引用本文:
张鄂, 陈单丹, 王守锋, 刘文. 基于生物合成的双环硫肽类抗生素结构改造进展[J]. 有机化学,
2020, 40(10): 3120-3131.
doi:
10.6023/cjoc202005071 Citation:
Zhang E, Chen Dandan, Wang Shoufeng, Liu Wen. Progress in Structural Modification of Bicyclic Thiopeptide Antibiotics Based on Biosynthesis[J]. Chinese Journal of Organic Chemistry,
2020, 40(10): 3120-3131.
doi:
10.6023/cjoc202005071
Shandong Provincial Key Laboratory of Fluorine Chemistry and Chemical Materials, School of Chemistry and Chemical Engineering, University of Jinan, Jinan 250022
b.
State Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032
c.
Huzhou Center of Bio-Synthetic Innovation, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Huzhou, Zhejiang 313000
Received Date:
26 May 2020 Revised Date:
20 June 2020 Available Online:
25 October 2020
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 31972850, 21750004, 21520102004), the Shandong Key Research Program (No. 2019GSF108223), the Chinese Academy of Sciences (Nos. QYZDJ-SSW-SLH037, XDB20020200), the Science and Technology Commission of Shanghai Municipality (No. 17JC1405100), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (No. 2017303), and the K. C. Wong Education Foundation
Abstract:
Thiopeptide antibiotics are a class of natural products with polythiophene (oxazol) polypeptides, which are rich in sulfur and highly modified. They are produced by secondary metabolism of microorganisms and have good biological activities. Developing thiopeptides into clinic is currently a challenge partly due to their low aqueous solubility and associated poor bioavailability. On the basis of understanding the mechanism of their biosynthesis, the method of obtaining thiopeptide analogues via reasonable bioengineering has become the research focus of biologists. In this paper, the advances in structural modifications of bicyclic thiopeptides by taking thiostrepton and nosiheptide as representatives are reviewed.
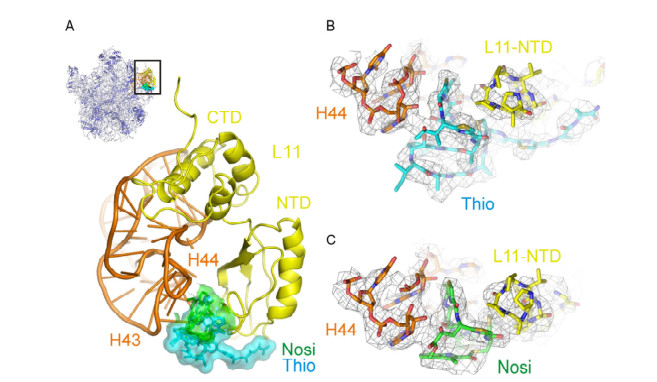
Figure 2.
Antibacterial mechanisms of thiopeptides
(A) Interface view of the 50S subunit with the thiopeptide binding site boxed. Enlargement of boxed region reveals that the thiopeptides Thio (cyan) and Nosi (green) bind within a cleft between the N-terminal domain (NTD) of r-protein L11 (yellow) and the 23S rRNA H43 and H44 (orange). (B) Difference electron density maps with Thio, L11-NTD, and H44 colored as in A). (C) Difference electron density maps with Nosi, L11-NTD, and H44 colored as in A)
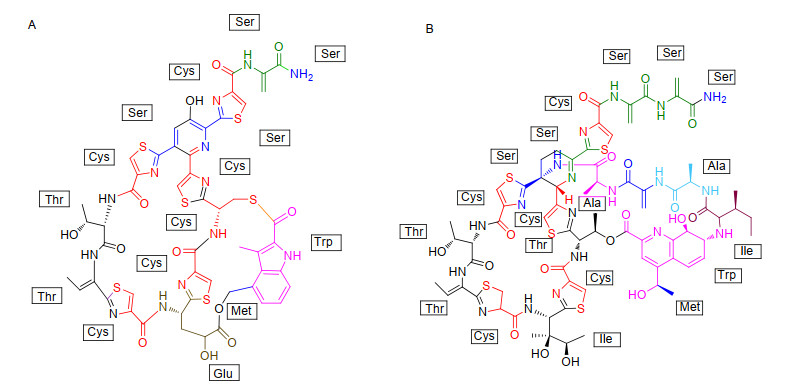
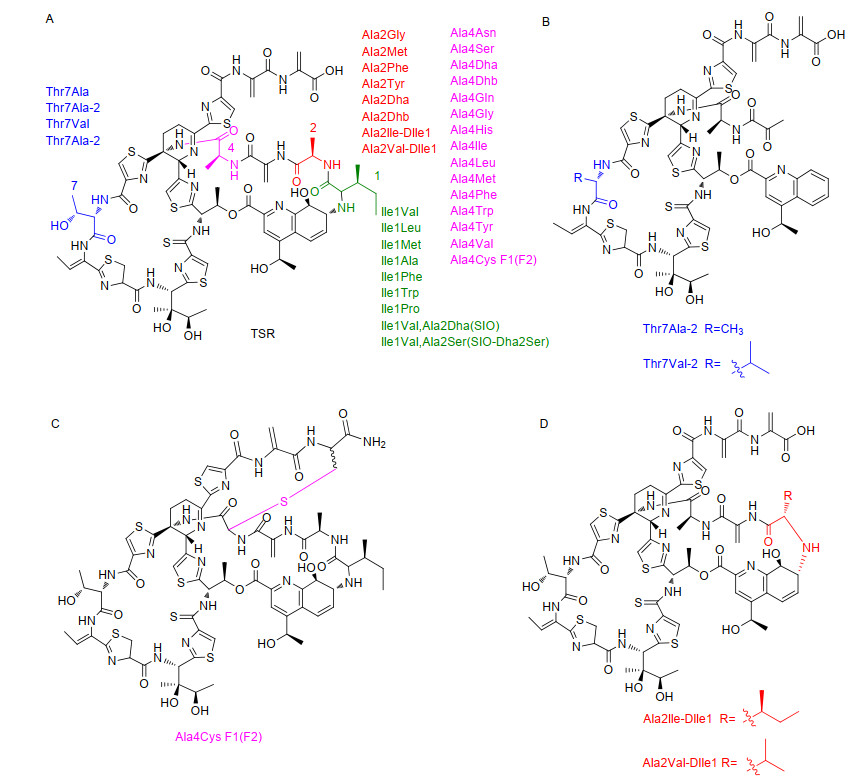
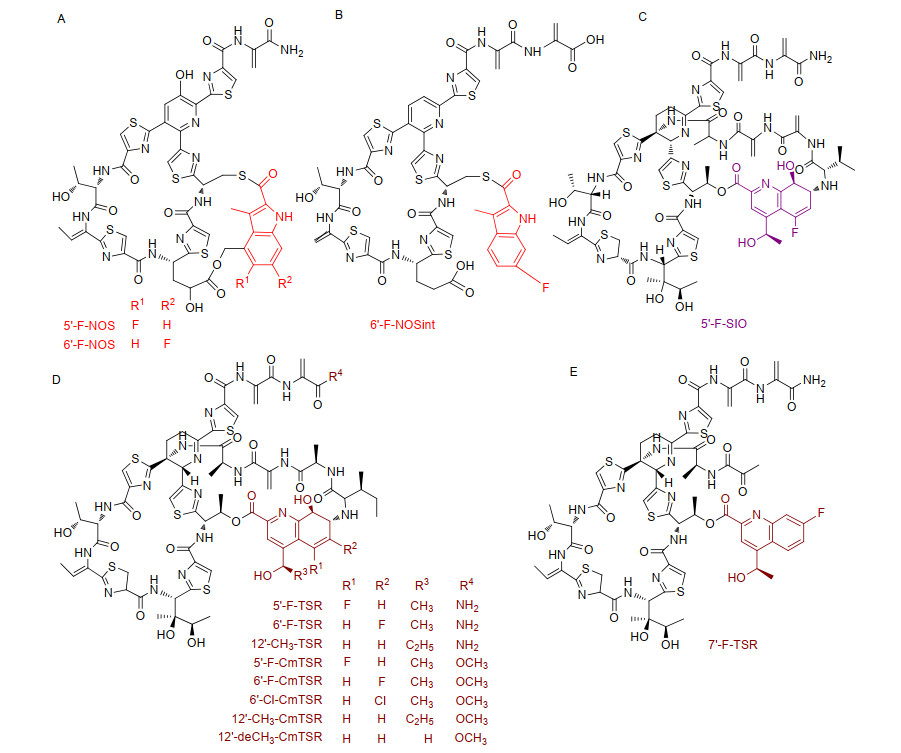
Figure 7.
TSR analogues obtained by site-directed mutation of TSR precursor peptide. The point mutation of isoleucine at position 1 is shown in green, the point mutation of alanine at position 2 is shown in red, the point mutation of alanine at position 4 is shown in plum red, and the point mutation of threonine at position 7 is shown in blue
Dedicated to Professor Henry N. C. Wong on the occasion of his 70th birthday.
[1]
Arnison, P. G.; Bibb, M. J.; Bierbaum, G.; Bowers, A. A.; Bugni, T. S.; Bulaj, G.; Camarero, J. A.; Campopiano, D. J.; Challis, G. L.; Clardy, J.; Cotter, P. D.; Craik, D. J.; Dawson, M.; Dittmann, E.; Donadio, S.; Dorrestein, P. C.; Entian, K. D.; Fischbach, M. A.; Garavelli, J. S.; Goransson, U.; Gruber, C. W.; Haft, D. H.; Hemscheidt, T. K.; Hertweck, C.; Hill, C.; Horswill, A. R.; Jaspars, M.; Kelly, W. L.; Klinman, J. P.; Kuipers, O. P.; Link, A. J.; Liu, W.; Marahiel, M. A.; Mitchell, D. A.; Moll, G. N.; Moore, B. S.; Muller, R.; Nair, S. K.; Nes, I. F.; Norris, G. E.; Olivera, B. M.; Onaka, H.; Patchett, M. L.; Piel, J.; Reaney, M. J.; Rebuffat, S.; Ross, R. P.; Sahl, H. G.; Schmidt, E. W.; Selsted, M. E.; Severinov, K.; Shen, B.; Sivonen, K.; Smith, L.; Stein, T.; Sussmuth, R. D.; Tagg, J. R.; Tang, G. L.; Truman, A. W.; Vederas, J. C.; Walsh, C. T.; Walton, J. D.; Wenzel, S. C.; Willey, J. M.; van der Donk, W. A. Nat. Prod. Rep.2013, 30, 108. doi: 10.1039/C2NP20085F
Vandeputte, J.; Dutcher, J. D. Antibiot. Annu.1955~1956, 560.
[11]
Trejo, W. H.; Dean, L. D.; Pluscec, J.; Meyers, E.; Brown, W. E. J. J. Antibiot (Tokyo) 1977, 30, 639. doi: 10.7164/antibiotics.30.639
[12]
(a) Anderson, B.; Hodgkin, D. C.; Viswamitra, M. A. Nature1970, 225, 233. (b) Hensens, O. D.; Albers-Schönberg, G. J. Antibiot (Tokyo) 1983, 36, 814. (c) Hensens, O. D.; Albers-Schönberg, G. J. Antibiot (Tokyo) 1983, 36, 832. (d) Hensens, O. D.; Albers-Schonberg, G.; Anderson, B. F. J. Antibiot (Tokyo) 1983, 36, 799.
[13]
Benazet, F.; Cartier, M.; Florent, J.; Godard, C.; Ninet, L. Experientia1980, 36, 414. doi: 10.1007/BF01975121
[14]
Depaire, H.; Thomas, J. P.; Brun, A.; Olesker, A.; Lukacs, G. Tetrahedron Lett.1977, 18, 1397. doi: 10.1016/S0040-4039(01)93054-0
Rogers, M. J.; Cundliffe, E.; Mccutchan, T. F. Antimicrob. Agents Chemother.1998, 42, 715. doi: 10.1128/AAC.42.3.715
[17]
(a) Naidu, B. N.; Sorenson, M. E.; Zhang, Y.; Kim, O. K.; Matiskella, J. D.; Wichtowski, J. A.; Connolly, T. P.; Li, W.; Lam, K. S.; Bronson, J. J. Bioorg. Med. Chem. Lett.2004, 14, 5573. (b) Zhang, C.; Herath, K.; Jayasuriya, H.; Ondeyka, J. G.; Zink, D. L.; Occi, J.; Birdsall, G.; Venugopal, J.; Ushio, M.; Burgess, B. J. Nat. Prod.2009, 72, 841. (c) Zhang, C.; Occi, J.; Masurekar, P.; Barrett, J. F.; Zink, D. L.; Smith, S.; Onishi, R.; Ha, S.; Salazar, O.; Genilloud, O. J. Am. Chem. Soc.2008, 130, 12102.
[18]
(a) Xing, Y.; Draper, D. E. Biochemistry1996, 35, 1581. (b) Blyn, L. B.; Risen, L. M.; Griffey, R. H.; Draper, D. E. Nucleic Acids Res.2000, 28, 1778. (c) Wimberly, B. T.; Guymon, R.; McCutcheon, J. P.; White, S. W.; Ramakrishnan, V. Cell1999, 97, 491. (d) Lentzen, G.; Klinck, R.; Matassova, N.; Aboul-ela, F.; Murchie, A. I. Chem. Biol.2003, 10, 769. (e) Harms, J. M.; Wilson, D. N.; Schluenzen, F.; Connell, S. R.; Fucini, P. Mol. Cell2008, 30, 26. (f) Bo, T. P.; Leviev, I.; Mankin, A. S.; Garrett, R. A. J. Mol. Biol.1998, 276, 391. (g) Gonzalez, R. L.; Chu, S.; Puglisi, J. D. RNA (New York, N. Y.) 2007, 13, 2091.
Houck, D. R.; Chen, L. C.; Keller, P. J.; Beale, J. M.; Floss, H. G. J. Am. Chem. Soc.1988, 110, 5800. doi: 10.1021/ja00225a035
[21]
Houck, D. R.; Chen, L. C.; Keller, P. J.; Beale, J. M.; Floss, H. G. J. Am. Chem. Soc.1993, 115, 7992. doi: 10.1021/ja00071a009
[22]
Yu, Y.; Duan, L.; Zhang, Q.; Liao, R.; Ding, Y.; Pan, H.; Evelyn, W. P.; Tang, G. L.; Shen, B.; Liu, W. ACS Chem. Biol.2009, 4, 855. doi: 10.1021/cb900133x
[23]
(a) Liao, R.; Duan, L.; Lei, C.; Pan, H.; Ding, Y.; Zhang, Q.; Chen, D.; Shen, B.; Yu, Y.; Liu, W. Chem. Biol.2009, 16, 141. (b) Kelly, W. L.; Pan, L.; Li, C. J. Am. Chem. Soc.2009, 131, 4327.
[24]
Morris, R. P.; Leeds, J. A.; Naegeli, H. U.; Oberer, L.; Memmert, K.; Weber, E.; LaMarche, M. J.; Parker, C. N.; Burrer, N.; Esterow, S.; Hein, A. E.; Schmitt, E. K.; Krastel, P. J. Am. Chem. Soc.2009, 131, 5946. doi: 10.1021/ja900488a
[25]
Hudson, G. A.; Zhang, Z.; Tietz, J. I.; Mitchell, D. A.; van der Donk, W. A. J. Am. Chem. Soc.2015, 137, 16012. doi: 10.1021/jacs.5b10194
[26]
Zhang, Q.; Li, Y. X.; Chen, D. D.; Yu, Y.; Duan, L.; Shen, B.; Liu, W. Nat. Chem. Biol.2011, 7, 154. doi: 10.1038/nchembio.512
[27]
Sicoli, G.; Mouesca, J. M.; Zeppieri, L.; Amara, P.; Martin, L.; Barra, A. L.; Fontecilla Camps, J. C.; Gambarelli, S.; Nicolet, Y. Science2016, 351, 1320. doi: 10.1126/science.aad8995
[28]
Qiu, Y. P.; Du, Y. N.; Zhang, F.; Liao, R. J.; Zhou, S. X.; Peng, C.; Guo, Y. L.; Liu, W. J. Am. Chem. Soc.2017, 139, 18186. doi: 10.1021/jacs.7b11367
[29]
Qiu, Y. P.; Du, Y. N.; Wang, S. F.; Zhou, S. X.; Guo, Y. L.; Liu, W. Org. Lett.2019, 21, 1502. doi: 10.1021/acs.orglett.9b00293
[30]
Wang, B.; LaMattina, J. W.; Marshall, S. L.; Booker, S. J. J. Am. Chem. Soc.2019, 141, 5788. doi: 10.1021/jacs.8b13157
Liu, W. Y.; Ma, M.; Xue, Y. J.; Liu, N.; Wang, S. Z.; Chen, Y. J. ChemBioChem2013, 14, 573. doi: 10.1002/cbic.201200681
[33]
Wang, S. Z.; Zheng, X. L.; Pan, Q.; Chen, Y. J. RSC Adv.2016, 6, 94643. doi: 10.1039/C6RA20302G
[34]
Kwok, M. M.; Myatt, S. S.; Marson, C. M.; Coombes, R. C.; Constantinidou, D.; Lam, W. F. Mol. Cancer Ther.2008, 7, 2022. doi: 10.1158/1535-7163.MCT-08-0188
[35]
Li, C.; Zhang, F.; Kelly, W. L. Chem. Commun.2012, 48, 558. doi: 10.1039/C1CC14281J
[36]
Zhang, F.; Li, C.; Kelly, W. L. ACS Chem. Biol.2015, 11, 415.
[37]
Zhang, F.; Kelly, W. L. ACS Chem. Biol.2015, 10, 998. doi: 10.1021/cb5007745
[38]
Guo, H.; Wang, J.; Li, Y. M.; Yu, Y.; Zheng, Q. F.; Wu, J. Q.; Liu, W. Chem. Sci.2014, 5, 240. doi: 10.1039/C3SC52015C
[39]
Duan, P. P.; Zheng, Q. F.; Lin, Z.; Wang, S. F.; Chen, D. D.; Liu, W. Org. Chem. Front.2016, 3, 1254. doi: 10.1039/C6QO00320F
[40]
Liu, W. Y.; Xue, Y. J.; Ma, M.; Wang, S. Z.; Liu, N.; Chen, Y. J. ChemBioChem2013, 14, 1544. doi: 10.1002/cbic.201300427
(A) Interface view of the 50S subunit with the thiopeptide binding site boxed. Enlargement of boxed region reveals that the thiopeptides Thio (cyan) and Nosi (green) bind within a cleft between the N-terminal domain (NTD) of r-protein L11 (yellow) and the 23S rRNA H43 and H44 (orange). (B) Difference electron density maps with Thio, L11-NTD, and H44 colored as in A). (C) Difference electron density maps with Nosi, L11-NTD, and H44 colored as in A)
Figure 7
TSR analogues obtained by site-directed mutation of TSR precursor peptide. The point mutation of isoleucine at position 1 is shown in green, the point mutation of alanine at position 2 is shown in red, the point mutation of alanine at position 4 is shown in plum red, and the point mutation of threonine at position 7 is shown in blue

 下载:
下载:

 下载:
下载:










