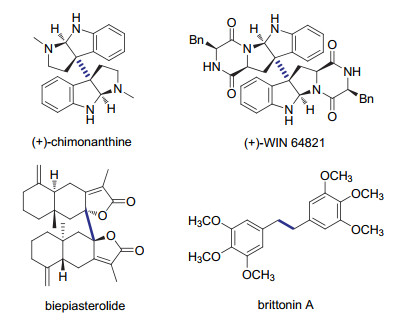
图 1.
具有对称结构的生物活性分子
Figure 1.
Representative examples of symmetric bioactive molecules

形成碳碳键的偶联反应在有机合成研究中具有重要地位. 2010年诺贝尔化学奖被授予给理查德-赫克、根岸英一和铃木彰, 以表彰他们在偶联反应中的贡献.目前, 形成C(sp2)—C(sp2)键的偶联反应已获得广泛关注和深入探索[1].与之相对应的过渡金属催化形成C(sp3)—C(sp3)键的反应则更具挑战.其主要原因包括: (1)过渡金属与烷基卤化物发生氧化加成的速率缓慢; (2)发生氧化加成后形成的烷基-金属络合物在发生转金属步骤前可能会先发生β-氢消除得到副产物.目前已有很多工作针对交叉偶联中的该类问题做出改进, 得到了较好的结果.除了交叉偶联反应[2-6], 形成C(sp3)—C(sp3)的自偶联反应同样有重要的应用.例如很多天然产物和生物活性分子中经常存在以C(sp3)—C(sp3)为中心的对称结构(图 1), 利用自偶联反应不仅可以极大地简化合成步骤, 而且可能同时实现对两个手性中心的精确控制.正因为这些原因, 条件温和、适用性广且具有立体选择性的C(sp3)—C(sp3)自偶联受到越来越多的关注[7-9].
目前氧化偶联[10]、类Wittig反应[11]、Suzuki-Miyaura偶联[12]、还原偶联[13]及电化学方法[14]均已被用于形成C(sp3)—C(sp3)键的自偶联反应中.其中, 还原偶联反应通常以易得的有机卤化物为底物, 应用较为广泛.由于结构复杂的烷基卤代物可以方便地制得, 还原偶联反应已经被用于复杂分子的全合成中.金属参与的还原偶联反应最早可以追溯到Wurtz反应, 该反应利用单质钠促进烷基卤代物的二聚.在后续发展的改良型Wurtz反应中, 烷基卤代物在金属单质、格氏试剂或金属锂试剂的存在下现场转化为相应的烷基金属物种.其与体系中的另一分子烷基卤代物发生亲核取代反应, 即可制得还原偶联产物[15-18].由于高活性金属物种的使用, 该方法通常适用于不含活性官能团的简单小分子的二聚反应.过渡金属催化的偶联反应是实现惰性卤代物还原二聚反应的有效方法.尽管可能存在氧化加成速率缓慢, 加成后易发生β-氢消除等不利因素, 通过对过渡金属及配体的调节, 目前人们已经发展了多种高效的还原自偶联反应体系.在一些反应中, C(sp3)—C(sp3)键形成于高价过渡金属物种的还原消除过程, 给控制偶联过程的立体选择性带来机遇.由于这些过渡金属参与及催化的反应过程具有优良的官能团兼容性, 一些方法已经被应用于天然产物的全合成中以快速构建复杂的对称结构[15-18].近年来, 其他相关领域的研究成果也被应用于过渡金属参与的还原偶联反应, 为该研究领域进一步发展注入了新的活力.例如, 基于光氧化还原催化、离子液体及无机纳米材料的反应体系, 进一步提升了还原偶联反应的效率及选择性.在此背景下, 本综述首先按照过渡金属分类, 系统总结钴、镍、铜、铑、钛等金属参与或催化的还原偶联反应.之后, 探讨近来发展的光介导还原偶联反应体系, 并对还原偶联反应在天然分子及高分子合成方面的应用进行详细介绍.
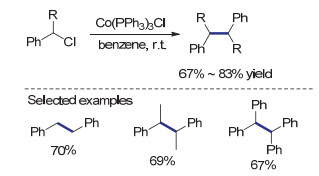
可参与C(sp3)—C(sp3)还原自偶联反应的底物主要为苄基及烯丙基卤代物.当经还原形成自由基物种时, 苄基及烯丙基的共轭体系可有效稳定有效稳定自由基从而减少因“自由基-溶剂分子”作用而产生的副反应, 使偶联反应可以稳定、高效地进行[19].利用低价态金属作为单电子还原剂, 攫取卤代烃中的卤原子是形成自由基中间体的有效手段. 1981年, Yamada等[13]报道了使用Co(PPh3)3Cl作为还原剂, 以苄基氯为底物的还原偶联反应.反应过程中, 低价的钴物种攫取苄基氯的氯原子形成苄基自由基, 苄基自由基重组即可形成目标二聚产物(Scheme 1).该反应温和快速, 在室温条件下反应1.5 h即可以70%的产率制得还原自偶联产物1, 2-二苯乙烷. Co(PPh3)3Cl制备简单[20], 所介导的还原偶联反应条件温和且官能团兼容性好, 因而在天然产物全合成中得到广泛应用.
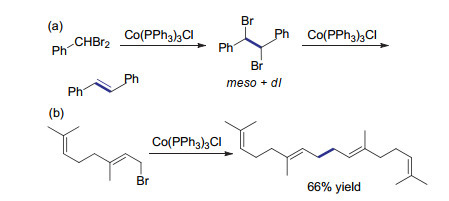
1984年, Yamada课题组[21]进一步将该方法拓展至烯丙基及苄基溴代物的还原偶联反应.由于使用了反应活性更高的溴代物作为底物, 反应时间由原来的1.5 h缩短至15 min而不影响反应的转化率.当使用二溴及三溴甲基苯作为底物与足量Co(PPh3)3Cl反应, 可分别获得反式二苯基乙烯和二苯基乙炔.作者在机理研究的过程中发现, 二溴甲基苯的还原偶联产物在双键形成前, 首先得到meso与dl两种非对映异构的邻二溴代物.在进一步还原形成二苯基乙烯的过程中, 由于meso型的自由基比dl型更稳定, 因此只得到反式产物(Scheme 2a).值得指出的是, 当烯丙基溴代物为底物时, 双键的构型在反应后得到很好的保持, 因此该工作提供了一种合成双键构型确定的非共轭多烯的有效方法(Scheme 2b).
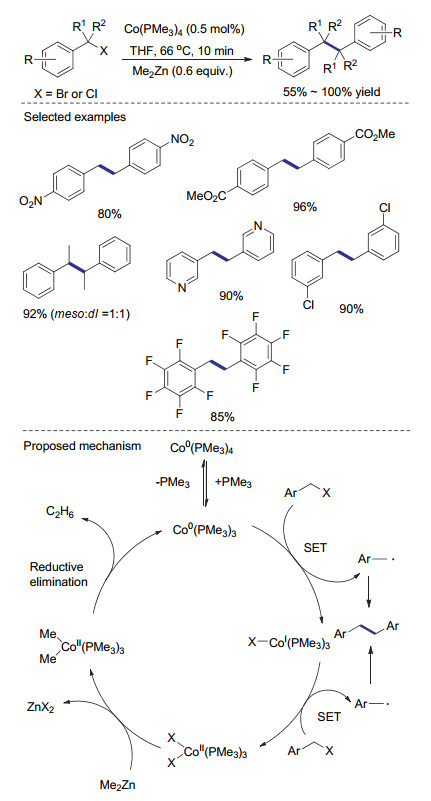
如果利用一些还原剂将上述反应生成的二价钴物种现场还原成低价钴就有可能实现钴催化的还原偶联过程. 2016年, Petit课题组[22]发现当利用二甲基锌作为还原剂时, 在催化量Co(PPh3)4存在下, 苄基溴代物可以快速高效地转化为相应的二聚产物.通过机理研究, 作者认为在该催化过程中, 一分子三甲基膦配体解离后形成的零价钴络合物通过两次单电子转移生成苄基自由基及二溴化钴.其中, 苄基自由基经二聚得到偶联产物, 而二价钴物种在二甲基锌存在下被还原, 再生零价钴物种, 从而完成催化循环(Scheme 3).相较于使用化学计量的低价钴物种作为还原剂, 该催化方法的原子经济性明显提升.由于使用较为温和的二甲基锌作为还原剂, 该反应的官能团兼容性良好.酯基及硝基等吸电子基团在还原偶联过程中均可得到保留且相应产物的产率不受明显影响.值得指出的是, 当反应溶剂为甲苯时, 二甲基锌直接与苄基溴化物发生亲核取代反应形成乙基苯; 而使用四氢呋喃作为溶剂则可抑制这个副反应途径, 提高自偶联反应的效率.
除了二甲基锌, 金属锰也是一种高效的还原剂, 可以用于低价钴还原剂的再生. 2016年, Gosmini课题组[23]报道了CoBr2催化的烷基溴代物的还原偶联反应(Scheme 4).反应利用过量金属锰作为还原剂, 无需含磷配体, 仅用适量的吡啶和三氟乙酸作为添加剂促进反应进行.作者在反应机理研究中发现, 当使用溴甲基环丙烷作为底物时得到开环偶联产物1, 7-辛二烯.如果在还原偶联反应中添加四甲基哌啶氮氧化物(TEMPO)等自由基捕获剂, 则没有目标二聚产物生成.这些发现说明反应催化循环中可能有自由基物种参与.作者推测在反应过程中, 由金属锰还原得到的零价钴物种可能夺取底物中的溴原子形成烷基自由基.烷基自由基与金属钴物种经过一系列转化得到双烷基钴物种, 其经还原消除可再生零价钴, 从而实现催化循环.值得指出的是, 当加入碘化钠作为添加剂时, 烷基氯代物、烷基对甲苯磺酸酯、烯丙基碳酸酯及醋酸酯等惰性底物也能顺利参与该还原偶联反应.

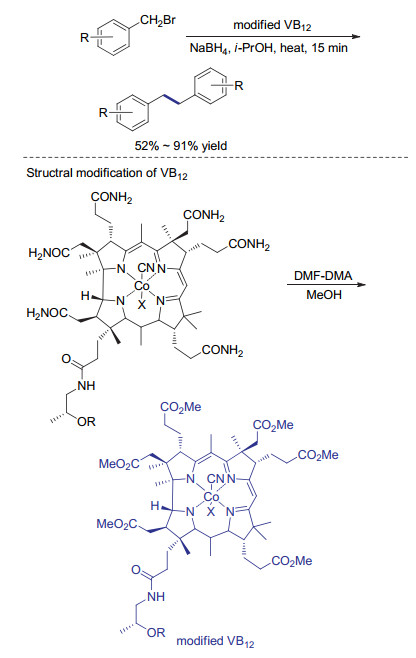
钴元素在生命体中广泛存在, 含有金属钴的维生素B12作为一种酶的辅因子, 可以催化异构化、甲基转移及脱卤等多种反应.这些独特的反应特性都与维生素B12中卟啉大环稳定的金属钴中心有关, 因此维生素B12的催化活性也引起了合成化学家的广泛兴趣[24]. 2001年, Pertrovic课题组[25]发现, 当使用锌粉作为还原剂时, 维生素B12可以有效催化溴代烷基醇的还原自偶联反应, 得到结构较为复杂的多羟基烷基醇类化合物. 2002年, Donk课题组[26]使用柠檬酸钛(Ⅲ)作为还原剂在乙醇和水混合溶液中成功实现了维生素B12催化苄基卤代物的还原偶联反应.为了提高维生素B12在有机溶剂中的溶解度, 2014年, Gryko课题[27]对维生素B12进行改造, 引入疏水基团提高催化剂脂溶性(Scheme 5).当以易得的硼氢化钠为还原剂, 该维生素B12类似物作为催化剂时, 各种苄基溴代物可以高效地转化为相应的还原自偶联产物.与上面介绍的低价态钴物种攫取溴原子形成苄基自由基的方式有所不同, 维生素B12类似物中的一价钴具有很强的亲核性, 可与苄基溴代物发生取代反应生成烷基钴物种.该中间体中的碳-钴键容易发生均裂得到偶联反应需要的苄基自由基[28].作者在研究中还发现, 尽管含吸电子基团的底物在该催化体系中反应效率较低, 增加催化剂的负载量可以大幅提高产率, 使硝基、氰基等易被还原的官能团在反应过程中不受影响.
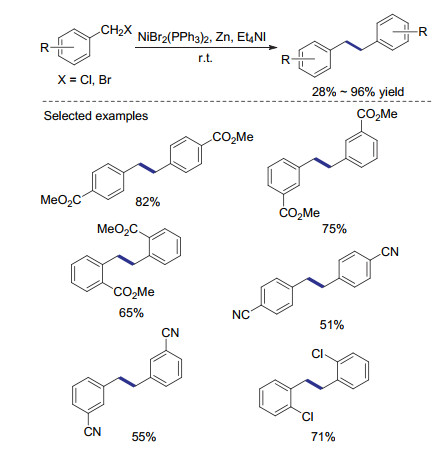
除钴外, 过渡金属镍在还原偶联反应中也具有很好的催化效果. 1985年, Oda课题组[29]报道了基于锌粉及NiCl2(PPh3)2的催化体系, 用于卤代物的还原偶联反应(Scheme 6).该方法适用范围广, 无论活性较高的苄基溴代物还是活性较低的烷基溴代物, 都可以顺利转变为还原偶联产物.除使用锌粉作为还原剂外, 添加四乙基碘化铵对该反应的成功至关重要.根据控制对照实验, 作者发现碘离子的存在可大幅提高偶联反应的产率及反应速率.当反应在室温进行时, 不添加碘负离子偶联产物的产率只有40%, 而添加碘化锂可使二聚反应的产率提高至91%.相较于碘化锂, 四乙基碘化铵较好的溶解性使其可以在多种不同溶剂中使用.该反应主要副反应为还原脱卤, 对于含氰基、甲氧基的底物, 该副反应更易发生, 导致偶联产率偏低.
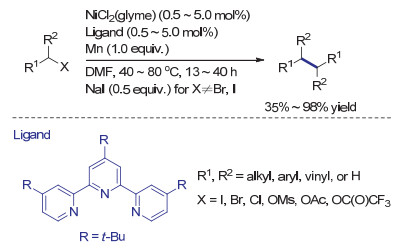
2010年, Weix课题组[30]报道了适用于各种惰性烷基卤代物还原偶联反应的镍催化(Scheme 7).在筛选了一系列含氮配体后, 作者发现使用三联吡啶的效果最佳, 该发现与Fu, Hu等[31-32]的相关研究一致.强极性非质子性溶剂可以有效促进反应(不同溶剂对反应促进能力大小为DMF>DMAC>DMSO>NMP~DMI~DMPU~2-butanol>THF>MeCN), 其中DMF是该反应的最优溶剂.当使用化学计量的锰作为还原剂, 底物1-溴辛烷的还原偶联产率可达98%.值得一提的是, 该反应不需要在氮气氛围或手套箱中进行, 在空气中进行时反应产率也可达到96%.另外, 该镍催化还原偶联体系对常见的氮保护基团和羰基都具有良好的兼容性.当二级有机溴代物作为底物时, 反应产率良好但立体选择性不佳, 所得产物中的两种非对映异构体比例为1:1.对于活性较低的1-溴-环戊烷, 催化剂的负载量需从0.5 mol%提高至2 mol%才能得到较好的产率.当在反应体系中加入0.5 equiv.的碘化钠时, 反应可以应用于更为惰性的烯丙基酯类底物.作者认为, 碘代钠的作用有可能是促进镍催化剂的还原, 现场将三氟乙酸酯转变为活性更高的碘代物.
同年, Huang等[33]报道了使用钳形镍络合物作为催化剂的苄基溴化物还原偶联反应.与基于Co(PPh3)3Cl的反应机理类似, 一价的镍络合物攫取苄基溴代物的溴原子后得到二价镍物种及苄基自由基, 苄基自由基经二聚得到目标自偶联产物.体系中生成的二价镍物种可被锌粉还原从而实现催化循环(Scheme 8).当反应的底物为二级苄基溴代物时, 通过该方法得到的非对映异构体的偶联产物比例为1:1, 说明自由基偶联过程没有立体选择性.
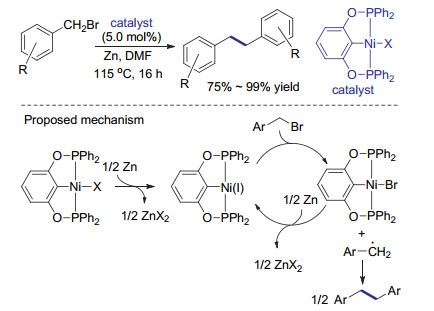
由于镍催化剂也可用于涉及自由基机理的还原偶联, 且苄基溴代物与烯丙基溴代物容易形成相对稳定的自由基, 镍催化的还原偶联可能不是简单地通过镍零价/二价物种的催化循环形成C(sp3)—C(sp3)键. 2013年, Renaud课题组[34]发现, [NiCl(1-napht)(PPh3)2]既可催化苄基溴化物的还原偶联反应, 也可以催化Suzuki-Miyaura偶联反应.但是, 当他们尝试使用对溴甲基溴苯和苯硼酸为底物, 将上述两种偶联反应串联, 以实现一锅法制备1, 2-二联苯基乙烷时, 却只能得到还原偶联的产物.作者推断还原偶联反应和Suzuki-Miyaura偶联过程中具有催化活性的镍物种有所不同, 当反应体系有残留氧气, 可以观察到副产物苯甲醛的生成.因此作者认为, 反应过程中可能生成了苄基自由基, 其经氧气氧化得到苯甲醛.基于上述发现, 他们提出该催化体系中经还原得到的零价镍物种可能先与底物作用, 释放的苄基自由基同时被氧化为一价镍物种.该物种与底物发生氧化加成得到三价镍物种, 与烷基金属试剂发生配体交换后, 经还原消除得到二聚产物(Scheme 9).
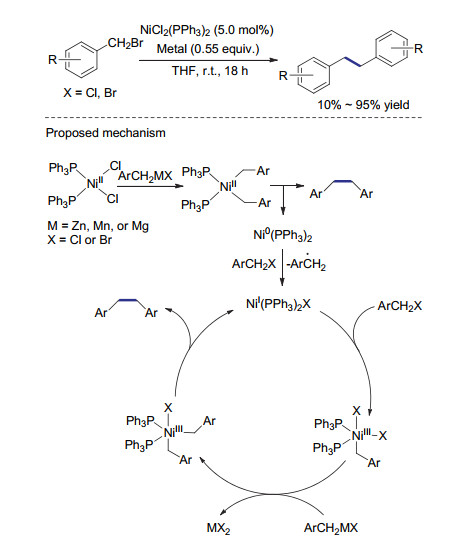
2017年, Liu课题组[35]发展了一种不需要配体参与的镍催化还原偶联体系.该体系使用氯化镍为催化剂, 钐粉为还原剂.当在体系中添加与钐有配位作用的六甲基磷酰三胺时, 苄基溴还原偶联的产率可提高至95% (Scheme 10).该反应具有良好的官能团兼容性, 含有硝基、羧基等活性官能团的底物也可以被顺利地转化还原偶联产物.值得一提的是, 该体系也适用于烷基氯代物、三氟甲磺酸苯酚酯、甲基磺酸苯酚酯等惰性底物.当1-溴乙基苯作为底物时, 该还原偶联反应表现出优秀的立体选择性, 只得到meso构型的产物.这种比较少见的立体选择性可能与反应的独特反应机制有关.首先, 金属钐通过单电子转移与底物作用形成有机钐化合物时伴随着底物的消旋.在还原消除过程中, 将偶联的两部分交错排列以减小位阻, 从而仅得到meso构型的偶联产物.尽管该催化体系使用较为昂贵的金属钐作为还原剂, 其优秀的官能团兼容及立体选择性使其在复杂天然产物合成方面具有良好的应用前景.
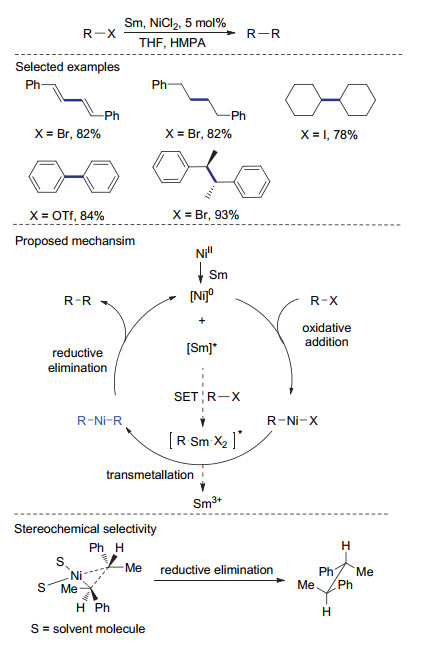
除了利用锌、铝、锰、钐[36-37]等金属单质, 有机金属试剂也可以作为镍催化偶联反应的还原剂. 2016年, Zhao课题组[38]也报道了以甲基格氏试剂为还原剂的镍催化还原偶联反应.当以苄基溴作为底物时, 反应以81%产率制得1, 2-二苯乙烷.
与活性较高的卤代物相比, 醇衍生物的直接还原偶联反应更具挑战. 2019年, Osaka课题组[39]基于相似的反应机理, 实现了烷基对甲苯磺酸酯的还原偶联反应.该反应以镍络合物与维生素B12为催化剂, 以锰为还原剂, 在室温DMF中反应24 h, 对大多数惰性对甲苯磺酸酯底物可获得60%~90%的产率.对于由二级醇衍生的对甲苯磺酸酯底物, 反应未表现出立体选择性, 得到两种近乎等量的非对映异构偶联产物.
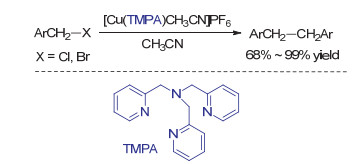
与钴镍等金属类似, 低价的金属铜络合物也可以介导卤代物的还原偶联过程. 1991年, Karlin课题组[40]使用四齿氮配体稳定的低价铜络合物[Cu(TMPA)CH3CN]PF6高效实现了苄基及烯丙基卤代物的还原偶联反应(Scheme 11).反应过程中一价铜络合物作为卤素负离子受体, 反应后可以分离得到相应的二价铜络合物[CuX(TMPA)CH3CN]PF6.当1, 2-二氯甲基苯与足量的还原剂反应时主要获得反式1, 2-二苯乙烯.由于烯烃存在时没有观察到环丙烷产物的生成, 作者推测1, 2-二苯乙烯的生成主要经历分步还原而非卡宾物种的二聚.
与金属钴及镍的还原偶联过程类似, 利用化学计量的还原性金属再生低价铜物种有可能实现铜催化的还原偶联反应. 1998年, Chan等[41]利用金属锰作为还原剂实现非活化卤代烃的还原偶联反应. 2007年, Li等[42]发现当用铁粉作为还原剂可在水溶液体系中实现铜催化的苄基卤代物的还原偶联反应.反应不需要配体, 廉价易得的碘化亚铜、溴化亚铜及氯化亚铜都具有良好的催化效果.由于是非均相反应, 搅拌效率对反应的产率具有较大影响. 2012年, Shekarriz等[43]发现铁纳米颗粒是更为优良的还原剂, 可以进一步提高偶联反应效率, 在室温条件下实现各种芳基及烷基氯代物的二聚反应.金属钐具有优良的单电子转移特性, 被广泛应用于各种还原反应. 2019年, Liu等[37]利用金属钐作为还原剂实现了氯化亚铜催化的苄基卤代物的还原偶联反应.值得一提的是, 该催化体系也可以用于芳基溴代物的还原偶联反应, 说明反应可能不是通过自由基二聚反应机理进行.
离子液体由于其高沸点、高极性、易于回收等特点已经在有机合成中得到广泛引用. 2011年Lu等发现基于二氯合铜负离子的离子液体在卤代物的还原偶联反应中有很好的催化活性.反应以锌作为还原剂, 兼容硝基、氰基、羰基等吸电子官能团, 也可用于活性较低的苄基氯代物的还原偶联.作者推测, 反应中一价的铜物种与底物作用形成苄基自由基, 自由基结合得到二聚产物(Scheme 12).该反应的优点在于不需要使用配体, 且催化剂在反应之后可回收再利用[44].
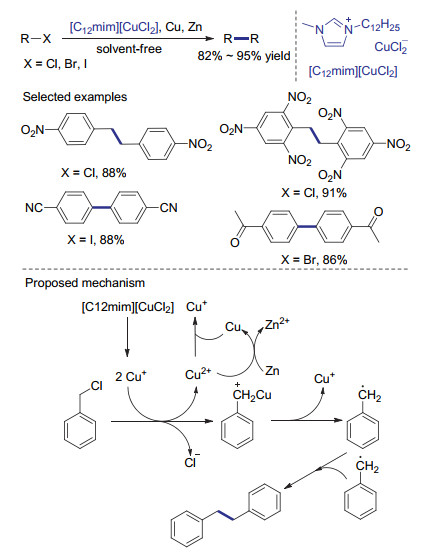
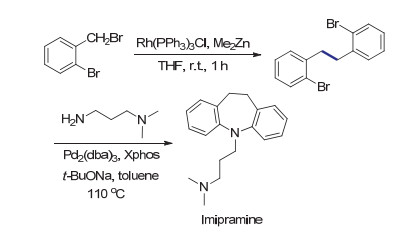
金属铑催化剂已经广泛用于1, 4-加成, 氢酰基化及碳环化等各种反应中, 并表现出独特及高效的反应活性.尽管铑催化的碳碳键形成反应有很多报道, 反应的中心一般是sp及sp2杂化碳原子.基于金属铑催化构建C(sp3)—C(sp3)键的例子仍然很少. 2014年, Ando等[45]报道了Rh(PPh3)3Cl催化苄基溴化物的还原偶联反应.该催化体系中使用了与Petit等的工作中相同的二甲基锌作为还原剂.相对温和的二甲基锌也使该还原偶联反应具有优秀的官能团兼容性.很多简单的苄基溴底物可以定量转化为二聚产物.当向反应体系中鼓入氧气时, 作者发现反应不能进行且原料可被定量回收, 因此推测该反应主要历程可能没有自由基参与.结合之前的相关报道及机理研究, 作者认为反应中一价的铑催化剂与二甲基锌反应得到甲基铑物种, 之后与苄基卤代物发生氧化加成, 得到的三价铑物种再次与甲基锌反应, 经还原消除得到关键中间体苄基铑物种.其与另一分子苄基卤代物发生氧化加成及还原消除制得偶联产物(Scheme 13).当底物为1-溴乙基苯时, 该反应得到的两种非对映异构偶联产物的比例为1:1, 说明还原消除步骤没有明显的立体选择性.作者认为当使用二乙基锌作为还原剂时, 生成的乙基铑中间体可能会发生β-氢消除形成铑-氢物种, 从而发生脱卤氢化的副反应, 降低还原偶联反应的效率.与这种设想一致, 当使用不含有β-氢原子的二苯基锌作为还原剂时, 反应同样以高产率获得还原偶联产物.
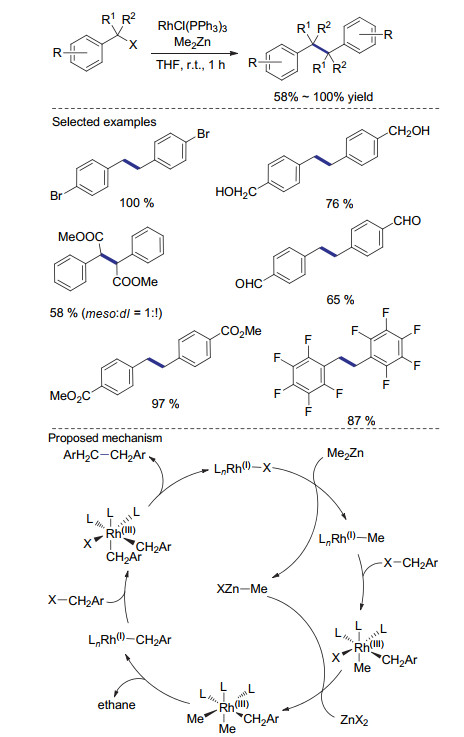
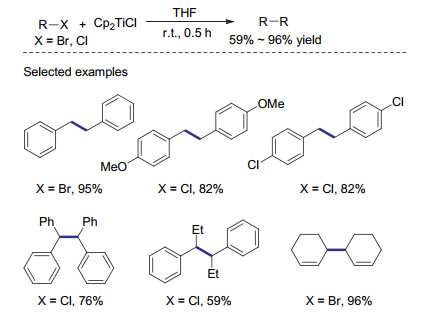
双(环戊二烯基)氯化钛是一种较为绿色的单电子转移试剂, 常常被应用于自由基聚合和天然产物等领域. 1990年, Qian课题组[46]报道了基于Cp2TiCl对苄基及烯丙基卤代物的还原偶联反应.在这个反应中, 作者利用异丙基氯化镁, 将易得的Cp2TiCl2现场还原为Cp2TiCl, 用以还原偶联反应.各种苄基及烯丙基卤代物都可在室温快速转变为相应的还原偶联产物.由于存在异丙基格氏试剂与卤代物直接亲核取代的副反应途径, 反应需要利用化学计量的钛络合物才能得到满意的收率(Scheme 14).值得指出的是, 反应得到的四价钛络合物在不经分离的情况下可以再次被异丙基氯化镁还原为Cp2TiCl, 从而实现钛络合物的重复利用.
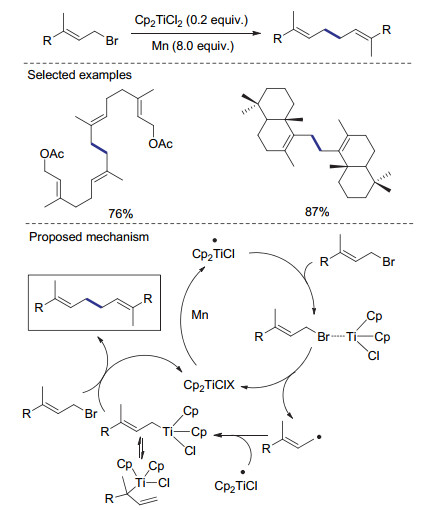
在这一发现的基础上, 2007年Barrero课题组[47]利用金属锰作为还原剂实现了钛催化的烯丙基溴代物的还原自偶联反应(Scheme 15).通过机理研究作者认为, Cp2TiCl2首先被金属锰还原为三价钛物种, 该物种攫取底物中的溴原子形成自由基, 快速与三价钛物种结合形成四价钛物种, 之后与烯丙基溴底物发生亲核取代反应制得还原偶联产物.在这一反应过程中, 形成的自由基的稳定性及反应活性对反应途径有重要影响.当苄基溴代物作为底物时, 由于苄基自由基与三价钛物种结合较慢, 反应也可能通过苄基自由基直接结合的反应途径进行.由于该反应具有很好的官能团兼容性, 作者将其成功应用于onocerane triterpene的合成中[48].
光作为一种易于获得的能量形式, 目前已经被越来越广地应用于各种有机化学反应.由于光的量子特性, 当其与分子作用可以产生热化学条件下难于达到的激发状态, 从而实现独特的反应性质及催化过程.使用Mn2(CO)10作为还原剂的有机卤代物自由基偶联方法于1999年由Parsons等[49]报道.在光照条件下, Mn2(CO)10中的锰锰键发生断裂, 形成五羰基合锰.该络合物具有自由基性质, 可以攫取有机卤代物的卤原子产生烷基自由基物种.之后经烷基自由基偶联即可得到还原二聚产物.由于产生烷基自由基的条件十分温和, 该反应可兼容羟基、羧基、三甲基硅基等较为活泼的官能团, 因而在复杂天然产物的全合成中具有良好的应用前景.值得指出的是, 通过调节两种结构不同的苄基卤代物的比例, 利用该方法也可以较高效地制备交叉还原偶联产物(Scheme 16).在还原偶联反应中, 通常需要使用化学计量的Mn2(CO)10作为还原剂, 因其价格昂贵, 难于应用于较大规模的合成中.
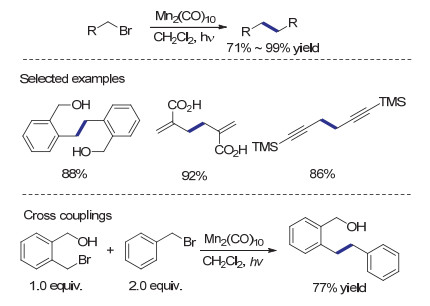
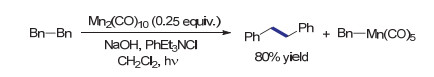
2004年Parson课题组[50]发现还原偶联中产生的副产物Br—Mn(CO)5在碱性两相反应条件下可以再生Mn2(CO)10.基于这一发现, 作者成功利用催化量的Mn2(CO)10, 高效实现了苄基溴代物的还原自偶联反应(Scheme 17).有趣的是, 作者在反应体系中观察到Bn—Mn(CO)5的生成, 随反应时间延长, 其逐渐转变为还原自偶联产物.这一现象说明Mn—C键有可能缓慢均裂, 形成偶联反应所需的苄基自由基.
与上面提到的反应过程类似, 一些基于钌[51]、铜[52]、钴[53]、锆[54]等金属的络合物在光的介导下也可以将电子转移给苄基溴代物, 从而实现还原偶联反应.若金属络合物的激发态具有较长的寿命及合适的氧化还原电势, 则有可能实现光氧化还原催化的还原偶联反应.
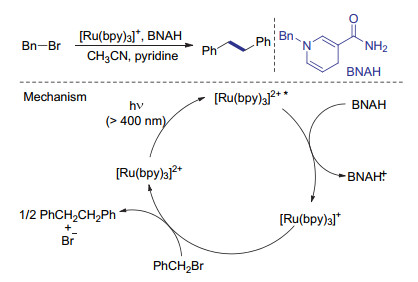
1984年Tanaka课题组[51]发现, 当利用[Ru(bpy)3]2+作为光敏剂, 1-苄基-1, 4-二氢烟酰胺(BNAH)作为还原剂时, 可以将苄基溴高效转化为还原偶联产物(Scheme 18).而当反应体系中不加入[Ru(bpy)3]2+时主要得到还原氢化产物.作者认为激发态的[Ru(bpy)3]2+是一种很强的还原剂, 可以将苄基自由基还原为负离子, 从而避免发生BNAH参与的自由基链式反应过程.
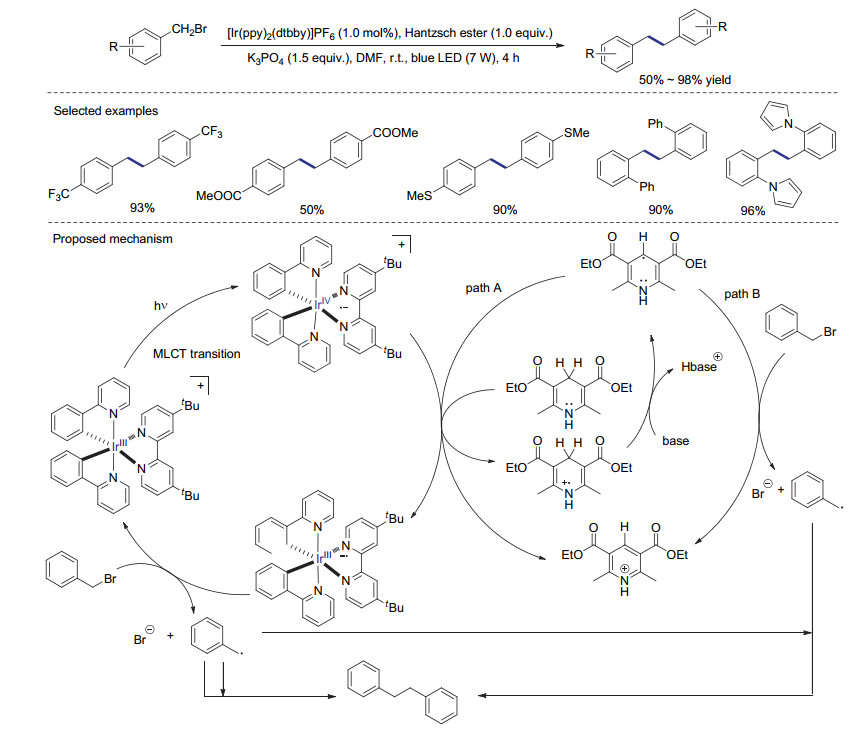
2016年, You课题组[55]实现了可见光催化的苄基溴代物还原偶联反应.作者基于细致的反应机理研究认为, 激发态的汉斯酯及金属铱络合物都可以作为催化过程的还原性物种, 用于产生苄基自由基中间体, 从而确保促进自由基偶联反应高效进行(Scheme 19).值得一提的是, 该反应的光化学量子产率高达20%, 单个光子可以诱导两个电子的转移过程.
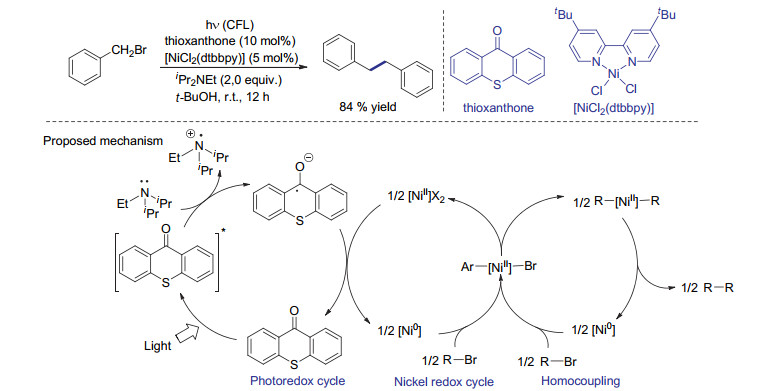
光氧化还原过程不仅可被用于涉及自由基机理的C(sp3)—C(sp3)键形成, 也可被用于介导过渡金属催化剂价态的变化. 2016年, Murakami等[56]报道了使用硫杂蒽酮作为光敏剂的镍催化芳基溴化物或苄基溴化物还原偶联反应.该催化体系包含三个催化循环, 即光氧化还原循环、镍氧化还原循环和自偶联催化循环.在这个反应中, 光激发的电子转移不是用于形成苄基自由基, 而是用于硫杂蒽酮的氧化还原循环, 进一步介导二价镍的还原.在自偶联催化循环中, 两个二价镍物种发生配体交换得到双芳基或双烷基镍物种, 之后经还原消除得到自偶联产物(Scheme 20).
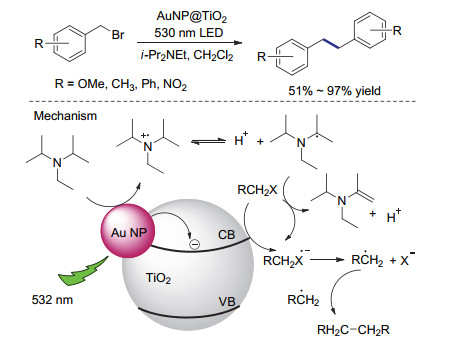
除了利用均相的光氧化还原催化剂, 无机纳米粒子也被用来催化卤代物的还原偶联反应. 2015年Scaiano等[57]报道了基于金纳米颗粒的非均相光催化体系对苄基溴化物进行还原偶联反应.该方法基于金纳米颗粒的表面等离子体带对可见光区具有较强吸收及金表面易于自由基相互作用的特点, 将金纳米颗粒负载于二氧化钛表面, 用作催化剂.反应以过量的二异丙基乙基胺作为还原剂, 在532 nm波长光照下得到了较好的偶联效果.对硝基溴甲基苯作为底物时, 偶联产率可以达到83%.基于控制对照实验, 作者推测光首先激发金纳米颗粒, 将电子注入二氧化钛的导带, 转移给底物之后形成苄基自由基物种(Scheme 21).同时, 二异丙基乙基胺作为牺牲试剂提供电子, 将金还原至初始价态.值得注意的是, 反应过程中生成的烷基胺自由基去质子之后也是较强的还原剂, 可以诱导底物发生还原偶联反应.由于经历苄基自由基中间体, 反应中常常会产生少量脱卤氢化的副产物.
天然产物中常常存在以C(sp3)—C(sp3)键为中心的对称结构.在氧化酶的存在下, 生物分子特定位置的碳氢键可以高效转化为碳自由基, 经自由基二聚反应获得多样化的天然产物.由于碳氢键在有机分子中广泛存在, 利用碳氢键选择性氧化偶联构筑复杂的对称分子结构仍然极具挑战.相比而言, 基于卤代物的还原自偶联反应可以对反应的位点进行准确的控制, 已经成为合成具有对称结构的天然产物的有效手段[58-61].例如在Ando等[45]报道的三环抗抑郁药物imipramine的合成中, 作者利用廉价易得的邻溴甲基溴苯通过铑催化的还原偶联反应, 成功制备了具有对称结构的中间体.还原偶联反应良好的官能团兼容性使芳基溴结构得以保留, 使后续的Buchwald-Hartwig胺化成环反应成为可能(Scheme 22).
生物活性分子中经常存在多个手性中心, 因此在天然产物全合成过程中偶联反应的立体选择性十分重要. Co(PPh3)3Cl是一种高效的还原剂, 已经被用来促进各种还原偶联反应.当烯丙基卤代物作为底物时, 反应的区域选择性良好, 同时双键的立体构型可以得到保持.这些优良的反应性质引起了合成化学家的广泛兴趣, 并将被应用于复杂天然产物的全合成中. 2003年, Baldwin等[62]报道了利用自由基自偶联策略对倍半萜内酯biatractylolide和biepiasterolide进行仿生化合成, 其中化合物biatractylolide具有显著的降血压功效.在利用模板底物对二聚反应的还原剂进行筛选的过程中, 作者发现常用的金属锌及铜等还原剂不能有效促进氯代内酯的还原偶联反应.而当利用Co(PPh3)3Cl作为还原剂时, 可以以57%的产率顺利得到目标产物(Scheme 23a).与通常经历自由基二聚机理的还原偶联反应有所不同, 该转化表现出优秀的立体选择性.二聚产物中dl与meso构型比例可以达到98:2.这可能是与底物分子自身的三维结构有关[63].
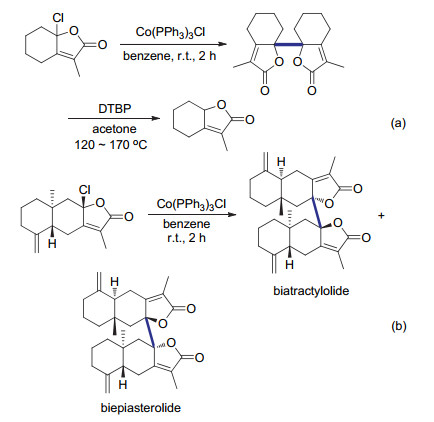
此外, 作者也尝试了其他的自由基生成方法用于实现自由基二聚反应.尽管利用化学计量的DTBP(双特丁基过氧化物)进行单电子氧化的方法也可以制得目标产物(Scheme 23a), 反应的产率及非对映选择性相对较低.这可能是由于同面的甲基基团阻碍攫氢过程.同时, 剧烈的反应条件及反应所伴随的Michael加成及聚合等副反应限制了该方法在合成复杂天然产物中的应用[64].与之对比, 当使用Co(PPh3)3Cl作为还原剂时, 可以在室温利用偶联前体快速制得对倍半萜内酯类化合物biatractylolide和biepiasterolide (Scheme 23b).与模板反应的高立体选择性不同, 产物中非对映异构体biatractylolide和biepiasterolide比例为1:3, 说明底物的结构对这类还原偶联反应的立体选择性影响很大.
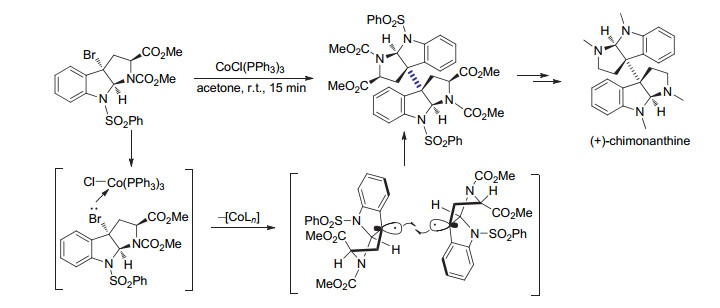
2007年, Movassaghi等[65]将该还原偶联策略用于天然产物(+)-Chimonanthine的合成(Scheme 24).在反应条件筛选的过程中, 作者发现溶剂种类及反应物浓度对偶联反应效率有较大影响.与以往经常使用的芳烃类溶剂苯相比, 利用丙酮作为溶剂可以更为有效促进形成并环中间体的自偶联反应.在克级规模的反应中, 利用Co(PPh3)3Cl作为还原剂室温条件下15 min即可以60%产率制得目标二聚产物, 而且原料中的手性中心在反应过程中能得到很好的保留.作者也对还原剂进行了尝试.除Co(PPh3)3Cl外, 只有利用Mn2(CO)10作为还原剂时可以观测到目标偶联产物的生成.由于很难抑制脱溴还原的副反应, 即使经过细致的条件优化, 反应的产率只有32%, 其效率远低于基于Co(PPh3)3Cl的反应过程.由于可以用于实现具有挑战性的叔碳手性中心的直接链接, 该合成策略不断被应用于各种生物活性分子的合成中[66].
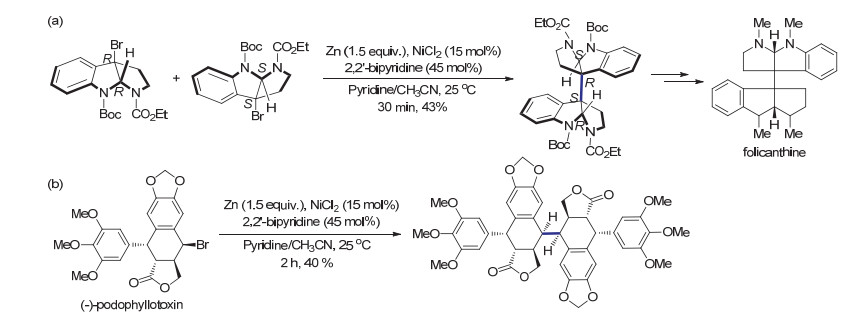
除了利用化学计量的Co(PPh3)3Cl作为还原剂, 过渡金属镍催化的还原自偶联反应也被应用于天然产物的全合成中. 2013年, Wang等[67]报道了使用镍催化惰性有机溴代物的室温还原偶联.该反应以2, 2-联吡啶为催化剂配体, 锌作为还原剂.与基于Co(PPh3)3Cl的还原偶联过程有所不同, 该反应的非对映异构体选择性良好, 通常只得到meso构型的产物.这可能是反应过程中经历了烷基基团从高价镍物种上还原消除的反应机制有关, 因而可以得到与自由基直接偶联不同的立体选择性.作者进一步将该反应过程应用于复杂分子的合成中, 实现了生物碱folicanthine的全合成(Scheme 25a).该反应过程也被用于抗癌药物前体(-)-podophyllotoxin的立体选择性二聚(Scheme 25b), 为生物活性木脂素的合成及应用研究奠定了基础. 2014年, Oguri等[68]也报道了使用1, 2-二苯基膦乙烷为配体, 锰为还原剂的镍催化还原偶联反应.该反应基于低价态的镍攫取溴形成苄基自由基的机理, 同样实现了生物活性分子chimonanthine及其类似物的合成.
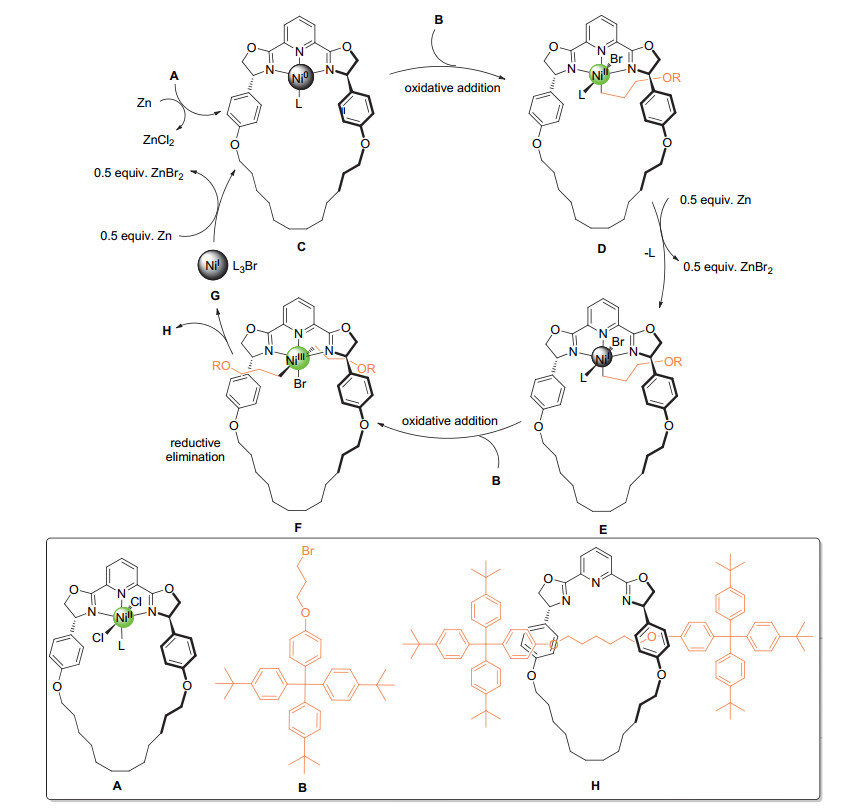
轮烷在超分子研究和纳米机器人开发等领域具有重要意义.轮烷的高效合成通常需要两个构筑单元上都有永久性的识别单元, 从而确保链装单元可以从环状单元中穿插而过.这种合成策略尽管高效, 却对两种构筑单元的结构具有较高要求, 一定程度上限制了轮烷结构的多样性. 2010年, Leigh课题组[69]基于惰性有机卤代物的还原偶联, 巧妙地将环状双唑啉型吡啶(pybox)配体与烷基卤代物组合, 成功合成新型轮烷分子.该工作利用一端具有较大位阻基团的溴代物B为底物, 通过还原自偶联反应组装轮烷分子.镍络合物在反应中不仅起催化作用, 作为合成过程的模板.催化循环中, 零价镍物种C与一分子底物发生氧化加成后形成的二价镍物D, 经单质锌还原形成连有B主体结构的一价镍物种E.该一价镍物种与另一分子底物再次发生氧化加成形成的三价镍物种F, 经还原消除形成新的C(sp3)—C(sp3)键并形成轮烷结构H (Scheme 26).由于该合成策略基于惰性有机卤代物还原偶联, 轮烷链状部分的长短可以利用不同卤代烃底物进行精确控制.值得一提的是, 利用pybox及三联吡啶作为配体的镍催化体系底物适用范围广, 各种烷基溴代物都可以高效地转化为相应的还原偶联产物.
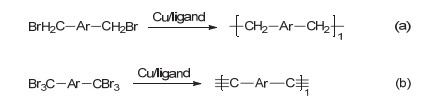
派瑞林[Parylene或PPX (poly-p-xylylene)]因为其极低的介电常数和气体透过率成为一种极其优良的保护材料.目前工业上PPX的制备主要通过真空化学气相沉积来实现, 需要利用专用设备, 制备成本较高[70].对二溴甲基苯的还原自偶联反应也是制备PPX的一种有效策略. 2016年, Wang课题组[71]基于铜参与的苄基自由基偶联反应, 成功制备了一系列PPX衍生物(Scheme 27a).在过量的单质铜和配体存在条件下, 各种结构的二溴代物经还原得到自由基物种, 通过自由基偶联构建结构多样的聚合物.当底物分子中含有酯基及砜基时, 可以得到主链含有烷基芳基交替结构的聚酯和聚砜.该方法也可以用于合成聚苯撑乙烯和聚苯撑乙炔[72, 73]类共轭聚合物(Scheme 27b).
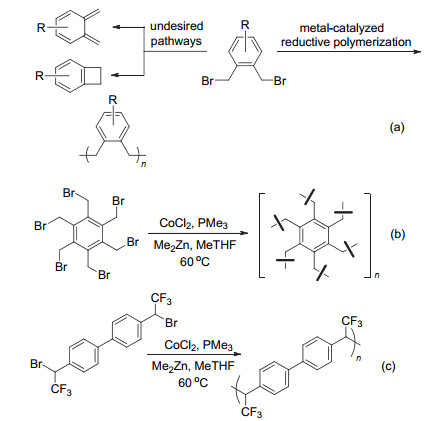
尽管PPX具有优良的物理性能, 其低溶解度也给加工应用带来了一些困难.作为PPX的结构类似物, POX (poly-o-xylylene)是一种在多种有机溶剂中具有良好溶解度的聚合物, 有可能应用于制备高性能涂层.然而, 由于单体二聚等副反应的存在, POX很难通过真空气相沉积方法及其他传统方法制得, 大大限制了其研究应用.尽管关于POX的合成有一些报道, 但通常基于多步反应利用结构较为特殊的单体[74]. 2020年, Zhao课题组[75]基于邻二苄溴的还原自偶联反应, 实现了POX及多种衍生物的合成(Scheme 28a).该方法利用基于钴的还原偶联体系[22], 以二甲基锌作为还原剂, 不仅可制备POX, 也可用于PPX及各种共聚物的制备.由于该聚合反应可以兼容芳基溴结构, 利用Suzuki-Miyaura偶联反应可以方便地对聚合物进行结构修饰, 引入所需的功能基团.当底物为六溴甲基苯时, 利用该反应可以制得高比表面积的超交联多孔聚合物(Scheme 28b).值得一提的是, 还原偶联策略也被应用于聚合物的后续交联, 目前已经被用于制备可泡沫化的防火聚氯乙烯材料[76].另一方面, 含氟聚合物具有独特的物理及化学特性, 在材料科学中广受关注.如前所述, Petit, Ando, You等报道的还原偶联反应所用底物中的氟原子一般连在芳环上, 对还原偶联反应的速率及选择性影响有限.当底物中的氟烷基与卤原子连在同一个碳原子上时, 在还原偶联反应中易发生还原氢化及β-氟消除等副反应.尽管与还原二聚产物结构相似的化合物可以通过其他途径制得, 反应效率通常较低, 难于用于聚合物的合成[77]. 2020年, Zhao课题组[76]发现, 在二氯化钴的催化下可以实现三氟甲基取代的苄基溴代物的还原聚合物反应(Scheme 28c).所制得的含氟聚合物具有很高的热稳定性, 并可以溶于常见的有机溶剂中.这些优良特性使这类新型的含氟聚合物在疏水涂料方面具有良好的应用前景.
目前, 通过有机卤化物的还原自偶联反应形成C(sp3)—C(sp3)键的研究已取得了较大进展.很多烷基卤代物在过渡金属催化条件下可以以近乎定量的收率转化为还原偶联产物.通过选择较为温和的还原剂, 硝基、氰基、乙酰基等活性官能团在反应后可以保留, 使得分子的后续转化成为可能.在反应的可持续发展方面, 钴、镍等廉价金属的使用及催化剂回收再利用, 使该C(sp3)—C(sp3)成键方法适用于规模化合成.还原自偶联的合成策略在天然产物及药物合成方面也极具应用前景, 在合成具有对称结构的分子时可以大幅简化合成步骤.尽管目前部分偶联过程中原料的立体化学信息得以保留, 但适用广泛的立体选择性还原自偶联反应的实现依然存在挑战.通过开发新型手性催化剂, 借助高效的光氧化还原体系, 有可能进一步拓展还原偶联的应用范围, 实现两个手性中心立体化学的精确控制.另一方面, 通过形成C(sp3)—C(sp3)键构建非对称有机分子, 可以加速天然产物及生物活性分子的合成.在目前已报道的C(sp3)—C(sp3)还原偶联反应中, 当使用比例差异较大的两种有机卤化物作为底物, 可以实现交叉偶联反应, 非对称的产物产率有时可以达到70%.然而, 反应中同时会生成大量不需要的还原自偶联产物, 降低了反应的原子经济性.因此, 提高C(sp3)—C(sp3)还原交叉偶联反应的选择性是该领域未来的研究重点.这类反应的成功实施将显著提高复杂天然分子的合成效率.
Wu, X. F.; Anbarasan, P.; Neumann, H.; Beller, M. Angew. Chem. Int. Ed. 2010, 49, 9047. doi: 10.1002/anie.201006374
李祯龙, 金健, 黄莎华, 有机化学, 2020, 40, 563. doi: 10.6023/cjoc201910031Li, Z. L.; Jin, J.; Huang, S. H. Chin. J. Org. Chem. 2020, 40, 563(in Chinese). doi: 10.6023/cjoc201910031
董奎, 刘强, 吴骊珠, 化学学报, 2020, 78, 299.Dong, K.; Liu, Q.; Wu, L. Z. Acta Chim. Sinica 2020, 78, 299(in Chinese).
李娅琼, 范玉航, 贾乾发, 有机化学, 2020, 39, 350. doi: 10.6023/cjoc201806038Li, Y. Q.; Fan, Y. H.; Jia, Q. F. Chin. J. Org. Chem. 2019, 39, 350(in Chinese). doi: 10.6023/cjoc201806038
Liu, L.; Xi, Z. F. Chin. J. Chem. 2019, 36, 1213.
Wang, K.; Kong, W. Q. Chin. J. Chem. 2018, 36, 247. doi: 10.1002/cjoc.201700745
Voloshchuk, T.; Farina, N. S.; Wauchope, O. R.; Kiprowska, M.; Haberfield, P.; Greer, A. J. Nat. Prod. 2004, 67, 1141. doi: 10.1021/np049899e
Buschleb, M.; Dorich, S.; Hanessian, S.; Tao, D.; Schenthal, K. B.; Overman, L. E. Angew. Chem. Int. Ed. 2016, 55, 4156. doi: 10.1002/anie.201507549
Kirchhoff, J. H.; Dai, C.; Fu, G. C. Angew. Chem. Int. Ed. 2002, 41, 1945. doi: 10.1002/1521-3773(20020603)41:11<1945::AID-ANIE1945>3.0.CO;2-7
Hua, S. K.; Hu, Q. P.; Ren, J. M. Zeng, B. B. Synthesis 2013, 45, 518. doi: 10.1055/s-0032-1316841
Tran, U. P. N.; Hock, K. J.; Gordon, C. P.; Koenigs, R. M.; Nguyen, T. V. Chem. Commun. 2017, 53, 4950. doi: 10.1039/C7CC02033C
Du, F.; Zhou, Q.; Liu, D.; Fang, T.; Shi, Y.; Du, Y.; Chen, G. Synlett 2018, 29, 779. doi: 10.1055/s-0036-1591892
Yamada, Y.; Momose, D. Chem. Lett. 1981, 10, 1277. doi: 10.1246/cl.1981.1277
Barhdadi, R.; Courtinard, C.; Nedelec, J. Y.; Troupel, M. Chem. Comm. 2003, 12, 1434.
Schriver, G. W. Tetrahedron Lett. 1988, 29, 1521. doi: 10.1016/S0040-4039(00)80341-X
Albrecht, M. Synthesis 1996, 230.
Shimizu, T.; Tanaka, K.; Paudel, A.; Yamato, T. J. Chem. Res. 2009, 570.
Gozhina, O. V.; Thomassen, I. K.; Lejon, T. Synth. Commun. 2013, 43, 1867. doi: 10.1080/00397911.2012.675459
Zhang, W.; Li, A. Nat. Chem. 2017, 9, 198. doi: 10.1038/nchem.2734
Aresta, M.; Rossi, M.; Sacco, A. Inorg. Chim. Acta 1969, 3, 227; doi: 10.1016/S0020-1693(00)92484-8
Momose, D.; Iguchi, K.; Sugiyama, T.; Yamada, Y. Chem. Pharm. Bull. 1984, 32, 1840.
Fallon, B. J.; Corce, V.; Amatore, M.; Aubert, C.; Chemla, F.; Ferreira, F.; Perez-Luna, A.; Petit, M. New J. Chem. 2016, 40, 9912. doi: 10.1039/C6NJ03265F
Cai, Y.; Qian, X.; Gosmini, C. Adv. Synth. Catal. 2016, 15, 358.
Giedyk, M.; Goliszewska, K.; Gryko, D. Chem. Soc. Rev. 2015, 44, 3391. doi: 10.1039/C5CS00165J
Petrovic, Z.; Mojsilovic, B.; Bugarcic, Z. M. J. Mol. Catal. A:Chem. 2001, 170, 267. doi: 10.1016/S1381-1169(00)00557-4
Shey, J.; McGinley, C. M.; McCauley, K. M.; Dearth, A. S.; Young, B. T.; Donk, W. A. J. Org. Chem. 2002, 67, 837. doi: 10.1021/jo0160470
Giedyk, M.; Fedosov, S. N.; Gryko, D. Chem. Commun. 2014, 50, 4674.
Schrauzer, G. N.; Grate, J. H. J. Am. Chem. Soc. 1981, 103, 541. doi: 10.1021/ja00393a009
Iyoda, M.; Sakaitani, M.; Otsuka, H.; Oda, M. Chem. Lett. 1985, 14, 127. doi: 10.1246/cl.1985.127
Prinsell, M. R.; Everson, D. A.; Weix, D. J. Chem. Commun. 2010, 46, 5743. doi: 10.1039/c0cc01716g
Zhou, J.; Fu, G. J. Am. Chem, Soc. 2003. 125. 14726. doi: 10.1021/ja0389366
Vechorkim, O. Proust, V. Hu, X. J. Am. Chem. Soc. 2009, 131, 9756.
Chen, T.; Yang, L.; Li, L.; Huang, K. W. Tetrahedron 2012, 68, 6152. doi: 10.1016/j.tet.2012.05.075
Mboyi, C. D.; Gaillard, S.; Mabaye, M. D.; Pannetier, N.; Renaud, J. L. Tetrahedron 2013, 69, 4875. doi: 10.1016/j.tet.2013.04.073
Liu, Y. J.; Xiao, S. H.; Qi, Y.; Du, F. Chem. Asian J. 2017, 12, 673. doi: 10.1002/asia.201601712
Nayak, M. K.; Mukhi, P.; Mohanty, A. Rana, S. S.; Arora, R.; Narjinari, H.; Roy, S. J. Chem. Sci. 2019, 131, 59.
Liu, Y. J.; Zhang, D. M.; Xiao, S. H.; Qi, Y.; Liu, S. F. Chem. Asian J. 2019, 8, 858.
Teo, W. J.; Wang, Z.; Xue, F.; Andy Hor, T. S.; Zhao, J. Dalton Trans. 2016, 45, 7312. doi: 10.1039/C6DT00252H
Komeyama, K.; Tsunemitsu, R.; Michiyuji, T.; Yoshida, H.; Osaka, I. Molecules 2019, 24, 1458.
Jacobson, R. R.; Tyklar, Z.; Karlin, K. D. Inorg. Chim. Acta 1991, 181, 111.
Ma, J.; Chan, T. H. Tetrahedron Lett. 1998, 39, 2499. doi: 10.1016/S0040-4039(98)00348-7
Liu, J.; Li, B. Synth. Commun. 2007, 37, 3273. doi: 10.1080/00397910701483340
Shekarriz, M.; Adib, M.; Biabani, T.; Taghipoor, S. J. Chem. Res. 2012, 29.
Hu, Y. L.; Li, F.; Gu, G. L. Catal. Lett. 2011, 141, 467. doi: 10.1007/s10562-010-0535-5
Sato, K.; Inoue, Y.; Mori, T.; Sakaue, A.; Taruo, A.; Omote, M.; Kumadaki, I.; Ando, A. Org. Lett. 2014, 16, 3756. doi: 10.1021/ol501619w
Qian, Y.; Li, G.; Huang, Y. Z. J. Organomet. Chem. 1990, 381, 29.
Barrero, A. F.; Herrador, M. M.; Moral, J. F. Q.; Arteaga, P.; Akssira, M.; Hanbali, F. E.; Artega, J. F.; Dieguez, H. R.; Sanchez, E. M. J. Org. Chem. 2007, 72, 2251.
Barrero, A. F.; Herrador, M. M.; Moral, J. F. Q.; Arteaga, P.; Arteaga, J. F.; Piedra, M.; Sanchez, E, M, Org. Lett. 2005, 7, 2301. doi: 10.1021/ol050335r
Gilbert, B. C.; Lindsay, C. I.; McGrail, P. T.; Parsons, A. F.; Whittaker, D. T. E. Synth. Commun. 1999, 29, 2711. doi: 10.1080/00397919908086433
Huther, N.; McGrail, P. T.; Parsons, A. F. Eur. J. Org. Chem. 2004, 1740.
Hironaka, K.; Fukuzumi, S.; Tanaka, T. J. Chem. Soc., Perkin Trans. 21984, 1705.
Kern, J. M.; Sauvage, J. P. J. Chem. Soc., Chem. Commun. 1987, 546.
Fukuzumi, S.; Ishikawa, K.; Tanaka, T. Organometallics 1987, 6, 358. doi: 10.1021/om00145a020
Zhang, Y.; Petersen, J. L.; Milsmann, C. Organometallics 2018, 37, 4488.
Park, G.; Yi, S. Y.; Jung, J.; Cho, E. J.; You. Y. Chem. Eur. J. 2016, 22, 17790. doi: 10.1002/chem.201603517
Masuda, Y.; Ishida, N.; Murakami, M. Eur. J. Org. Chem. 2016, 5822.
Lanterna, A. E.; Elhage, A.; Scaiano, J. C. Catal. Sci. Technol. 2015, 5, 4336.
Vrettou, M.; Gray, A. A.; Brewer, A. R. E.; Barret, A. G. M. Tetrahedron 2007, 63, 1487.
Gentry, E. C. G.; Rono, L. J.; Hale, M. E.; Matsuura, R.; Knowles, R. R.; J. Am. Chem. Soc. 2018, 140, 3394. doi: 10.1021/jacs.7b13616
Lathrop, S. P.; Pompeo, M. T.; Chang, W. T. T.; Movassaghi, M. J. Am. Chem. Soc. 2016, 138, 7763. doi: 10.1021/jacs.6b04072
Ding, M.; Liang, K. J.; Pan, Rui.; Zhang, H. B.; Xia, C. F. J. Org. Chem. 2015, 80, 10309.
Bagal, S. K.; Adlington, R. M.; Baldwin, J. E.; Marquez. R. J. Org. Chem. 2004, 69, 9100.
Bagal, S. K.; Adlington, R. M.; Marquez, R.; Cowley, A. R.; Baldwin, J. E. Tetrahedron Lett. 2003, 44, 4993.
Bagal, S. K.; Adlington, R. M.; Baldwin, J. E.; Marquez, R. Cowley, A. Org. Lett. 2003, 5, 3049. doi: 10.1021/ol035022f
Movassaghi, M.; Schmidt, M, A. Angew. Chem. Int. Ed. 2007, 46, 3725. doi: 10.1002/anie.200700705
Iwasa, E.; Hamashima, Y.; Fujishiro, S.; Higuchi, E.; Ito, A.; Yoshida, M.; Sodeoka, M. J. Am. Chem. Soc. 2010, 132, 4078. doi: 10.1021/ja101280p
Peng, Y.; Luo, L.; Yan, C. S.; Zhang, J. J.; Wang, Y. W. J. Org. Chem. 2013, 78, 10960.
Wada, M.; Murata, T.; Oikawa, H.; Oguri, H. Org. Biomol. Chem. 2014, 12, 298.
Goldup, S. M.; Leigh, D. A.; McBurney, R. T.; McGonigal, P. R.; Plant, A. Chem. Sci. 2010, 1, 383.
Gorham, W. F. J. Poly. Sci. 1966, 4, 3027. doi: 10.1002/pol.1966.150041209
Liu, Z.; Wang, Q. RSC Adv. 2016, 6, 39568. doi: 10.1039/C6RA02669A
Liu, Z.; Wang, Q. Polymer 2017, 100, 56.
Liu, Z.; Wang, Q. ACS Macro Lett. 2018, 7, 604.
Zhu, R.; Swager, T. M. J. Am. Chem. Soc. 2018. 140. 5211. doi: 10.1021/jacs.8b01106
Chen, S.; Zhao, Y. Chin. J. Chem. 2020, 38, 953.
Pike, R. D.; Starnes Jr, W. H.; Jeng, J. P.; Bryant, W. S.; Kourtesis, P.; Adams, C. W.; Bunge, S. D.; Kang, Y. M.; Kim, A. S.; Kim, J. H.; Macko, J. A.; O'Brien, C. P. Macromolecules 1997, 30, 6957. doi: 10.1021/ma9707749
Gao, B.; Zhao, Y. C.; Ni, C. F.; Hu, J. B. Org. Lett. 2014, 16, 102. doi: 10.1021/ol403083e

图式 1 Co(PPh3)3Cl参与的苄基氯还原偶联反应
Scheme 1 Co(PPh3)3Cl-mediated reductive homocouplings of benzyl chlorides

图式 2 (a) Co(PPh3)3Cl参与的二溴甲基苯还原偶联和(b) Co-(PPh3)3Cl参与的烯丙基溴代物还原偶联
Scheme 2 (a) Reductive homocoupling of (dibromomethyl)benzene with Co(PPh3)3Cl, and (b) reductive homocoupling of allylic bromides with Co(PPh3)3Cl

图式 3 四(三甲基膦)钴催化的还原自偶联反应及推测机理
Scheme 3 Co(PMe3)4-catalyzed reductive homocouplings and the proposed mechanism

图式 4 钴催化惰性有机卤化物的还原自偶联反应及其推测机理)
Scheme 4 Cobalt-catalyzed reductive homocouplings of inert organic halides and the proposed mechanism

图式 5 维生素B12改性以用于苄溴的还原自偶联反应
Scheme 5 Structurally-modified vitamin B12 for reductive coupling of benzyl halides

图式 8 钳形镍络合物催化还原自偶联反应的推测机理
Scheme 8 Proposed mechanism for the reductive homocouplings catalyzed by a pincer-type nickel complex

图式 9 Renaud课题组镍催化还原自偶联及推测机理
Scheme 9 Renaud's Ni-catalyzed reductive homocoupling and its proposed mechanism

图式 10 钐作为还原剂的镍催化还原偶联反应
Scheme 10 Ni-catalyzed reductive coupling using Sm as the reducing reagent

图式 12 使用离子液体和铜/锌的还原自偶联
Scheme 12 Reductive homocoupling based on ionic liquid and Cu/Zn

图式 13 二甲基锌作为还原剂的铑催化还原自偶联反应及其推测机理
Scheme 13 Rh-catalyzed reductive homocouplings using Me2Zn as the reducing reagent and the proposed mechanism

图式 15 Barrero等报道的钛催化还原自偶联反应的可能机理
Scheme 15 Proposed mechanism for Barrero's Ti-catalyzed reductive homocouplings

图式 16 以Mn2(CO)10为还原剂的还原偶联反应
Scheme 16 Reductive couplings using Mn2(CO)10 as the reductant

图式 18 钌络合物介导的光催化还原偶联反应
Scheme 18 Photoredox catalytic reductive couplings mediated by a ruthenium complex

图式 19 铱络合物介导的光催化还原偶联反应
Scheme 19 Photocatalytic reductive couplings mediated by iridium catalysts

图式 20 C(sp3)—C(sp3)键形成于还原消除的光催化反应
Scheme 20 Photocatalytic reductive homocouplings with C(sp3)—C(sp3) bond formed by reductive elimination

图式 21 二氧化钛负载的金纳米颗粒用于光催化还原偶联反应及其推测机理
Scheme 21 Photocatalytic reductive homocouplings promoted by TiO2-supported gold nanoparticles and the proposed mechanism

图式 23 通过还原自偶联反应制备biatractylolide和biepiasterolide
Scheme 23 Synthesis of biatractylolide and biepiasterolide via reductive homocoupling

图式 24 (+)-Chimonanthine合成中的还原自偶联反应
Scheme 24 Reductive homocouplings for the synthesis of (+)-chimonanthine

图式 25 (a) 通过还原自偶联合成folicanthine (b)通过还原自偶联进行(-)-podophyllotoxin的二聚
Scheme 25 (a) Synthesis of folicanthine via reductive homocoupling (b) Dimerization of (-)-podophyllotoxin via reductive homocoupling

图式 26 通过还原自偶联反应合成新型轮烷分子
Scheme 26 Preparation of novel rotaxanes via reductive homocoupling

图式 27 通过铜介导的还原偶联制备POX和PPE类聚合物
Scheme 27 Preparation of POXs and PPEs via copper-catalyzed reductive elimination

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:






