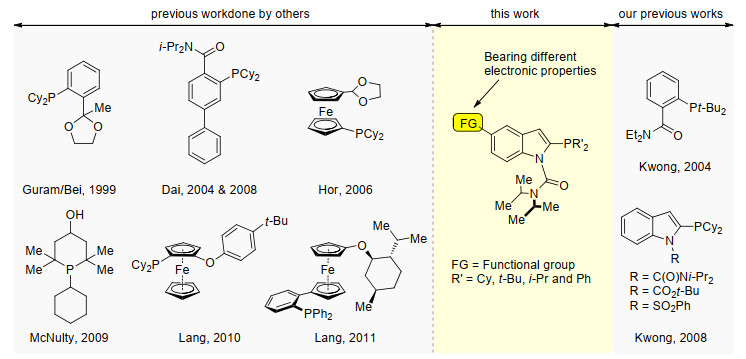
Figure 1.
Selected examples of hemilabile P, O-type ligands particularly for Pd-catalyzed Suzuki-Miyaura coupling of aryl chlorides and present study
Facile One-Pot Assembly of New 5-Substituted P, O-Type Indolylphosphine Ligands for Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling of Aryl Chlorides
Man Pan Leung , Chung Chiu Yeung , Pui Ying Choy , Chau Ming So , Fuk Yee Kwong

Palladium-catalyzed cross-coupling is one of the most important synthetic tools for constructing aromatic carbon-carbon bonds. Among Suzuki-Miyaura, Negishi, Kumada, Stille, Hiyama cross-couplings and others in relevance, [1] Suzuki-Miyaura reaction is found to be the most useful synthetic method, for instance of their widespread applications in pharmaceutical, material, and agricultural syntheses.[2] The rule of thumb of enhancing the catalytic efficiency for a coupling reaction is to employ ancillary ligands equipping with appropriate steric and electronic environments.[3] A number of brilliant chemists have put huge effort towards the design and modification of phosphine ligands, such as Fu, [4] Hartwig, [5] Buchwald, [6] Ackermann, [7] Beller, [8] Kwong, [9] Stradiotto, [10] Tang, [11] Zhang, [12] Ma, [13] Lipshutz, [14] Kapdi[15] and other groups.[16] Tailor-made phosphine ligands aiming at tackling difficult Suzuki-Miyaura coupling are generally electron-rich and steric bulky, in which they are actually devised to favorably cater the oxidative addition (O.A.) and reductive elimination (R.E.) steps in a catalytic cycle, respectively. The unsaturation of a Pd/ligand complex is believed to offer more spatial availability for initial substrate binding, and allow the following oxidative addition to kick start. While bulky monophosphine ligands would generate the coordinative unsaturated Pd complex, they have limited adjustability for stabilizing the highly active yet unstable low-valent and low-coordinate Pd metal center, especially at a high reaction temperature or under prolonged reaction time.[1a-1c, 17] The decomposition of this labile Pd complex often leads to the formation of inactive Pd black and therefore suppresses the ultimate catalytic performance. In order to deal with this problematic issue, we are interested to develop a coping strategy of crafting a weakly coordinating group to the ligand skeleton. The proximal hard donor moiety is conceivable to realize the complementary stabilization of the highly active palladium center and hence a better Pd catalyst longevity would be resulted.[18]
Hemilabile P, O-type ligand embodies both hard oxygen and soft phosphorus donor atoms.[19] The hard oxygen atom weakly binds to the soft Pd center in providing extra metal complex stability and also allows possible dissociation for offering vacant site whenever needed.[17, 19-20] The first successful example of P, O-type ligand for promoting Suzuki-Miyaura coupling of aryl chlorides was reported by Guram/Bei and co-workers in 1999 (Figure 1).[21] Later, a ferrocenyl ligand embedded an acetal group was disclosed by Hor and co-workers, [22] in which this ligand was found to be effective in the cross-coupling of aryl chlorides (Figure 1). The X-ray crystal structures of Guram/Bei's and Hor's Pd-ligand complexes were shown to have P—Pd—O coordination. Our research group also expressed interest in developing new series of P, O-type ligand bearing hemilabile amido group for cross-coupling reaction.[23] These ligands exhibited high efficacy in aromatic C—C and C—N bond-forming processes even under low catalyst loading. An independent investigation of P, O-type hemilabile ligands bearing amido group was also reported by Dai and co-workers in 2004.[24] Such series of ligands were later modified by a self-assisted molecular editing process. In 2009, McNulty and co-workers[25] reported a new trialkylphosphine associated with a proximal hydroxyl group for cross-coupling of aryl chlorides. An interesting ligand effect was postulated that the remote 4-hydroxyl substituent was suggested to stabilize the Pd complex in a boat conformation, while it could be easily isomerized to regenerate the active monodentate Pd complex in a chair conformer. Recently, an optically active ferrocenyl ligand was reported by Lang and co-workers[26] for tackling the difficult steric hindered biaryl synthesis under Suzuki- Miyaura reaction conditions. Indeed, it is highly desirable if the tailor-made phosphines can be easily accessed and fine-tuned in a cost-effective and step-economical synthetic pathway. Inspired by our previous success in dealing with challenging cross-coupling reactions, [27] we are interested to further explore new P, O-type indolylphosphine ligands for Suzuki-Miyaura cross-coupling of aryl chlorides.
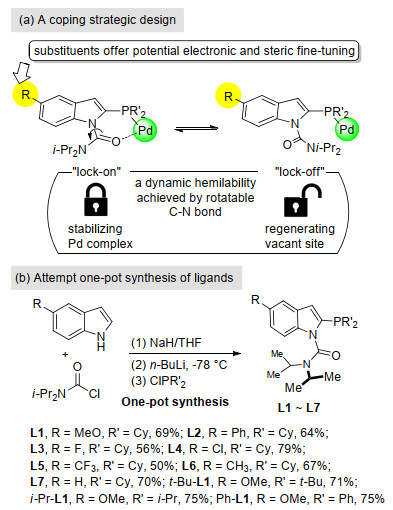
The newly designed ligand entities are illustrated to have a "lock-on & lock-off" switching ability, in which they are envisioned to offer awaiting coordination and thus to enhance catalyst longevity of the resulting unsaturated Pd complex (Figure 2a). The P donor electron-richness can be easily tuned to reach synergistic outcome by altering the substituent at the 5-position of indole core. We attempted to prepare a variety of 5-substituted indolylphosphines using a one-pot multi-step strategy (Figure 2b). These new ligands were directly assembled from readily available and inexpensive starting materials. Direct N—H deprotonation of commercially available 5-substituted indoles were attained by sodium hydride and the main ligand scaffolds were then generated by subsequent reaction with N, N- diisopropylcarbamoyl chloride. The desired ligands were afforded in good yields by the following lithiation and trapping with chlorodiphenylphosphine or chlorodialkylphosphines. It should be noted that the purification of target ligands was straightforwardly achieved by easily washing with methanol. No tedious crystallization process or column chromatographic purification step was required. Hence the possible industrial process synthesis is noted to be practicable.
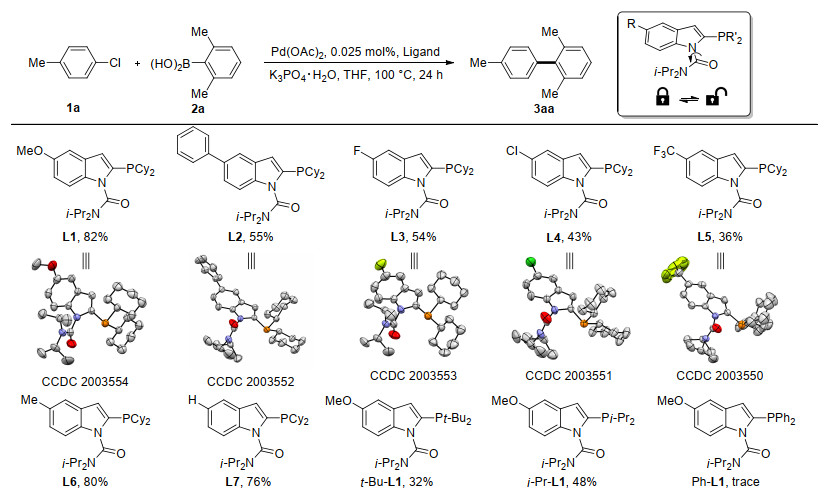
With the new P, O-type indolylphosphine ligands in hand, we first evaluated their efficacy in Suzuki-Miyaura reaction using non-activated 4-chlorotoluene and steric hindered 2, 6-dimethylphenylboronic acid as benchmark substrates (Scheme 1). It is interesting to show that the remote 5-phenyl group of the ligand had negative effect towards the cross-coupling efficiency (L2 vs. L7). The electron-withdrawing 5-halo-substuituted groups even further suppressed the reactivity (L3 and L4 vs. L7). These inferior results are probably due to the diminishing of electron-richness of P donor atom and hence decreasing the rate of O.A. of electrophile. Therefore, it is not surprised that the stronger electron-withdrawing 5-trifluoromethyl group afforded even a lower product yield (L5). On con- trary to the electron-deficient substituents, the incorporation of electron-donating 5-methyl or 5-methoxyl group increased the efficiency of the ligands (L1 and L6). The electron-rich moiety enhanced the election-richness of P donor and provided a stronger σ-donation to C—Cl σ* anti-bonding orbital, leading to the ease of bond breaking. The less steric bulky and electron-rich i-Pr-L1 and Ph-L1 showed decreasing trend on the activity when compared to L1. Nevertheless, further increase of the steric bulkiness and electron-richness did not give a better result (t-Bu-L1). To our delight, single crystals of ligands L1 to L5 were obtained for X-ray crystallographic analysis (Scheme 1). The carboamoyl oxygen points in a perpendicular direction towards the indole plane.
Reaction conditions: 4-chlorotoluene (1.0 mmol), 2, 6-dimethylphenylboronic acid (1.5 mmol), Pd:ligand (1:3), K3PO4•H2O (3 mmol), and THF (3 mL) were stirred at 100 ℃ for 24 h under N2. Isolated yields of 3aa were reported full-stop
Having identified the effective ligand, we next surveyed the substrate scope of aryl chlorides by employing the newly developed Pd/L1 catalyst system (Table 1). Non- activated aryl chlorides were found applicable under the catalyst loading of 0.025 mol% (Table 1, Entry 1). The catalyst loading for the activated aryl chlorides were able to be further reduced to 0.01 mol% in achieving good-to- excellent product yields (Table 1, Entries 7~9). To the best of our knowledge, L1 is one the most efficient P, O- type ligands for general Suzuki-Miyaura cross-coupling of aryl chlorides. Keto, aldehydo, ester and nitrile functionality were all well-tolerated under these reaction conditions (Table 1, Entries 5~9).
 下载:
导出CSV
下载:
导出CSV
 |
|||||
| Entry | ArCl | Ar'B(OH)2 | Product | Pd/mol% | Yieldb/% |
| 1 |  |
 |
 |
0.025 | 82 |
| 2 |  |
 |
 |
0.05 | 93 |
| 3 |  |
 |
 |
0.05 | 83 |
| 4 |  |
 |
 |
0.05 | 87 |
| 5 |  |
 |
 |
0.02 | 90 |
| 6 |  |
 |
 |
0.02 | 93 |
| 7 |  |
 |
 |
0.01 | 87 |
| 8 |  |
 |
 |
0.01 | 94 |
| 9 |  |
 |
 |
0.01 | 83 |
| a Reaction conditions: aryl chlorides (1.0 mmol), arylboronic acid (1.5 mmol), Pd:L1 (1:3), K3PO4•H2O (3 mmol), and THF (3 mL) were stirred at 100 ℃ for 24 h under N2. b Isolated yields were reported. Reaction times were not optimized for each substrate. | |||||
To further probe the generality of L1, a range of heteroaryl chlorides were then examined (Table 2). In fact, heteroaryl chlorides having adjacent nitrogen donor atom generally have deleterious to the Pd complex, in which the catalyst is rendered coordinative saturated. This situation becomes more vital especially under a very low catalyst concentration. Gratifyingly, P, O-type L1 still maintained comparable efficacy as the cases of simple chloroarenes. Pyridyl and quinolyl chlorides were applicable under these optimal reaction conditions (Table 2, Entries 1~5). It is worthy to note that free NH indole substrate was coupled smoothly under 0.05 mol% catalyst loading leading to 91% product yield (Table 2, Entry 6). No competitive C—N bond formation was observed for this entry. It is believed that the dynamic hemilability of L1 provides transient "lock-on" coordination to avoid excessive substrate binding scenario of the Pd complex by nitrogen-containing coupling partners.
下载:
导出CSV
 |
|||||
| Entry | ArCl | Ar'B(OH)2 | Product | Pd/mol% | Yieldb/% |
| 1 |  |
 |
 |
0.02 | 93 |
| 2 |  |
 |
 |
0.01 | 97 |
| 3 |  |
 |
 |
0.01 | 94 |
| 4 |  |
 |
 |
0.01 | 95 |
| 5 |  |
 |
 |
0.01 | 85 |
| 6 |  |
 |
 |
0.05 | 91 |
| a Reaction conditions: heteroaryl chlorides (1.0 mmol), arylboronic acid (1.5 mmol), Pd:L1 (1:3), K3PO4•H2O (3 mmol), and THF (3 mL) were stirred at 100 ℃ for 24 h under N2. b Isolated yields were reported. Reaction times were not optimized for each substrate. | |||||
In summary, we have established a facile synthetic pathway for assembling new P, O-type indolyl phosphines. This one-pot procedure allowed us to utilize readily available and inexpensive starting materials directly. By employing the newly developed Pd/L1 catalyst system, the Suzuki-Miyaura coupling of aryl/heteroaryl chlorides proceeded well. Particularly noteworthy is that the catalyst loading was found able to reach 0.01 mol% Pd. This record represents one of the lowest catalyst loadings achieved so far of P, O-type ligand for general Suzuki-Miyaura coupling of aryl chlorides. Further investigations and applications of this ligand series for tackling difficult and unexplored coupling processes are underway.
Unless otherwise noted, all the reagents were purchased from commercial suppliers and used without further purification. Tetrahydrofuran (THF) was freshly distilled from sodium and sodium benzophenone ketyl under nitrogen.[28] All bases were used as received without grinding. A new bottle of n-butyllithium was used (note: since the concentration of n-BuLi from old bottles may vary, a titration is highly recommended prior to use). All Pd-catalyzed cross-coupling reactions were performed in a resealable screw-capped Schlenk flask (approx. volume 20 mL) in the presence of a Teflon-coated magnetic stir bar. Thin-layer chromatography (TLC) was performed on precoated silica gel 60 F254 plates. Silica gel (70~230 and 230~400 mesh) was used for flash column chromatography. Melting points were recorded on an uncorrected instrument. 1H NMR spectra were recorded on a 400 MHz spectrometer. Spectra were referenced internally to the residual proton resonance in CDCl3 (δ 7.26) or to tetramethylsilane (TMS) (δ 0.00) as an internal standard. Chemical shifts (δ) were reported downfield from TMS. 13C NMR spectra were recorded on a 100 MHz spectrometer, and the spectra were referenced to CDCl3 (δ 77.0, the middle peak). 31P NMR spectra were referenced to the external 85% H3PO4. Mass spectrometry (EI-MS and ESI-MS) was performed on a mass spectrometer. High-resolution mass spectrometry (HRMS) was performed on a Q Exactive Focus Orbitrap mass spectrometer with an atmospheric-pressure chemical ionization (APCI) source. GC-MS analysis was conducted on a GCD system using a 30 m×0.25 mm HP-5MS column. Product GC yields were obtained on the basis of authentic sample/dodecane calibration standards from the GC-FID system.
2-(Dicyclohexylphosphino)-N, N-diisopropyl-5-meth- oxy-1H-indole-1-carboxamide (L1): The synthetic conditions of L1 were slightly modified from the literature.[23c] 5-Methoxy-1H-indole (10 mmol, 1.47 g) was dissolved in anhydrous THF (20 mL) and added dropwise to the flask charged with THF (100 mL) solution with NaH (60% in mineral oil, 11 mmol, 3.0 g). The reaction was allowed to stir for 30 min at room temperature. After cooling to 0 ℃, N, N-diisopropylcarbamoyl chloride in THF (20 mL) was added dropwise and the reaction mixtures were allowed to stir for another 2 h at r.t. After the completion of the reaction as confirmed by GC-MS analysis, the reaction mixtures were cooled to -78 ℃. n-BuLi (11 mmol) was added dropwise and the resulting mixtures were stirred for 1 h. Then the chlorodicyclohexylphosphine was added dropwise and the reaction was allowed to reach room temperature and stirred overnight. MeOH (15 mL) was added to quench the reaction. After the removing of solvent, dichloromethane (DCM) (350 mL) was added to the mixture, and washed by water (150 mL×3) and brine (150 mL). The organic aliquot was dried with MgSO4 and then concentrated to a saturated solution. The cold MeOH was added and the precipitates were generated. Simple filtration was carried out to afford the desired product L1 as a white solid (3.55 g, 69% yield). m.p. 142.3~142.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.24~1.92 (m, 34H), 3.43~3.52 (m, 2H), 3.87 (d, J=10.7 Hz, 1H), 6.67 (s, 1H), 6.89 (d, J=6.6 Hz, 1H), 7.08 (d, J=2.2 Hz, 1H), 7.21 (d, J=8.8 Hz, 1H), 7.23 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 20.3, 20.5, 20.7, 20.8, 26.3, 26.5, 27.1, 27.2, 28.9, 29.0, 29.2, 29.8, 30.0, 30.1, 33.8, 55.6, 101.4, 109.3, 109.4, 111.1, 113.0, 128.3, 132.2, 135.1, 135.3, 151.7, 154.6 (complex unresolved C—P splitting was observed); 31P NMR (202 MHz, CDCl3) δ: -21.84; HRMS calcd for C28H44N2O2P (M+H+): 471.3140, found 471.3150.
2-(Dicyclohexylphosphino)-N, N-diisopropyl-5-phenyl- 1H-indole-1-carboxamide (L2): Followed the general procedure of synthesis of L1. 5-Phenyl-1H-indole was used to obtain the desired product as a white solid (3.59 g, 64% yield). m.p. 188.1~189.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.26~1.94 (m, 34H), 3.20~3.60 (m, 2H), 6.80 (s, 1H), 7.33~7.39 (m, 2H), 7.47 (t, J=6.3 Hz, 3H), 7.67 (d, J=7.7 Hz, 2H), 7.83 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 20.7, 21.0, 26.3, 27.2, 30.0, 30.1, 33.9, 110.1, 110.5, 118.7, 122.5, 126.3, 127.2, 128.4, 128.6, 134.1, 135.6, 135.8, 136.4, 142.2, 151.5 (complex unresolved C—P splitting was observed); 31P NMR (202 MHz, CDCl3) δ: -21.90; HRMS calcd for C33H46N2OP (M+ H+): 517.3348, found 517.3355.
2-(Dicyclohexylphosphino)-5-fluoro-N, N-diisopropyl- 1H-indole-1-carboxamide (L3): Followed the general procedure of synthesis of L1. 5-Fluoro-1H-indole was used to obtain the desired product as a white solid (2.82 g, 56% yield). m.p. 157.3~159.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.24~1.92 (m, 34H), 3.42 (bs, 2H), 6.70 (s, 1H), 6.95~7.00 (m, 1H), 7.21~7.28 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 20.5, 21.0, 26.2, 27.1, 29.9, 30.0, 30.1, 33.7, 104.8, 105.0, 105.1, 109.4, 109.5, 110.7, 110.9, 111.0, 128.1, 128.2, 133.4, 133.5, 136.7, 136.9, 151.3, 157.0, 159.3 (complex unresolved C—P splitting was observed); 31P NMR (202 MHz, CDCl3) δ: -21.56; HRMS calcd For C27H41N2OFP (M+H+): 459.2941, found 459.2956.
5-Chloro-2-(dicyclohexylphosphino)-N, N-diisopropyl- 1H-indole-1-carboxamide (L4): Followed by the procedure of synthesis of L1. 5-Chloro-1H-indole was used to obtain the desired product as a white solid (4.11 g, 79% yield). m.p. 169.1~171.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.24~1.99 (m, 34H), 3.46~3.51 (m, 2H), 6.68 (s, 1H), 7.15~7.30 (m, 2H), 7.59~7.64 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 20.6, 21.1, 26.2, 27.1, 29.9, 30.0, 33.7, 109.0, 111.1, 119.6, 122.7, 126.0, 128.8, 135.1, 136.5, 136.7, 151.0 (complex unresolved C—P splitting was observed); 31P NMR (202 MHz, CDCl3) δ: -21.64; HRMS calcd for C27H41N2OClP (M+H+): 475.2645, found 459.2651.
2-(Dicyclohexylphosphino)-N, N-diisopropyl-5-(trifluo- romethyl)-1H-indole-1-carboxamide (L5): Followed the general procedure of synthesis of L1. 5-(Trifluoromethyl)- 1H-indole was used to obtain the desired product as a white solid (2.75 g, 50% yield). m.p. 191.1~192.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 0.91~2.20 (m, 34H), 3.31~3.61 (m, 2H), 6.85 (s, 1H), 7.39~7.48 (m, 2H), 7.95 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 20.3, 20.7, 20.8, 21.0, 26.2, 26.5, 26.8, 27.2, 27.3, 27.4, 28.9, 29.0, 30.1, 30.2, 30.4, 33.7, 110.3, 110.5, 118.1, 118.2, 119.1, 122.7, 123.0, 123.8, 126.5, 127.2, 137.4, 137.6, 138.0, 150.8 (complex unresolved C—P splitting was observed); 31P NMR (202 MHz, CDCl3) δ: -21.79; HRMS calcd for C28H41N2O- F3P (M+H+): 509.2909, found 509.2914.
2-(Dicyclohexylphosphino)-N, N-diisopropyl-5-methyl- 1H-indole-1-carboxamide (L6): Followed the general procedure of synthesis of L1. 5-Methyl-1H-indole was used to obtain the desired product as a white solid (3.34 g, 67% yield). m.p. 162.3~163.1 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.44~1.76 (m, 30H), 1.80~2.05 (m, 2H), 2.31~2.33 (m, 2H), 2.47 (s, 3H), 3.48~3.53 (m, 2H), 6.69 (s, 1H), 7.04 (d, J=9.0 Hz, 1H), 7.22 (d, J=8.4 Hz, 1H), 7.40 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 20.5, 20.8, 21.2, 21.3, 25.6, 26.0, 26.3, 26.4, 26.7, 30.6, 30.8, 37.1, 37.4, 37.5, 37.8, 48.5, 48.7, 108.7, 108.8, 110.0, 119.8, 119.9, 120.2, 124.1, 128.2, 129.6, 135.0, 135.1, 137.5, 137.7, 151.8 (complex unresolved C—P splitting was observed); 31P NMR (202 MHz, CDCl3) δ: -25.20; HRMS calcd for C28H44N2OP (M+H+): 455.3191, found 455.3208.
2-(Di-tert-butylphosphino)-N, N-diisopropyl-5-methoxy-1H-indole-1-carboxamide (t-Bu-L1): Followed the general procedure of synthesis of L1. Di-tert-butylchloropho- sphine was used to obtain the desired product as a white solid (3.29 g, 71% yield). m.p. 128.6~129.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.11~1.70 (m, 30H), 3.60~3.75 (m, 2H), 3.87 (s, 3H), 6.87~6.95 (m, 2H), 7.08 (d, J=2.5 Hz, 1H), 7.18~7.22 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 20.8, 21.2, 30.5, 30.6, 32.8, 33.0, 48.5, 55.6, 101.6, 102.7, 104.5, 111.3, 111.7, 112.9, 113.3, 113.6, 125.7, 128.6, 129.5, 130.9, 132.2, 132.3, 136.0, 136.2, 151.2, 154.6, 155.1 (complex unresolved C—P splitting was observed); 31P NMR (202 MHz, CDCl3) δ: 9.43; HRMS calcd for C24H40N2O2P (M+H+): 419.2827, found 419.2840.
2-(Diisopropylphosphino)-N, N-diisopropyl-5-methoxy-1H-indole-1-carboxamide (i-Pr-L1): Followed the general procedure of synthesis of L1. Chlorodiisopropylphosphine was used to obtain the desired product as a white solid (3.26 g, 75% yield). m.p. 110.1~111.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.11~1.60 (m, 26H), 2.18 (s, 2H), 3.45~3.51 (m, 2H), 3.87 (s, 3H), 6.67 (d, J=0.4 Hz, 1H), 6.87~6.90 (m, 1H), 7.06 (d, J=2.4 Hz, 1H), 7.21 (d, J=8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 19.7, 19.9, 20.6, 24.1, 55.7, 101.6, 109.3, 109.4, 111.1, 113.2, 128.3, 132.2, 132.3, 135.4, 135.6, 151.2, 154.6 (complex unresolved C—P splitting was observed); 31P NMR (202 MHz, CDCl3) δ: -13.36; HRMS calcd for C22H36N2O2P (M+ H+): 391.2514, found 391.2499.
2-(Diphenylphosphino)-N, N-diisopropyl-5-methoxy-1H-indole-1-carboxamide (Ph-L1) Followed by the procedure of synthesis of L1. Chlorodiphenylphosphine was used to obtain a desired product as white solid (3.27 g, 65% yield). m.p. 186.1~187.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.34 (d, J=6.4 Hz, 12H), 3.50~3.57 (m, 2H), 3.83 (s, 3H), 6.23 (s, 1H), 6.89~6.91 (m, 1H), 6.97 (d, J=2.4 Hz, 1H), 7.23 (d, J=8.8 Hz, 1H), 7.34~7.45 (m, 10H); 13C NMR (100 MHz, CDCl3) δ: 20.6, 20.7, 21.2, 48.5, 48.8, 55.6, 55.7, 102.0, 111.3, 112.4, 113.6, 125.7, 128.3, 128.4, 128.5, 128.8, 133.1, 133.2, 133.4, 133.6, 135.9, 136.0, 136.7, 136.8, 151.2, 154.8 (complex unresolved C—P splitting was observed); 31P NMR (202 MHz, CDCl3) δ: -27.76; HRMS calcd for C28H32N2O2P (M+H+): 459.2201, found 459.2216.
Pd(OAc)2 (2.3 mg, 0.010 mmol) and ligand (0.030 mmol) were loaded into an oven-dried Schlenk tube equipped with a Teflon-coated magnetic stir bar. The tube was evacuated and flushed with nitrogen for three cycles. Precomplexation was applied by adding freshly distilled THF into the tube for stirring about 1 to 2 min until all the metal and ligand were dissolved in the solvent. Aryl chloride (1.0 mmol), arylboronic acid (1.5 mmol), and K3PO4• H2O (3.0 mmol) were loaded into another Schlenk tube, and the system was further evacuated and flushed with nitrogen for three times. The corresponding amount of precomplexation solution was added, following by the addition of THF to final volume of 3 mL. The tube was stirred at room temperature for 5 min and then placed into a preheated oil bath (100 ℃) for 24 h. After the completion of reaction, the reaction tube was allowed to cool to room temperature and quenched with water and diluted with EtOAc. The organic layer was separated and the aqueous layer was washed with diethyl ether twice. The combined organic layer was concentrated under reduced pressure. The crude products were purified by flash column chromatography on silica gel (230~400 mesh) to afford the desired product.
2, 4', 6-Trimethyl-1, 1'-biphenyl (3aa; Table 1, Entry 1)[29] Rf=0.5 (pure hexane); 1H NMR (400 MHz, CDCl3) δ: 2.10 (s, 6H), 2.47 (s, 3H), 7.09 (d, J=8.0 Hz, 2H), 7.15~7.19 (m, 3H), 7.29 (d, J=8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 20.8, 20.8, 21.2, 21.2, 126.9, 127.2, 128.8, 129.1, 135.9, 136.1, 138.0, 141.8.
2-Methoxy-2'-methyl-1, 1'-biphenyl (Table 1, Entries 2 and 3):[30] Rf=0.3 (EtOAc/Hexane, V:V=1:50); 1H NMR (400 MHz, CDCl3) δ: 2.23 (s, 3H), 3.84 (s, 3H), 7.04~7.12 (m, 2H), 7.23~7.35 (m, 5H), 7.41~7.43 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 19.8, 19.8, 55.2, 110.6, 120.4, 125.3, 127.2, 128.5, 129.5, 129.9, 130.8, 130.9, 136.7, 138.6, 156.5.
4'-Methoxy-2-methyl-1, 1'-biphenyl (Table 1, Entry 4):[31] Rf=0.2 (EtOAc/Hexane, V:V=1:50); 1H NMR (400 MHz, CDCl3) δ: 2.38 (s, 3H), 3.88 (s, 3H), 7.05 (d, J=9.2 Hz, 2H), 7.32~7.36 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 20.5, 55.2, 113.5, 125.7, 127.0, 129.9, 130.2, 130.3, 134.4, 135.5, 141.6, 158.3.
[1, 1'-Biphenyl]-3-carbonitrile (Table 1, Entry 5):[32] Rf=0.4 (EtOAc/Hexane, V:V=1:50); 1H NMR (400 MHz, CDCl3) δ: 7.31~7.53 (m, 6H), 7.67~7.71 (m, 1H), 7.74 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 112.8, 118.6, 126.9, 128.3, 129.0, 129.5, 130.5, 130.5, 131.9, 138.7, 142.3.
1-(3', 5'-Dimethyl-[1, 1'-biphenyl]-3-yl)ethan-1-one (Table 1, Entry 6):[23c] Rf=0.3 (pure hexane); 1H NMR (400 MHz, CDCl3) δ: 2.41 (s, 6H), 2.67 (s, 3H), 7.05 (s, 1H), 7.25 (s, 2H), 7.52 (d, J=12.4 Hz, 1H), 7.79 (d, J=8.4 Hz, 1H), 7.93 (d, J=8.4 Hz, 1H), 8.20 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 21.2, 26.6, 124.4, 126.8, 126.9, 128.8, 129.3, 131.6, 137.4, 138.9, 140.0, 141.8, 198.0.
1-(2'-Methoxy-[1, 1'-biphenyl]-4-yl)ethan-1-one (Table 1, Entry 7):[33] Rf=0.5 (EtOAc/Hexane, V:V=1:9); 1H NMR (400 MHz, CDCl3) δ: 2.61 (s, 3H), 3.80 (s, 3H), 6.97~7.03 (m, 2H), 7.30~7.34 (m, 2H), 7.62 (d, J=4.0 Hz, 2H), 7.98 (d, J=6.4 Hz); 13C NMR (100 MHz, CDCl3) δ: 26.5, 55.5, 111.3, 120.9, 128.0, 129.4, 129.4, 130.6, 135.4, 143.5, 156.4, 197.7.
2'-Methyl-[1, 1'-biphenyl]-4-carbaldehyde (Table 1, Entry 8)[23c] Rf=0.5 (EtOAc/Hexane, V:V=1:9); 1H NMR (400 MHz, CDCl3) δ: 2.30 (s, 3H), 7.24~7.31 (m, 4H), 7.50 (d, J=6.4 Hz, 2H), 7.94 (d, J=6.4 Hz, 2H), 10.07 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 20.2, 125.1, 127.3, 129.3, 129.4, 129.8, 130.5, 134.5, 134.9, 140.4, 148.2, 191.8.
Methyl 2'-methyl-[1, 1'-biphenyl]-3-carboxylate (Table 1, Entry 9):[17] Rf=0.3 (pure hexane); 1H NMR (400 MHz, CDCl3) δ: 2.30 (s, 3H), 3.97 (s, 3H), 7.24~7.31 (m, 4H), 7.43 (d, J=8.2Hz, 2H), 8.14 (d, J=8.2Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 20.6, 52.3, 126.1, 127.3, 128.3, 128.4, 129.5, 129.5, 130.4, 130.5, 130.6, 133.9, 135.5, 141.1, 142.5, 167.3.
3-(o-Tolyl)pyridine (Table 2, Entry 1):[23c] Rf=0.4 (EtOAc/Hexane, V:V=1:9); 1H NMR (400 MHz, CDCl3) δ: 2.27 (s, 3H), 7.22~7.30 (m, 5H), 7.58 (d, J=7.8Hz, 1H), 8.54~8.57 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 20.2, 122.9, 126.0, 128.6, 129.7, 130.5, 135.4, 136.3, 137.4, 138.0, 148.0, 149.6.
2-(o-Tolyl)quinoline (Table 2, Entry 2):[23c] Rf=0.5 (EtOAc/Hexane, V:V=1:9); 1H NMR (400 MHz, CDCl3) δ: 2.57 (s, 3H), 7.81~7.98 (m, 5H), 8.14~8.15 (m, 2H), 8.17 (d, J=8.4 Hz, 1H), 8.37 (d, J=6.4 Hz, 1H), 9.12 (d, J=4.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 19.3, 119.4, 125.1, 126.3, 126.7, 126.8, 127.9, 129.0, 129.5, 129.8, 135.8, 135.8, 138.5, 141.6, 160.8.
2-(Naphthalen-1-yl)pyridine (Table 2, Entry 3):[34] Rf=0.3 (pure hexane); 1H NMR (400 MHz, CDCl3) δ: 7.51 (m, 1H), 7.52~7.57 (m, 6H), 7.57~7.65 (m, 2H), 7.75 (m, 1H), 7.92~7.94 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 121.7, 124.8, 125.0, 125.4, 125.6, 126.2, 127.2, 128.1, 128.6, 130.9, 133.7, 136.1, 138.3, 149.2, 159.0.
2-(2-Methoxyphenyl)pyridine (Table 2, Entry 4)[35] Rf=0.4 (EtOAc/Hexane, V:V=1:9); 1H NMR (400 MHz, CDCl3) δ: 3.81 (s, 3H), 6.97~7.17 (m, 1H), 7.33~7.37 (m, 1H), 7.64~7.67 (m, 1H), 7.78~7.82 (m, 1H), 8.69~8.70 (m, 3H), 8.70 (d, J=1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 55.3, 111.2, 120.8, 121.4, 124.9, 128.7, 130.9, 135.3, 149.2.
3-(2-Methoxyphenyl)pyridine (Table 2, Entry 5):[36] Rf=0.4 (EtOAc/Hexane, V:V=1:9); 1H NMR (400 MHz, CDCl3) δ: 3.81 (s, 3H), 6.97~7.17 (m, 4H), 7.33~7.36 (m, 1H), 7.64~7.67 (m, 1H), 7.78~7.82 (m, 1H), 8.64~8.70 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 55.3, 111.2, 120.8, 121.4, 124.9, 128.3, 129.7, 130.5, 135.3, 143.1, 155.9, 156.7.
5-(o-Tolyl)-1H-indole (Table 2, Entry 6):[9b] Rf=0.3 (EtOAc/Hexane, V:V=1:9); 1H NMR (400 MHz, CDCl3) δ: 2.34 (s, 3H), 6.61~6.62 (m, 1H), 7.16~7.29 (m, 2H), 7.29~7.35 (m, 5H), 7.43~7.62 (m, 1H), 8.35 (bs, J=8.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 20.6, 29.7, 102.4, 102.8, 110.4, 111.9, 120.1, 121.1, 122.3, 129.8, 124.6, 125.5, 125.6, 126.7, 127.8, 130.1, 130.3, 133.6, 134.8, 135.7, 143.1.
Supporting Information 1H NMR, 13C NMR original spectra of the target compounds and the X-ray crystallographic analysis of ligand L1~L7. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
For selected books, see:
(a) Negishi, E. Handbook of Organopalladium Chemistry for Organic Synthesis, Vols. 1~2, Wiley-Interscience, New York, 2002.
(b) de Meijere, A.; Diederich, F. Metal-Catalyzed Cross-Coupling Reactions, Vols. 1~2, Wiley-VCH, Weinheim, 2004.
(c) Ackermann, L. Modern Arylation Methods, Wiley-VCH, Weinheim, 2009. For selected reviews, see:
(d) Corbet, J.-P.; Mignani, G. Chem. Rev. 2006, 106, 2651.
(e) Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V. Angew. Chem., Int. Ed. 2012, 51, 5062.
(f) Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Chem. Rev. 2018, 118, 2249.
(g) Zhang, Y.-F.; Shi, Z.-J. Acc. Chem. Res. 2019, 52, 161.
(a) Magano, J.; Dunetz, J. R. Chem. Rev. 2011, 111, 2177.
(b) Biteau, N.; Hervin, V.; Roy, V.; Agrofoglio, L. A. In Palladium-Catalyzed Modification of Nucleosides, Nucleotides and Oligonucleotides, Vol. 1, Eds.: Kapdi, A. R.; Maiti, D.; Sanghvi, Y. S., Elsevier, Amsterdam, Netherlands, 2018, p. 37.
(c) Devendar, P.; Qu, R.-Y.; Kang, W.-M.; He, B.; Yang, G.-F. J. Agric. Food Chem. 2018, 66, 8914.
For selected book, see:
(a) Stradiotto, M.; Lundgren, R. J. Ligand Design in Metal Chemistry, John Wiley & Sons, Hoboken, NJ, 2016. For selected articles, see:
(b) Valente, C.; Calimsiz, S.; Hoi, K. H.; Mallik, D.; Sayah, M.; Organ, M. G. Angew. Chem., Int. Ed. 2012, 51, 3314.
(c) Izquierdo, F.; Manzini, S.; Nolan, S. P. Chem. Commun. 2014, 50, 14926.
(d) Wong, S. M.; Yuen, O. Y.; Choy, P. Y.; Kwong, F. Y. Coord. Chem. Rev. 2015, 293-294, 158.
(e) Li, C.; Chen, D.; Tang, W. Synlett 2016, 2183.
Fu, G. C. Acc. Chem. Res. 2008, 41, 1555. doi: 10.1021/ar800148f
Hartwig, J. F. Synlett 1997, 329.
(a) Surry, D. S.; Buchwald, S. L. Angew. Chem., Int. Ed. 2008, 47, 6338.
(b) Martin, R.; Buchwald, S. L. Acc. Chem. Res. 2008, 41, 1461.
Ackermann, L.; Potukuchi, H. K.; Althammer, A.; Born, R.; Mayer, P. Org. Lett. 2010, 12, 1004. doi: 10.1021/ol1000186
Zapf, A.; Beller, M. Chem. Commun. 2005, 431.
(a) So, C. M.; Lau, C. P.; Kwong, F. Y. Angew. Chem., Int. Ed. 2008, 47, 8059.
(b) So, C. M.; Chow, W. K.; Choy, P. Y.; Lau, C. P.; Kwong, F. Y. Chem.-Eur. J. 2010, 16, 7996.
Lundgren, R. J.; Hesp, K. D.; Stradiotto, M. Synlett 2011, 2443.
(a) Tang, W.; Capacci, A. G.; Wei, X.; Li, W.; White, A.; Patel, N. D.; Savoie, J.; Gao, J. J.; Rodriguez, S.; Qu, B.; Haddad, N.; Lu, B. Z.; Krishnamurthy, D.; Yee, N. K.; Senanayake, C. H. Angew. Chem., Int. Ed. 2010, 49, 5879.
(b) Zhao, Q.; Li, C.; Senanayake, C. H.; Tang, W. Chem.-Eur. J. 2013, 19, 2261.
Liu, D.; Gao, W.; Dai, Q.; Zhang, X. Org. Lett. 2005, 7, 4907. doi: 10.1021/ol051844w
Li, P.; Lu, B.; Fu, C.; Ma, S. Adv. Synth. Catal. 2013, 355, 1255. doi: 10.1002/adsc.201300207
Handa, S.; Andersson, M. P.; Gallou, F.; Reilly, J.; Lipshutz, B. H. Angew. Chem., Int. Ed. 2016, 55, 4914. doi: 10.1002/anie.201510570
(a) Bhilare, S.; Gayakhe, V.; Ardhapure, A. V.; Sanghavi, Y. S.; Schulzke, C.; Borozdina, Y.; Kapdi, A. R. RSC Adv. 2016, 6, 83820.
(b) Girase, T. R.; Kapdi, A. R. Chem. Asian J. 2019, 14, 2611.
For Hiesro and Doucet's selected reference, see:
(a) Hierso, J.-C.; Fihri, A.; Amardeil, R.; Meunier, P.; Doucet, H.; Santelli, M.; Donnadieu, B. Organometallics 2003, 22, 4490. For Hoshi and Hagiwara's selected reference, see:
(b) Hoshi, T.; Nakazawa, T.; Saitoh, I.; Mori, A.; Suzuki, T.; Sakai, J.-I.; Hagiwara, H. Org. Lett. 2008, 10, 2063. For Yu and Liu's selected reference, see:
(c) Mao, S.-L.; Sun, Y.; Yu, G.-A.; Zhao, C.; Han, Z.-J.; Yuan, J.; Zhu, X.; Yang, Q.; Liu, S.-H. Org. Biomol. Chem. 2012, 10, 9410. For Doherty's selected reference, see:
(d) Doherty, S.; Smyth, C. H.; Knight, J. G.; Hashmi, S. A. K. Nat. Protocols 2012, 7, 1870. For Fossey's selected reference, see:
(e) Zhao, Y.; van Nguyen, H.; Male, L.; Craven, P.; Buckley, B. R.; Fossey, J. S. Organometallics 2018, 37, 4224.
Wong, S. M.; So, C. M.; Chung, K. H.; Lau, C. P.; Kwong, F. Y. Eur. J. Org. Chem. 2012, 4172.
For selected references, see:
(a) Wang, Z.; Guo, W. In Homogeneous Catalysis for Unreactive Bond Activation, Ed.: Shi, Z., John Wiley & Sons, Hoboken, NJ, 2015, p. 1.
(b) Braunstein, P.; Naud, F. Angew. Chem., Int. Ed. 2001, 40, 680.
(c) Weng, Z.; Teo, S.; Hor, T. S. A. Acc. Chem. Res. 2007, 40, 676.
(d) Zhang, W.-H.; Chien, S. W.; Hor, T. S. A. Coord. Chem. Rev. 2011, 255, 1991.
(e) Guram, A. S. Org. Process Res. Dev. 2016, 20, 1754.
Kwong, F. Y.; Chan, A. S. C. Synlett 2008, 1440.
(a) Old, D. W.; Wolfe, J. P.; Buchwald, S. L. J. Am. Chem. Soc. 1998, 120, 9722.
(b) Amatore, C.; Fuxa, A.; Jutand, A. Chem.-Eur. J. 2000, 6, 1474.
(a) Bei, X.; Crevier, T.; Guram, A. S.; Jandeleit, B.; Powers, T. S.; Turner, H. W.; Uno, T.; Weinberg, W. H. Tetrahedron Lett. 1999, 40, 3855.
(b) Bei, X.; Turner, H. W.; Weinberg, W. H.; Guram, A. S.; Petersen, J. L. J. Org. Chem. 1999, 64, 6797.
Teo, S.; Weng, Z.; Hor, T. S. A. Organometallics 2006, 25, 1199. doi: 10.1021/om050791j
(a) Kwong, F. Y.; Lam, W. H.; Yeung, C. H.; Chan, K. S.; Chan, A. S. C. Chem. Commun. 2004, 1922.
(b) Chen, G.; Lam, W. H.; Fok, W. S.; Lee, H. W.; Kwong, F. Y. Chem. Asian J. 2007, 2, 306.
(c) So, C. M.; Yeung, C. C.; Lau, C. P.; Kwong, F. Y. J. Org. Chem. 2008, 73, 7803.
(a) Dai, W.-M.; Li, Y.; Zhang, Y.; Lai, K. W.; Wu, J. Tetrahedron Lett. 2004, 45, 1999.
(b) Dai, W.-M.; Zhang, Y. Tetrahedron Lett. 2005, 46, 1377.
(c) Dai, W.-M.; Li, Y.; Zhang, Y.; Yue, C.; Wu, J. Chem.-Eur. J. 2008, 14, 5538.
Ullah, E.; McNulty, J.; Robertson, A. Tetrahedron Lett. 2009, 50, 5599. doi: 10.1016/j.tetlet.2009.07.088
(a) Schaarschmidt, D.; Lang, H. Eur. J. Inorg. Chem. 2010, 4811.
(b) Schaarschmidt, D.; Lang, H. ACS Catal. 2011, 1, 411.
(a) Chow, W. K.; So, C. M.; Lau, C. P.; Kwong, F. Y. J. Org. Chem. 2010, 75, 5109.
(b) Wong, P. Y.; Chow, W. K.; Chung, K. H.; So, C. M.; Lau, C. P.; Kwong, F. Y. Chem. Commun. 2011, 47, 8328.
(c) To, S. C.; Kwong, F. Y. Chem. Commun. 2011, 47, 5079.
(d) Chow, W. K.; Yuen, O. Y.; So, C. M.; Wong, W. T.; Kwong, F. Y. J. Org. Chem. 2012, 77, 3543.
(e) Leung, M. P.; Choy, P. Y.; Lai, W. I.; Gan, K. B.; Kwong, F. Y. Org. Process Res. Dev. 2019, 23, 1602.
Armarego, W. L. F. Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, UK, 2017.
Choy, P. Y.; Yuen, O. Y.; Leung, M. P.; Chow, W. K.; Kwong, F. Y. Eur. J. Org. Chem. 2020, 2846.
Lee, H. W.; Lam, F. L.; So, C. M.; Lau, C. P.; Chan, A. S. C.; Kwong, F. Y. Angew. Chem., Int. Ed. 2009, 48, 7436. doi: 10.1002/anie.200904033
Wu, Y.; Choy, P. Y.; Kwong, F. Y. Org. Biomol. Chem. 2014, 12, 6820. doi: 10.1039/C4OB01211A
Chung, K. H.; So, C. M.; Wong, S. M.; Luk, C. H.; Zhou, Z.; Lau, C. P.; Kwong, F. Y. Chem. Commun. 2012, 48, 1967. doi: 10.1039/c2cc15972d
McNulty, J.; Capretta, A.; Wilson, J.; Dyck, J.; Adjabeng, G.; Robertson, A. Chem. Commun. 2002, 1986.
So, C. M.; Lau, C. P.; Kwong, F. Y. Org. Lett. 2007, 9, 2795. doi: 10.1021/ol070898y
Mingji, D.; Liang, B.; Wang, C.; You, Z.; Xiang, J.; Dong, G.; Chen, J.; Yang, Z. Adv. Synth. Catal. 2004, 346, 1669. doi: 10.1002/adsc.200404165
Zhang, J.; Zhao, L.; Song, M.; Mak, T. C. W.; Wu, Y. J. Organomet. Chem. 2006, 691, 1301. doi: 10.1016/j.jorganchem.2005.11.027

Figure 1 Selected examples of hemilabile P, O-type ligands particularly for Pd-catalyzed Suzuki-Miyaura coupling of aryl chlorides and present study

Figure 2 Our rational ligand design and attempt one-pot synthesis of new P, O-type indolylphosphine ligands

Scheme 1 A survey of new P, O-type ligands for Suzuki-Miyaura coupling of aryl chlorides and the corresponding ORTEP representation of X-ray crystal structures (all hydrogens are omitted for clarity)
Reaction conditions: 4-chlorotoluene (1.0 mmol), 2, 6-dimethylphenylboronic acid (1.5 mmol), Pd:ligand (1:3), K3PO4•H2O (3 mmol), and THF (3 mL) were stirred at 100 ℃ for 24 h under N2. Isolated yields of 3aa were reported full-stop
Table 1. Palladium-catalyzed Suzuki-Miyaura coupling of aryl chloridesa
|
|||||
| Entry | ArCl | Ar'B(OH)2 | Product | Pd/mol% | Yieldb/% |
| 1 | |
|
|
0.025 | 82 |
| 2 | |
|
|
0.05 | 93 |
| 3 | |
|
|
0.05 | 83 |
| 4 | |
|
|
0.05 | 87 |
| 5 | |
|
|
0.02 | 90 |
| 6 | |
|
|
0.02 | 93 |
| 7 | |
|
|
0.01 | 87 |
| 8 | |
|
|
0.01 | 94 |
| 9 | |
|
|
0.01 | 83 |
| a Reaction conditions: aryl chlorides (1.0 mmol), arylboronic acid (1.5 mmol), Pd:L1 (1:3), K3PO4•H2O (3 mmol), and THF (3 mL) were stirred at 100 ℃ for 24 h under N2. b Isolated yields were reported. Reaction times were not optimized for each substrate. | |||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Palladium-catalyzed Suzuki-Miyaura coupling of heteroaryl chloridesa
|
|||||
| Entry | ArCl | Ar'B(OH)2 | Product | Pd/mol% | Yieldb/% |
| 1 | |
|
|
0.02 | 93 |
| 2 | |
|
|
0.01 | 97 |
| 3 | |
|
|
0.01 | 94 |
| 4 | |
|
|
0.01 | 95 |
| 5 | |
|
|
0.01 | 85 |
| 6 | |
|
|
0.05 | 91 |
| a Reaction conditions: heteroaryl chlorides (1.0 mmol), arylboronic acid (1.5 mmol), Pd:L1 (1:3), K3PO4•H2O (3 mmol), and THF (3 mL) were stirred at 100 ℃ for 24 h under N2. b Isolated yields were reported. Reaction times were not optimized for each substrate. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们