原位脱除, 而是进一步导向后续的B—H键活化, 再经由氧化所得的Ir(Ⅴ)-B物种与分子内苯甲酸羧基作用最终构建B—O键.该反应中C(1)羧基导向基以及氧化剂的存在是连续脱氢偶联的关键, 羧基导向基在反应的最终步才进行脱除.
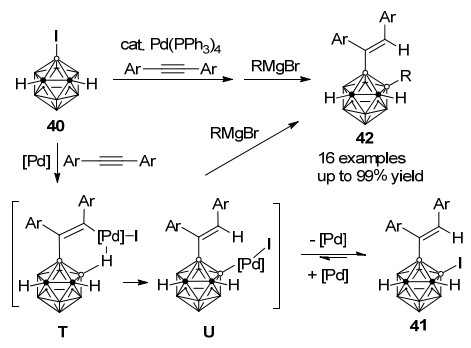
与Scheme 12中所示的一个碳顶点连有取代基的3-碘代-邻-碳硼烷28的反应不同, 炔烃可以直接插入3-碘代-邻-碳硼烷40与Pd(PPh3)4催化剂作用所得的B—Pd键, 而反应最终得到碘迁移至B(4)位产物41. DFT理论计算结果表明钯片段由exo-烯基碳向B(4)顶点迁移的过程(T→U)在热力学上十分有利(ΔG=-88.7 kJ/mol).而基于已知的碘代-邻-碳硼烷钯催化B—C偶联过程[37], 利用上述烯基化-碘迁移串联反应中回收的Pd(0)催化剂进一步活化新生成B(4)—I键, 通过向体系中加入不同的格氏试剂可以一锅法高效合成双官能团化邻-碳硼烷42 (Scheme 18).结果表明, 一系列烷基/烯丙基/炔基/芳基格氏试剂都能应用于此反应, 并以优秀的收率得到相应产物[38].
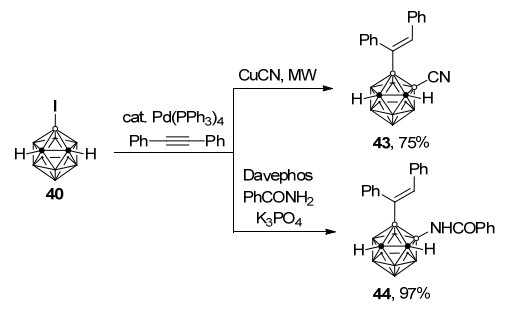
与此同时, 在碘迁移步骤后采用不同的偶联试剂也可以分别实现一锅法钯催化烯基化-氰基化/胺基化过程, 以较高至优秀的收率得到3-烯基-4-氰基/酰胺基-邻-碳硼烷43和44 (Scheme 19)[38].氘代实验表明, B(4)顶点的氘原子在反应后能以94%的效率迁移到烯基上, 此结果为反应中的钯迁移关键步骤提供了确凿的证据.该工作中高区域选择性双官能团化过程包含了对B—I、B(4)—H键的相继活化和由烯基碳至B(4)顶点钯迁移关键步骤, 成功发展了一锅法在碳硼烷硼顶点上引入两种不同取代基的新路径.
闭式十二硼烷阴离子[B12H12]2-同样具有二十面体结构, 分子中十二个BH顶点完全等价, 是苯的三维类似物.它具有电子离域、高化学及热稳定性、低毒性的特点, 在弱配位阴离子、配体、主-客体化学、催化、材料及医药领域用作重要的结构单元[39].通过各种官能团对其进行修饰, 可以为闭式十二硼烷的性质和应用研究提供一个新的平台. 2016年, Duttwyler课题组[40]发展了过渡金属催化十二硼烷阴离子区域选择性B—H键活化, 实现复杂的1, 2, 3-三取代(B—C/B—O/B—N)硼烷衍生物46的简单构筑(Scheme 20).作者首先合成了含有脲基导向基的十二硼烷阴离子45, 以Cu(OAc)2• H2O为氧化剂, 在[Cp*RhCl2]2催化下与炔烃发生烯基化-分子内环化串联反应, 以最高85%收率得到三取代的产物46.值得注意的是, 此反应条件温和, 在室温条件下即具有良好的反应普适性以及较好的区域选择性.
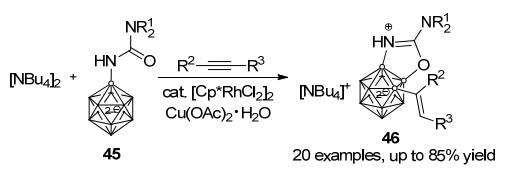
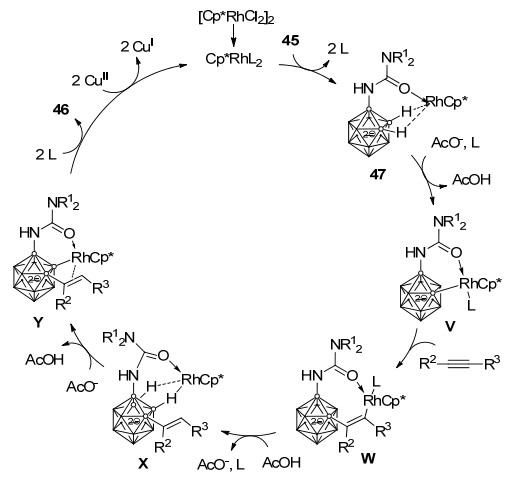
化合物45与化学计量的[RhCp*(CH3CN)3][SbF6]2反应, 可以高效生成中性含有抓氢键(agostic interaction)的Rh(Ⅲ)配合物47, 其结构经单晶X射线衍射证实.进一步通过控制实验对该反应的机理进行研究显示, 催化循环中首先[Cp*RhCl2]2催化剂解离为更活跃的单体CpRh*L2 (L=溶剂、Cl-或AcO-), 然后与硼烷阴离子45络合形成中间体47, 随后在AcO-作用下发生B—H键活化形成铑杂六元环中间体V, 反应物炔烃配位、插入得到中间体W, 随后质子化及金属Rh迁移形成中间体X. AcO-可以进一步促进第二次B—H键活化形成中间体Y, 最后通过还原消除得到产物46, Cu(Ⅱ)可以将Rh(I)再次氧化为Rh(Ⅲ)完成一次催化循环(Scheme 21)[40].
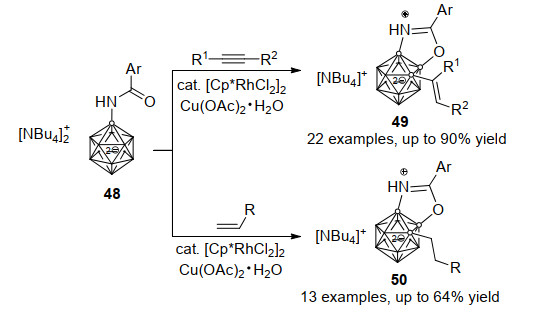
芳基酰胺作为导向基团同样可以促进闭式十二硼烷阴离子48与炔烃反应进行两个硼顶点的串联官能团化反应, 该反应官能团耐受性广, 最高能以90%的收率得到三取代的硼烷衍生物49. 48与芳基及烷基取代烯烃反应, 也可以得到类似的烷基化-环化产物50 (Scheme 22)[41].值得注意的是, 反应中并未观察到导向基中芳基C—H键活化的产物, 证明本反应体系对B—H和C—H具有很好的化学选择性, 而反应的第二步环化过程也具有完全的区域选择性.所合成的复杂十二硼烷阴离子衍生物将可以拓展硼笼阴离子在发光材料和药物化学领域应用研究.
本文综述了过渡金属促进的十二顶点碳硼烷与硼烷阴离子的复杂官能团化过程, 反应中涉及到多组分反应、串联反应或一锅法反应的系列过程, 具有高选择性、操作步骤简单、高效实用的优点, 为复杂硼烷衍生物的合成提供了新的方法和途径.目前, 过渡金属介导/催化的硼烷官能团化反应是合成复杂硼烷衍生物的重要方法之一, 尽管近年来过渡金属催化的B—H键活化研究成果显著, 但过渡金属催化的反应范围和机理仍有待进一步探索.另一方面, 这些高效B—H键活化反应可以很好地控制串联反应中的区域选择性, 而如何在系列串联反应中控制对映选择性而实现复杂手性硼烷化合物的不对称合成是今后该领域的一个重要研究方向[42], 具有广阔的发展空间, 并能进一步开拓官能团化碳硼烷及硼烷衍生物在医药、材料、催化等多个领域的应用前景.
(a) Grimes, R. N. Carboranes, 3rd ed., Academic Press, Amsterdam, 2016.
(b) Hosmane, N. S. Boron Science: New Technologies and Application, CRC Press, Boca Raton, FL, 2012.
(a) Scholz, M.; Hey-Hawkins, E. Chem. Rev. 2011, 111, 7035.
(b) Leśnikowski, Z. J. J. Med. Chem. 2016, 59, 7738.
(a) Hosmane, N. S.; Maguire, J. A. In Comprehensive Organometallic Chemistry Ⅲ, Vol. 3, Eds.: Crabtree, R. H.; Mingos, D. M. P., Elsevier, Oxford, 2007, Chapter 5.
(b) Xie, Z. Acc. Chem. Res. 2003, 36, 1.
(c) Yao, Z.-J.; Jin, G.-X. Coord. Chem. Rev. 2013, 257, 2522.
(d) Qiu, Z.; Ren, S.; Xie, Z. Acc. Chem. Res. 2011, 44, 299.
(e) Estrada, J.; Lavallo, V. Angew. Chem. Int. Ed. 2017, 56, 9906.
(f) El-Hellani, A.; Lavallo, V. Angew. Chem. Int. Ed. 2014, 53, 4489.
(g) Fisher, S. P.; Tomich, A. W.; Lovera, S. O.; Kleinsasser, J. F.; Guo, J.; Asay, M. J.; Nelson, H. M.; Lavallo, V. Chem. Rev. 2019, 119, 8262.
(h) Yao, Z.-J.; Yu, W.-B.; Lin, Y.-J.; Huang, S.-L.; Li, Z.-H.; Jin, G.-X. J. Am. Chem. Soc. 2014, 136, 2825.
(i) Gao, Y.; Guo, S.-T.; Cui, P.-F.; Aznarez, F.; Jin, G.-X. Chem. Commun. 2019, 55, 210.
(j) Cui, P.-F.; Gao, Y.; Guo, S.-T.; Lin, Y.-J.; Li, Z.-H.; Jin, G.-X. Angew. Chem. Int. Ed. 2019, 58, 8129.
(a) Jude, H.; Disteldorf, H.; Fischer, S.; Wedge, T.; Hawkridge, A. M.; Arif, A. M.; Hawthorne, M. F.; Muddiman, D. C.; Stang, P. J. J. Am. Chem. Soc. 2005, 127, 12131.
(b) Koshino, M.; Tanaka, T.; Solin, N.; Suenaga, K.; Isobe, H.; Nakamura, E. Science 2007, 316, 853.
(c) Dash, B. P.; Satapathy, R.; Gaillard, E. R.; Maguire, J. A.; Hosmane, N. S. J. Am. Chem. Soc. 2010, 132, 6578.
(d) Kung, C.-W.; Otake, K.; Buru, C. T.; Goswami, S.; Cui, Y.; Hupp, J. T.; Spokoyny, A. M.; Farha, O. K. J. Am. Chem. Soc. 2018, 140, 3871.
(e) Villagómez, C. J.; Sasaki, T.; Tour, J. M.; Grill, L. J. Am. Chem. Soc. 2010, 132, 16848.
(f) Qian, E. Q.; Wixtrom, A. I.; Axtell, J. C.; Saebi, A.; Rehak, P.; Han, Y.; Moully, E. H.; Mosallaei, D.; Chow, S.; Messina, M.; Wang, J.-Y.; Royappa, A. T.; Rheingold, A. L.; Maynard, H. D.; Kral, P.; Spokoyny, A. M. Nat. Chem. 2017, 9, 333.
(g) Saha, A.; Oleshkevich, E.; Viñas, C.; Teixidor, F. Adv. Mater. 2017, 29, 1704238.
(h) Guo, J.; Liu, D.; Zhang, J.; Zhang, J.; Miao, Q.; Xie, Z. Chem. Commun. 2015, 51, 12004.
(i) Jung, D.; Saleh, L. M. A.; Berkson, Z. J.; El-Kady, M. F.; Hwang, J. Y.; Mohamed, N.; Wixtrom, A. I.; Titarenko, E.; Shao, Y.; McCarthy, K.; Guo, J.; Martini, I. B.; Kraemer, S.; Wegener, E. C.; Saint-Cricq, P.; Ruehle, B.; Langeslay, R. R.; Delferro, M.; Brosmer, J. L.; Hendon, C. H.; Gallagher-Jones, M.; Rodriguez, J.; Chapman, K. W.; Miller, J. T.; Duan, X.; Kaner, R. B.; Zink, J. I.; Chmelka, B. F.; Spokoyny, A. M. Nat. Mater. 2018, 17, 341.
(j) Cui, P.-F.; Lin, Y.-J.; Li, Z.-H.; Jin, G.-X. J. Am. Chem. Soc. 2020, 142, 8532.
(a) Mukherjee, S.; Thilagar, P. Chem. Commun. 2016, 52, 1070.
(b) Núñez, R.; Tarrés, M.; Ferrer-Ugalde, A.; de Biani, F. F.; Teixidor, F. Chem. Rev. 2016, 116, 14307.
(c) Li, X.; Yan, H.; Zhao, Q. Chem. Eur. J. 2016, 22, 1888.
(a) Heying, T. L.; Ager, J. W.; Clark, S. L.; Mangold, D. J.; Goldstein, H. L.; Hillman, M.; Polak, R. J.; Szymanski, J. W. Inorg. Chem. 1963, 2, 1089.
(b) Fein, M. M.; Bobinski, J.; Mayes, N.; Schwartz, N.; Cohen, M. S. Inorg. Chem. 1963, 2, 1111.
(c) El-Zaria, M. E., Keskar, K., Genady, A. R., Ioppolo, J. A., McNulty, J.; Valliant, J. F. Angew. Chem. Int. Ed. 2014, 53, 5156.
(a) Wu, S.; Jones, M., Jr. Inorg. Chem. 1986, 25, 4802.
(b) Bregadze, V. I. Chem. Rev. 1992, 92, 209.
(c) Viñas, C.; Benakki, R.; Teixidor, F.; Casabó, J. Inorg. Chem. 1995, 34, 3844.
(d) Gomez, F. A.; Johnson, S. E.; Hawthorne, M. F. J. Am. Chem. Soc. 1991, 111, 5915.
(e) Gomez, F. A.; Hawthorne, M. F. J. Org. Chem. 1992, 57, 1384.
(f) Anderson, K. P.; Mills, H. A.; Mao, C.; Kirlikovali, K. O.; Axtell, J. C.; Rheingold, A. L.; Spokoyny, A. M. Tetrahedron 2019, 75, 187.
(a) Lu, J.-Y.; Wan, H.; Zhang, J.; Wang, Z.; Li, Y.; Du, Y.; Li, C.; Liu, Z.-T.; Liu, Z.-W.; Lu, J. Chem.-Eur. J. 2016, 22, 17542.
(b) Tang, C.; Xie, Z. Angew. Chem. Int. Ed. 2015, 54, 7662.
(c) Xie, Z. Sci. China Chem. 2014, 57, 1061.
(d) Coult, R.; Fox, M. A.; Gill, W. R.; Herbertson, P. L.; MacBride, J. A. H.; Wade, K. J. Organomet. Chem. 1993, 462, 19.
(a) Tang, C.; Zhang, J.; Xie, Z. Angew. Chem. Int. Ed. 2017, 56, 8642.
(b) Tang, C.; Zhang, J.; Zhang, J.; Xie, Z. J. Am. Chem. Soc. 2018, 140, 16423.
Lyu, H.; Zhang, J.; Yang, J.; Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2019, 141, 4219.
(b) Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2015, 137, 3502.
(c) Quan, Y.; Xie, Z. Angew. Chem. Int. Ed. 2016, 55, 1295.
(d) Zhang, X.; Yan, H. Chem. Sci. 2018, 9, 3964.
(e) Zhang, X.; Zheng, H.; Li, J.; Xu, F.; Zhao, J.; Yan, H. J. Am. Chem. Soc. 2017, 139, 14511.
(f) Cao, K.; Huang, Y.; Yang, J.; Wu, J. Chem. Commun. 2015, 51, 7257.
(g) Lin, F.; Yu, J.-L.; Shen, Y.; Zhang, S.-Q.; Spingler, B.; Liu, J.; Hong, X.; Duttwyler, S. J. Am. Chem. Soc. 2018, 140, 13798.
(h) Xu, T. T.; Cao, K.; Zhang, C. Y.; Wu, J.; Ding, L. F.; Yang, J. Org. Lett. 2019, 21, 9276.
(i) Xu, T. T.; Cao, K.; Zhang, C. Y.; Wu, J.; Jiang, L.; Yang, J. Chem. Commun. 2018, 54, 13603.
(a) Liang, X.; Shen, Y.; Zhang, K.; Liu, J.; Duttwyler, S. Chem. Commun. 2018, 54, 12451.
(b) Lin, F.; Shen, Y.; Zhang, Y.; Sun, Y.; Liu, J.; Duttwyler, S. Chem. Eur. J. 2018, 24, 551.
(c) Lyu, H.; Quan, Y.; Xie, Z. Angew. Chem. Int. Ed. 2015, 54, 10623.
(d) Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2014, 136, 15513.
(e) Mirabelli, M. G. L.; Sneddon, L. G. J. Am. Chem. Soc. 1988, 110, 449.
(f) Wu, J.; Cao, K.; Xu, T.-T.; Zhang, X.-J.; Jiang, L.; Yang, J.; Huang, Y., RSC Adv. 2015, 5, 91683.
(g) Shen, Y.; Pan, Y.; Zhang, K.; Liang, X.; Liu, J.; Spingler, B.; Duttwyler, S. Dalton Trans. 2017, 46, 3135.
(h) Shen, Y.; Zhang, K.; Liang, X.; Dontha, R.; Duttwyler, S. Chem. Sci. 2019, 10, 4177.
(i) Cheng, R.; Qiu, Z.; Xie, Z. Chem. Eur. J. 2020, 26, 7121.
(a) Chen, Y.; Au, Y. K.; Quan, Y.; Xie, Z. Sci. China Chem. 2019, 62, 74.
(b) Quan, Y.; Tang, C.; Xie, Z. Chem. Sci. 2016, 7, 5838.
Cheng, R.; Qiu, Z.; Xie, Z. Nat. Commun. 2017, 8, 14827. doi: 10.1038/ncomms14827
Qiu, Z.; Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2013, 135, 12192. doi: 10.1021/ja405808t
(a) Cui, C. X.; Zhang, J.; Qiu, Z.; Xie, Z. Dalton Trans. 2020, 49, 1380.
(b) Au, Y. K.; Lyu, H.; Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2020, 142, 6940.
(c) Lyu, H.; Quan, Y.; Xie, Z. Angew. Chem. Int. Ed. 2016, 55, 11840.
(d) Cao, K.; Xu, T.-T.; Wu, J.; Jiang, L.; Yang, J. Chem. Commun. 2016, 52, 11446.
(e) Li, C.-X.; Zhang, H.-Y.; Wong, T.-Y.; Cao, H.-J.; Yan, H.; Lu, C.-S. Org. Lett. 2017, 19, 5178.
Lyu, H.; Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2016, 138, 12727. doi: 10.1021/jacs.6b07086
Lyu, H.; Quan, Y.; Xie, Z. Chem. Eur. J. 2017, 23, 14866. doi: 10.1002/chem.201703006
(a) Quan, Y.; Qiu, Z.; Xie, Z. Chem. Eur. J. 2018, 24, 2795.
(b) Quan, Y.; Xie, Z. Chem. Soc. Rev. 2019, 48, 3660.
(c) Yu, W.-B.; Cui, P.-F.; Gao, W.-X.; Jin, G.-X. Coord. Chem. Rev. 2017, 350, 300.
(d) Zhang, X.; Yan, H. Coord. Chem. Rev. 2019, 378, 466.
Qiu, Z.; Ren, S.; Xie, Z. Acc. Chem. Res. 2011, 44, 299. doi: 10.1021/ar100156f
Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2009, 131, 2084. doi: 10.1021/ja809389k
Deng, L.; Chan, H.-S.; Xie, Z. J. Am. Chem. Soc. 2006, 128, 7728. doi: 10.1021/ja061605j
Ren, S.; Chan, H.-S.; Xie, Z. Organometallics 2009, 28, 4106. doi: 10.1021/om9002973
Ren, S.; Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2012, 134, 3242. doi: 10.1021/ja211485t
Ren, S.; Chan, H.-S.; Xie, Z. J. Am. Chem. Soc. 2009, 131, 3862. doi: 10.1021/ja900563u
Ren, S.; Qiu, Z.; Xie, Z. Organometallics 2012, 31, 4435. doi: 10.1021/om300202p
Ren, S.; Qiu, Z.; Xie, Z. Angew. Chem. Int. Ed. 2012, 51, 1010. doi: 10.1002/anie.201106212
Quan, Y.; Zhang, J.; Xie, Z. J. Am. Chem. Soc. 2013, 135, 18742. doi: 10.1021/ja410233e
Zhang, J.; Quan, Y.; Lin, Z.; Xie, Z. Organometallics 2014, 33, 3556. doi: 10.1021/om5004545
Cui, C.-X.; Ren, S.; Qiu, Z.; Xie, Z. Dalton Trans. 2018, 47, 2453. doi: 10.1039/C7DT04751G
Quan, Y.; Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2014, 136, 7599. doi: 10.1021/ja503489b
Wang, Z.; Ye, H.; Li, Y.; Li, Y.; Yan, H. J. Am. Chem. Soc. 2013, 135, 11289. doi: 10.1021/ja4047075
Zhang, R.; Zhu, L.; Liu, G.; Dai, H.; Lu, Z.; Zhao, J.; Yan, H. J. Am. Chem. Soc. 2012, 134, 10341. doi: 10.1021/ja303334t
Qiu, Z.; Xie, Z. J. Am. Chem. Soc. 2010, 132, 16085. doi: 10.1021/ja1058789
Lyu, H. Quan, Y.; Xie, Z. Chem. Sci. 2018, 9, 6390.
Au, Y.-K.; Lyu, H.; Quan, Y.; Xie, Z. J. Am. Chem. Soc. 2019, 141, 12855. doi: 10.1021/jacs.9b06204
Au, Y.-K.; Lyu, H.; Quan, Y.; Xie, Z. Chin. J. Chem. 2020, 38, 383. doi: 10.1002/cjoc.201900475
(a) Li, J.; Logan, C. F.; Jones, M. Jr. Inorg. Chem. 1991, 30, 4866.
(b) Zheng, Z.; Jiang, W.; Zinn, A. A.; Knobler, C. B.; Hawthorne, M. F. Inorg. Chem. 1995, 34, 2095.
(c) Harakas, G.; Vu, T.; Knobler, C. B.; Hawthorne, M. F. J. Am. Chem. Soc. 1998, 120, 6405.
(d) Viñas, C.; Barberà, G.; Oliva, J. M.; Teixidor, F.; Welch, A. J.; Rosair, G. M. Inorg. Chem. 2001, 40, 6555.
Ge, Y.; Zhang, J.; Qiu, Z.; Xie, Z. Angew. Chem. Int. Ed. 2020, 59, 4851. doi: 10.1002/anie.201914500
(a) Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. Chem. Eur. J. 2016, 22, 7437.
(b) Axtell, J. C.; Saleh, L. M. A.; Qian, E. A.; Wixtrom, A. I.; Spokoyny, A. M. Inorg. Chem. 2018, 57, 2333.
(c) Bolli, C.; Derendorf, J.; Keßler, M.; Knapp, C.; Scherer, H.; Schulz, C.; Warneke, J. Angew. Chem. Int. Ed. 2010, 49, 3536.
(d) Kirchmann, M.; Wesemann, L. Dalton Trans. 2008, 2144.
(e) Messina, M. S.; Axtell, J. C.; Wang, Y.; Chong, P.; Wixtrom, A. I.; Kirlikovali, K. O.; Upton, B. M.; Hunter, B. M.; Shafaat, O. S.; Khan, S. I.; Winkler, J. R.; Gray, H. B.; Alexandrova, A. N.; Maynard, H. D.; Spokoyny, A. M. J. Am. Chem. Soc. 2016, 138, 6952.
(f) Peryshkov, D. V.; Strauss, S. H. Inorg. Chem. 2017, 56, 4072.
(g) Wang, W.; Wang, X.; Cao, J.; Liu, J.; Qi, B.; Zhou, X.; Zhang, S.; Gabel, D.; Nau, W. M.; Assaf, K. I.; Zhang, H. Chem. Commun. 2018, 54, 2098.
(h) Assaf, K. I.; Ural, M. S.; Pan, F.; Georgiev, T.; Simova, S.; Rissanen, K.; Gabel, D.; Nau, W. M. Angew. Chem. Int. Ed. 2015, 54, 6852.
(i) Assaf, K. I.; Hennig, A.; Peng, S.; Guo, D.-S.; Gabel, D.; Nau, W. M. Chem. Commun. 2017, 53, 4616.
(j) Jung, D.; Saleh, L. M. A.; Berkson, Z. J.; El-Kady, M. F.; Hwang, J. Y.; Mohamed, N.; Wixtrom, A. I.; Titarenko, E.; Shao, Y.; McCarthy, K.; Guo, J.; Martini, I. B.; Kraemer, S.; Wegener, E. C.; Saint-Cricq, P.; Ruehle, B.; Langeslay, R. R.; Delferro, M.; Brosmer, J. L.; Hendon, C. H.; Gallagher-Jones, M.; Rodriguez, J.; Chapman, K. W.; Miller, J. T.; Duan, X.; Kaner, R. B.; Zink, J. I.; Chmelka, B. F.; Spokoyny, A. M. Nat. Mater. 2018, 17, 341.
Zhang, Y.; Sun, Y.; Lin, F.; Liu, J.; Duttwyler, S. Angew. Chem. Int. Ed. 2016, 55, 15609. doi: 10.1002/anie.201607867
Zhang, Y.; Wang, T.; Wang, L.; Sun, Y.; Lin, F.; Liu, J.; Duttwyler, S. Chem. Eur. J. 2018, 24, 15812. doi: 10.1002/chem.201803455
Cheng, R.; Li, B.; Wu, J.; Zhang, J.; Qiu, Z.; Tang, W.; You, S. L.; Tang, Y.; Xie, Z. J. Am. Chem. Soc. 2018, 140, 4508. doi: 10.1021/jacs.8b01754

图式 1 镍-介导碳硼炔、烯烃、炔烃的三组分环加成反应
Scheme 1 Nickel-mediated three-component cycloaddition reaction of carboryne, alkenes and alkynes

图式 2 锆、镍共同介导的碳硼炔与炔烃的[2+2+2]环加成反应
Scheme 2 Zirconium/nickel-co-mediated [2+2+2] cycloaddition reaction of carboryne and alkynes

图式 4 碳硼烷并锆杂环戊烯中间体与异腈及邻二碘苯的反应
Scheme 4 Reactions of carboranozirconacyclopentene intermediate with isocyanide and o-diiodobenzene

图式 5 锆、镍共同介导的碳硼炔与烯烃、炔烃的一锅法[2+2+2]环加成反应
Scheme 5 Zirconium/nickel-co-mediated one-pot [2+2+2] cycloaddition reaction of carboryne, alkenes and alkynes

图式 7 锆、镍共同介导的碳硼炔与烯烃、三甲硅基炔烃的一锅法[2+2+1]环加成反应
Scheme 7 Zirconium/nickel-co-mediated one-pot [2+2+1] cycloaddition reaction of carboryne, alkenes and trimethylsilylalkynes

图式 8 通过碳硼烷并锆杂环戊烷对系列碳硼烷并碳环和杂环衍生物的合成
Scheme 8 Synthesis of carborane-fused carbo- and heterocycles via zirconacyclopentane intermediate

图式 9 过渡金属介导[2+2+1]环加成与B—H键芳基化串联环化反应
Scheme 9 Transition-metal-co-mediated [2+2+1] cyclization and B—H arylation cascade cyclization

图式 10 通过B—H键活化合成B-冰片烯基取代邻-碳硼烷
Scheme 10 Synthesis of B-norbornyl o-carboranes via B—H activation

图式 11 钴介导邻-碳硼烷B—H键活化与环戊二烯基C—H键活化参与的三组分反应
Scheme 11 Co-mediated three-component reactions incorporating with o-carborane B—H activation and cyclopentadiene C—H activation

图式 12 钯、镍-共同催化的1, 3-脱氢-邻-碳硼烷与炔烃环加成反应
Scheme 12 Pd/Ni-cocatalyzed cycloaddition of 1, 3-dehydro-o- carborane with alkynes

图式 13 钯、镍-共同催化[2+2+2]环加成反应机理
Scheme 13 Proposed mechanism for Pd/Ni-cocatalyzed [2+ 2+2] cycloaddition

图式 14 铑催化B—H和C—H键串联活化合成氮杂多环并碳硼烷
Scheme 14 Synthesis of carborane-fused N-polyheterocycles by rhodium catalyzed cascade B—H and C—H activation

图式 15 铑催化B—H和C—H键串联活化环化机理
Scheme 15 Proposed mechanism for rhodium catalyzed cascade B—H/C—H activation and cyclization

图式 16 铱催化BH/CH和BH/NH串联脱氢交叉偶联合成碳硼烷并异喹啉酮
Scheme 16 Synthesis of carborano-isoquinolinones via iridium catalyzed cascade dehydrogenative cross-coupling of BH/CH and BH/NH

图式 17 铱催化BH/CH和BH/OH串联脱氢交叉偶联合成碳硼烷并香豆素
Scheme 17 Synthesis of carborano-coumarins via iridium catalyzed cascade dehydrogenative cross-coupling of BH/CH and BH/OH

图式 18 钯迁移实现3-碘代-邻-碳硼烷选择性一锅法双官能团化
Scheme 18 Selective one-pot bifunctionalization of 3-iodo-o- carborane by Pd migration

图式 19 钯催化一锅法烯基化-氰基化/酰胺基化过程
Scheme 19 Pd-catalyzed one-pot alkenylation-cyanation/ami- nation

图式 20 铑催化闭式十二硼烷阴离子经烯基化-环化串联反应合成复杂三取代硼烷衍生物
Scheme 20 Synthesis of trisubstituted closo-dodecaborates by Rh-catalyzed cascade alkenylation-annulation of closo-dodeca- borate anions

图式 21 铑催化双重B—H键活化及烯基化-环化串联反应机理
Scheme 21 Proposed mechanism for rhodium catalyzed cascade alkenylation-annulation through double B—H activation

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:


