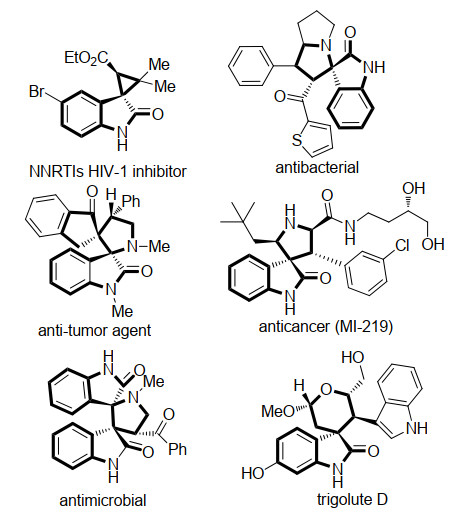
图 1.
含有螺环氧吲哚骨架的生物活性分子
Figure 1.
Biologically active molecules with spirooxindole skeleton

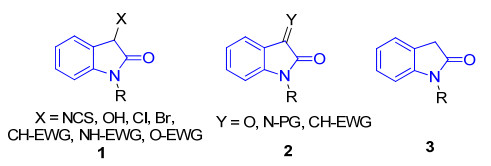
螺环化合物由于其广泛的生物学性质和药理学活性而受到越来越多合成和药物化学家的关注[1].其中, 高度官能化的螺环氧吲哚更是由于其显著和多样的生物活性而成为研究热点[2].如图 1所示, 这些具有多种螺环结构的氧吲哚骨架化合物, 已经被证明具有广泛的生物学性质和药理活性, 如抗感染、抗肿瘤、抗菌以及抗癌活性等[3].因此, 设计不对称催化合成结构多样的螺环氧吲哚化合物具有重要的意义.目前利用氧吲哚或者氧吲哚衍生物作为起始原料来构建螺环氧吲哚骨架被认为是最直接有效的方法[4].其中, 开发用于有机催化不对称合成螺环氧吲哚衍生物的基础合成子主要有三类(图 2).第一类是3-取代氧吲哚衍生物1, 作为启动串联反应试剂可与反应受体发生串联环化反应构建螺环氧吲哚骨架; 第二类是不饱和氧吲哚衍生物2, 如靛红、亚胺氧吲哚以及3-亚甲基氧吲哚等, 作为串联反应受体可与同时具有亲核及亲电位点的串联反应试剂发生连续的加成环化反应构建螺环氧吲哚骨架; 第三类是C-3位具有双亲核位点的2-吲哚酮3, 可与其他两组分或具有双亲电位点的化合物发生串联反应构建螺环氧吲哚骨架.
近年来, 不对称有机小分子催化反应作为一种新型的、强有力的、环境友好的有机合成方法在不对称有机合成化学中得到飞速发展, 已经成为继金属有机催化和酶催化之后的第三种不对称催化方法, 是合成及构建复杂手性有机分子骨架的重要手段[5].目前, 有机小分子催化剂主要包括手性磷酸、脯胺酸、氮杂卡宾、胍类化合物、伯胺、金鸡纳碱衍生物、手性双功能硫脲以及手性双功能方酰胺等催化剂.
手性方酰胺是近年来出现的一类新型有机小分子催化剂[6], 由于其特殊的结构和高的催化活性而备受关注[7].该类催化剂是由硫脲催化剂过渡而来的, 也是一类基于氢键诱导活化作用的有机小分子催化剂, 结构较硫脲更具有刚性, 同时由于双羰基的吸电子效应, 而具有更强的氢键供体活性, 可以作为一种双氢键催化剂的核心骨架单元, 表现出更强的酸性, 因而在不对称催化反应中表现出高催化活性和高选择性.方酰胺催化剂已经成功地应用于催化不对称Michael反应、Mannich反应、Aldol反应、Henry反应、Diels-Alder反应、Baylis-Hillman反应等有机催化反应中[8].另一方面, 目前, 不对称催化串联反应已经成为构建螺环化合物的一种高效方法, 该类反应是根据底物的多个反应位点, 可以发生两步及两步以上的串联反应, 构建环状化合物, 进而合成螺环化合物.因此, 发展方酰胺催化的不对称串联反应合成螺环氧吲哚化合物具有重要意义.
本文将综述2015年以来利用手性方酰胺催化剂, 催化不对称串联反应合成螺环氧吲哚衍生物的研究进展, 并根据不同的催化反应类型进行分类讨论, 阐明手性方酰胺催化剂在不对称催化串联反应合成螺环氧吲哚衍生物中的催化机理.
2011年, 袁伟成课题组[9]将异硫氰酸酯基团引入到氧吲哚的C-3位, 并利用手性硫脲催化剂催化其与酮类化合物的不对称串联反应合成新型手性螺环氧吲哚衍生物.自此, 3-异氰酸酯氧吲哚便成为构建吡咯烷基螺环吲哚酮的高效合成子, 被广泛地应用于不对称合成各种新型光学纯的螺环氧吲哚[10].
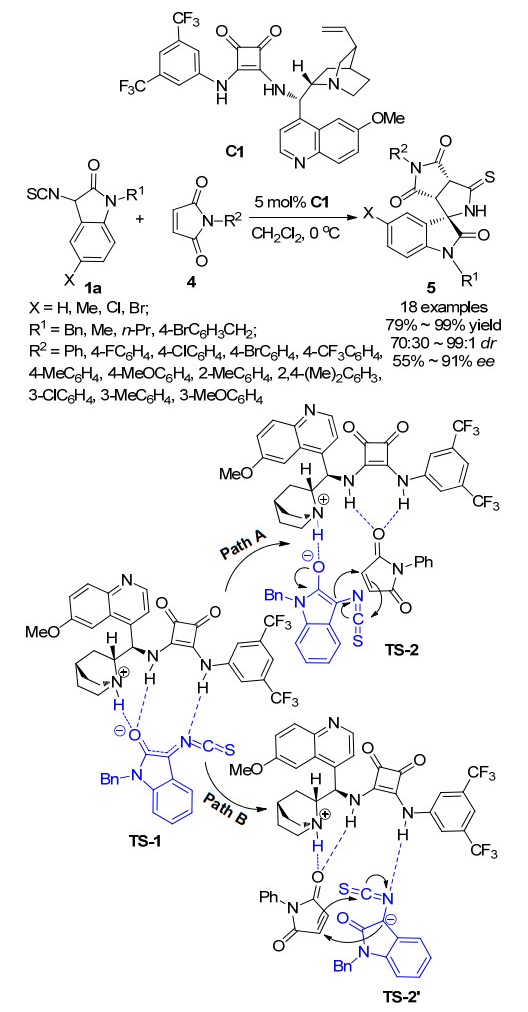
因此, 本课题组设想利用手性方酰胺催化剂, 催化3-异氰酸酯氧吲哚与不饱和烯烃发生不对称Michael/环化串联反应, 立体选择性地得到具有光学活性的吡咯烷基螺环氧吲哚.基于这一设想, 2016年, 本课题组[11]成功报道了一例3-异氰酸酯氧吲哚1a与马来酰亚胺4的不对称Michael/环化串联反应, 该策略首次利用双功能方酰胺C1为催化剂, 实现了吡咯烷基螺环氧吲哚5的高产率(79%~99%)、良好立体选择性(70:30~99:1 dr, 55%~91% ee)的合成(Scheme 1).值得注意的是, 为了与方酰胺催化剂进行比较, 我们尝试了硫脲催化剂, 然而, 结果并不令人满意, 硫脲催化剂在此反应中的立体选择性不如方酰胺催化剂(70:30 dr, 45% ee vs. 93:7 dr, 90% ee).根据实验结果以及前人的报道, 我们提出了两种可能的催化机理:首先双功能方酰胺的叔胺拔掉氧吲哚C-3位的质子形成活化中间态TS-1, 随后经历两种可能的催化路径, 路径A为双功能方酰胺的叔胺部分活化氧吲哚, 两个N—H键与丁二酰亚胺的羰基形成双氢键, 从而催化反应的发生并控制反应的立体选择性; 路径B为双功能方酰胺的叔胺与一个N—H键共同活化丁二酰亚胺, 另一个N—H键与3-异硫氰酸酯氧吲哚形成氢键, 从而催化反应的发生并控制反应的立体选择性.
随后, 利用方酰胺催化3-异氰酸酯氧吲哚与不饱和烯酮的不对称Michael/环化串联反应构建吡咯烷基螺环氧吲哚的报道逐渐增多(Scheme 2). 2016年, 施敏课题组[12]同样利用奎宁衍生的方酰胺催化剂C1, 以甲苯与水为混合溶剂, -20 ℃条件下实现了3-异氰酸酯氧吲哚1a与二亚苄基酮6的不对称Michael/环化串联反应, 构建了手性吡咯烷基螺环氧吲哚10, 最高取得了96%的ee值.当利用奎宁衍生的硫脲催化剂时, 产物的对映选择性急剧下降, 仅有54%.当溶剂为二氯甲烷或甲苯的单一溶剂时, 该反应不仅生成了吡咯烷基螺环氧吲哚10, 还生成了含硫杂环吡咯烷基螺环氧吲哚11.有趣的是, 当加入1 mmol的水溶剂时, 可以调控生成螺环氧吲哚10, 而不生成螺环氧吲哚11, 并大大提高了反应的收率.
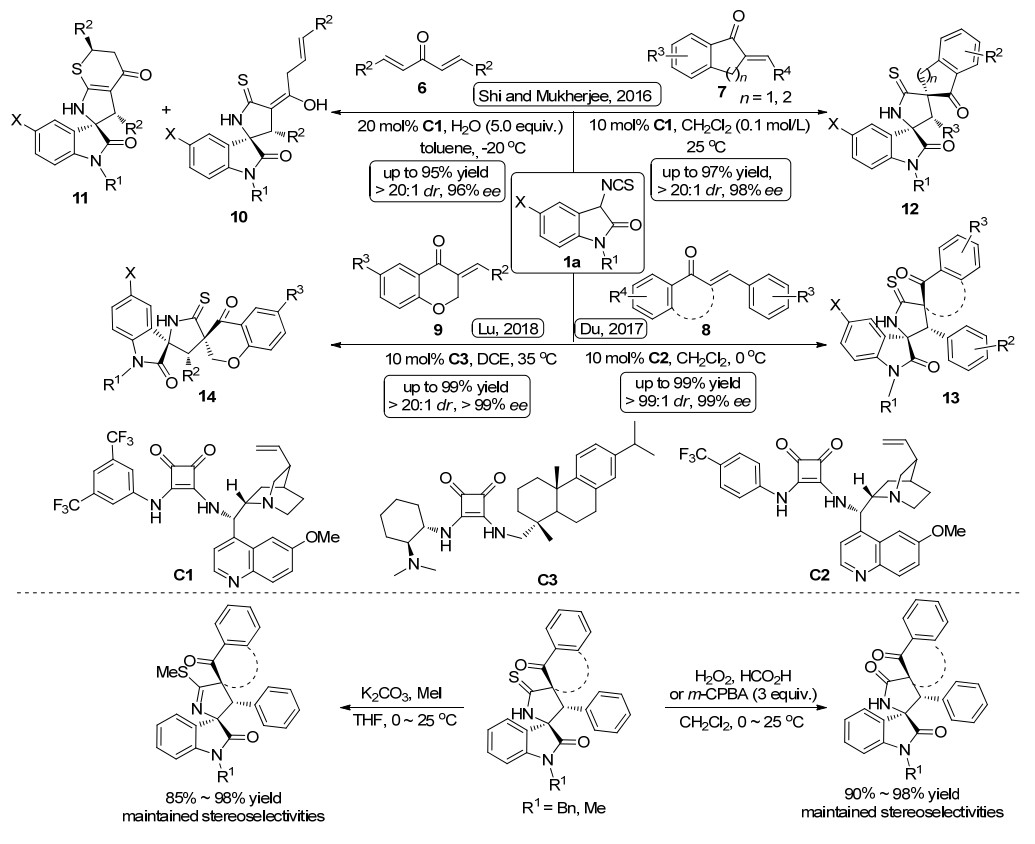
同年, Mukherjee课题组[13]报道了3-异氰酸酯氧吲哚1a与环外α, β-不饱和酮7的不对称Michael/环化串联反应, 同样在方酰胺催化剂C1的催化作用下, 以高达97%的收率, 99:1的er值, 大于20:1的dr值获得了具有连续三个手性中心, 两个螺季碳中心的双螺环吡咯烷基螺环氧吲哚12.同时, 我们课题组[14]也成功开发了3-异氰酸酯氧吲哚1a与α, β-不饱和酮8的不对称Michael/环化串联反应, 高产率、高选择性地得到光学纯吡咯烷基螺环氧吲哚13.我们利用方酰胺催化剂C2, 不仅对环外α, β-不饱和酮底物7(苯并环戊烯酮、苯并环己烯酮、苯并吡喃烯酮)进行底物适应性考察, 还对简单的查尔酮衍生物8进行底物适应性考察, 反应都具有很好的底物适应性, 仅有将苯环用烷基取代时, 反应的收率和立体选择性都有明显的下降, 分别下降到52%的收率, 74:26的dr值以及55%的ee值. 2018年, 鲁桂课题组[15]对方酰胺催化3-异氰酸酯氧吲哚1a与苯并吡喃烯酮9的不对称Michael/环化串联反应进行补充报道, 考察了更广泛的苯并吡喃烯酮底物.在方酰胺C3的催化作用下, 同样能够以出色的产率和立体选择性获得目标产物.以上三项工作都对产物进行硫甲基化及氧化的衍生化反应, 在立体选择性能够保持的情况下, 都有很好的转化率.
2018年, 本课题组[16]又成功实现了3-异氰酸酯氧吲哚1a与亚苄基噻唑烷酮15的不对称Michael/环化串联反应(Scheme 3a).经过条件筛选, 确立了以氢化奎宁衍生的方酰胺C4为催化剂, 二氯甲烷为溶剂, 5 mol%的催化剂用量, 室温下为该催化体系的最优条件.在最优反应条件下, 考察了反应底物的适应性, 无论是取代基的电子效应还是空间位阻, 反应都能够顺利的进行, 并以高达99%的产率, >99:1的dr值及>99%的ee值获得吡咯烷基螺环氧吲哚并噻唑烷酮衍生物16.仅当噻唑烷酮的N无保护基时, 反应无法进行; 而当底物为罗丹宁时, 反应的立体选择性明显下降(88:12 dr, 63% ee).
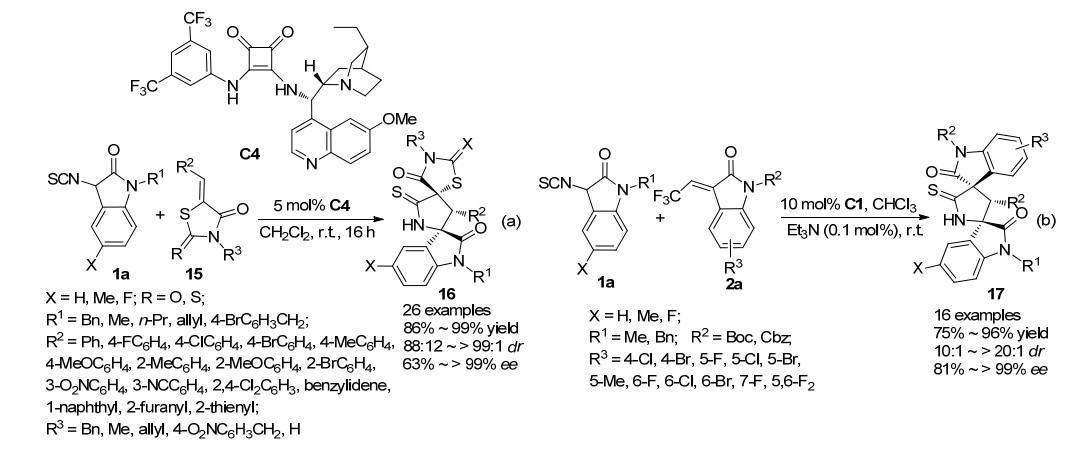
2018年, 鲁桂课题组[17]利用方酰胺催化剂C1, 促进3-异氰酸酯氧吲哚1a对3-三氟乙烯氧吲哚2a的不对称Michael/环化串联反应, 该反应活性较低.作者通过添加0.1 mol%的三乙胺促进反应的进行, 反应时间为0.5~7 d不等.通过对底物适应性的考察发现, 该反应的底物适应性也比较广泛, 取得了75%~96%的产率和81%~>99%的对映选择性(Scheme 3b).值得注意的是, 在以上所报道的工作中, 为了与方酰胺催化剂进行比较, 作者尝试了硫脲催化剂, 结果发现当反应受体为环内或直链烯酮时, 硫脲催化剂在此类反应中的立体选择性都不如方酰胺催化剂, 而当反应受体为环外烯酮时, 硫脲与方酰胺的催化效果差别不大.
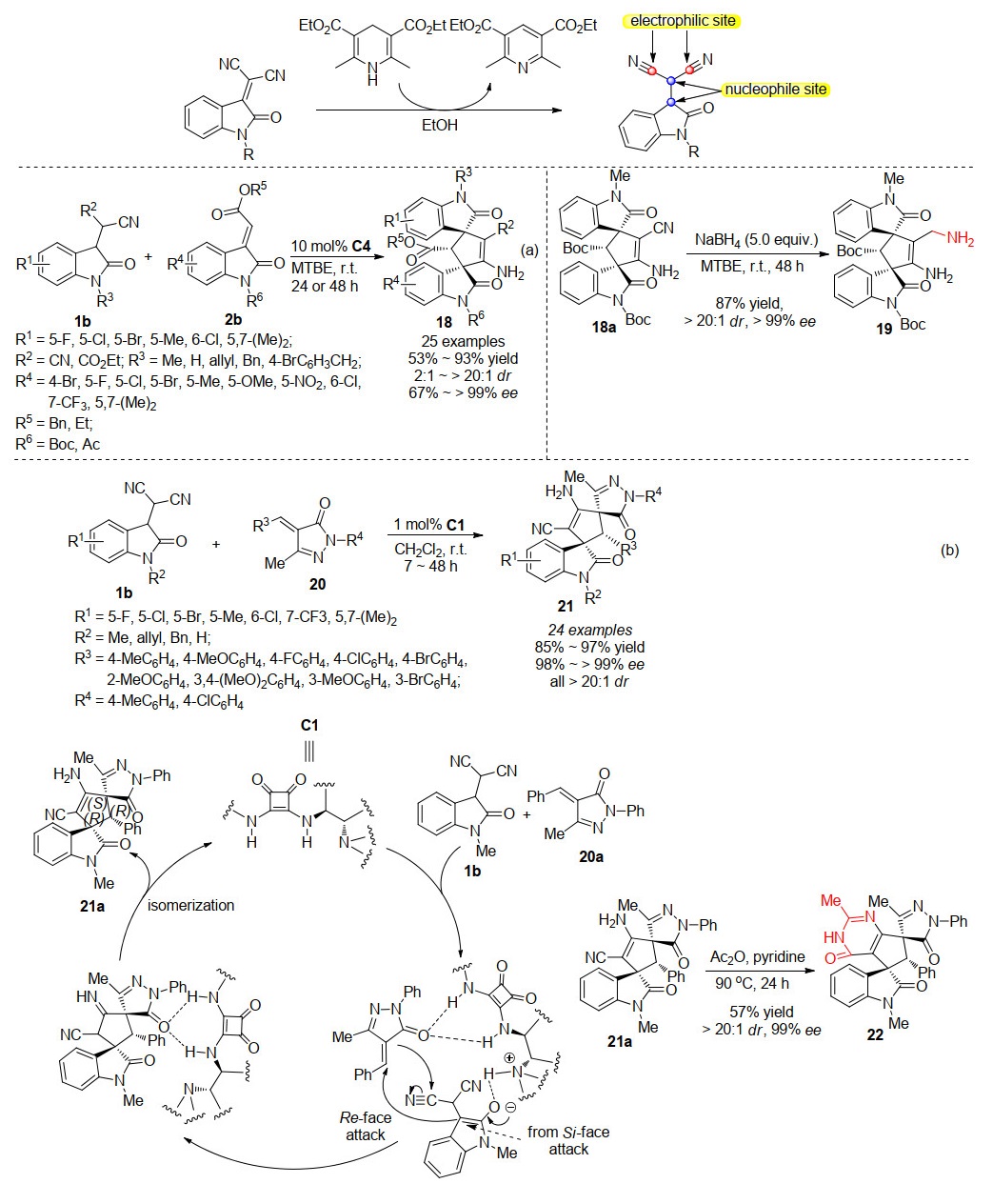
3-亚甲基丙二腈氧吲哚是一种简单易得的高活性亲电底物, 通过与各种串联试剂反应, 该底物已被广泛用于设计和合成各种螺氧杂吲哚骨架.而关于具有多个亲核及亲电位点的3-丙二腈吲哚在不对称催化反应中的应用却未见报道. 2019年, 本课题组[18]先后成功开发了两例关于3-丙二腈氧吲哚1b的不对称串联反应(Scheme 4).我们首先利用方酰胺催化剂C4, 催化3-丙二腈氧吲哚1b与3-亚甲基氧吲哚2b进行不对称Michael/环化串联反应合成螺环双氧吲哚18.反应能够以中等至良好的收率, 出色的立体选择性获得目标产物, 当3-丙二腈氧吲哚N上无保护基时, 反应的非对映选择性极具下降为2:1, 而当R2用乙酯基取代时, 反应活性有所下降, 反应时间延长为48 h.紧接着, 我们又开发了一例3-丙二腈氧吲哚1b与不饱和吡唑啉酮20不对称Michael/环化串联反应, 仅在1 mol%的方酰胺催化剂C1的作用下, 能以高达96%的产率, >20:1的dr, >99%的ee得到了螺环氧吲哚并吡唑啉酮骨架化合物21.以上两例报道说明了3-丙二腈氧吲哚底物不仅有很好的催化反应活性, 而且适应范围广泛, 是构建螺环氧吲哚骨架化合物的优良潜在合成子.当我们将反应进行克级规模放大以及衍生化反应实验时, 目标产物的产率和立体选择性都能很好地保持, 说明该策略具有良好潜在的应用价值.另外, 如Scheme 4b, 根据反应结果, 我们设想了一个合理的反应机理来更好地理解和设计3-丙二腈氧吲哚的不对称串联反应.
2019年, 鄢明课题组[19]发展了3-氨基氧吲哚1c与不饱和磺酰氟23在双功能方酰胺C1为催化剂, 二氯甲烷为溶剂, 室温条件下合成3, 3'-双取代氧吲哚24以及螺环氧吲哚25的有效合成方法.如Scheme 5所示, 当底物为乙烯基磺酰氟23a时, 反应只发生一步Michael加成, 得到3, 3'-双取代氧吲哚, 最高取得了99%的收率和99%的ee值.无论R1, R2, R3取代基如何变换, 都能获得很好的收率(70%~99%)和对映选择性(90%~99% ee), 只有R3取代基为四氢吡喃时, 反应的收率下降为46%.将分离提纯的3, 3'-双取代氧吲哚进一步用1, 8-二氮杂二环[5,4,0]十一碳-7-烯(DBU)处理, 得到关环产物螺环氧吲哚25a, 该步反应的收率为99%.当底物为亚苄基磺酰氟23b时, 直接得到Michael/环化串联反应产物螺环氧吲哚25b.然而, 该反应的收率和立体选择性都有明显的下降.

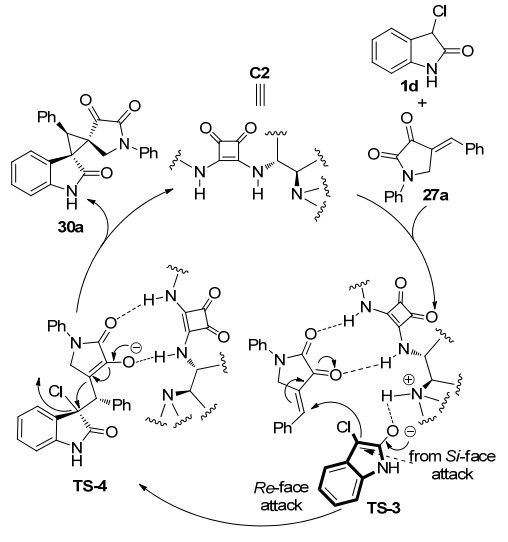
目前, 3-氯氧吲哚参与的不对称Michael/烷基化反应被认为是合成环丙烯基螺环氧吲哚衍生物的最有效方法之一. 2015年, 本课题组[20]报道了一例方酰胺催化的3-氯氧吲哚1d与不饱和吡唑啉酮20的不对称Michael/alkylation串联反应(Scheme 6).通过添加0.1 mmol的K2CO3促进烷基化反应的发生, 实现了不对称串联反应合成环丙烯基螺环氧吲哚并吡唑啉酮衍生物28.在条件优化过程中, 发现手性有机催化剂与底物相结合的方式可能与最佳优势构象存在一定的矛盾, 因此无法达到非常令人满意的立体选择性(2.4:1~87:13 dr, 40%~74% ee).在此基础上, 我们进一步筛选了其他骨架底物, 分别于2019年[21]和2020年[22]先后报道了3-氯氧吲哚1d与5-烯基噻唑酮26以及2, 3-二羰基吡咯烷27的不对称Michael/烷基化串联反应.通过反应条件优化, 成功构建了手性方酰胺C4与无机碱Na2CO3的催化体系, 以及手性方酰胺C2与无机碱NH4HCO3的催化体系, 高效高选择性的合成了环丙烯基螺环氧吲哚并噻唑酮衍生物29以及环丙烯基螺环氧吲哚并吡咯烷酮衍生物30.当我们将底物3-氯氧吲哚换作3-溴氧吲哚1e时, 反应仍能高效地进行, 以80%的收率, 95%的ee值, >25:1的dr值得到产物.然而有趣的是, 经过核磁以及高效液相谱图对比, 我们发现该体系催化生成的产物为环丙烯基螺环氧吲哚并吡咯烷酮衍生物30的非对映异构体30'.
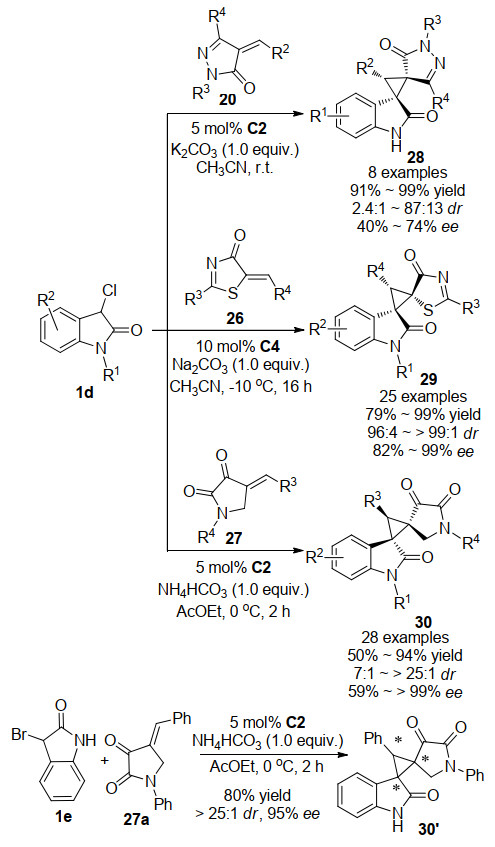
对于反应的立体化学选择性, 我们总结了之前的研究结果, 提出了一个合理的反应机理.如Scheme 7所示, 双功能方酰胺的叔胺拔掉氧吲哚C-3位的质子而活化供体并与氧吲哚羰基形成氢键, 两个酰胺的NH与吡咯烷二酮的羰基形成双氢键达到过渡态TS-3, 同时氧吲哚从Si向吡咯烷酮的Re面进攻, 发生一次Micheal加成反应形成过渡态TS-4, 随后在无机碱的活化作用下, 吡咯烷二酮的α碳进攻氧吲哚的C-3位, Cl离子离去, 发生烷基化反应合成环丙烯基螺环氧吲哚并吡咯烷酮衍生物30.
2016年, 本课题[23]发现α-亚烷基琥珀酰亚胺31在手性方酰胺催化条件下, 可与3-亚甲基氧吲哚2b发生不对称Michael/Michael串联反应(Scheme 8).在最初的研究中, 以不带保护基的α-亚烷基琥珀酰亚胺为底物, 反应无法进行, 当以苄基或苯基保护的α-亚烷基琥珀酰亚胺为底物时, 只有痕量的反应发生.由于叔丁氧羰基(Boc)往往能活化反应底物, 因此我们尝试使用Boc保护的α-亚烷基琥珀酰亚胺作为底物, 反应能够顺利地进行, 并以57%的收率, 76:24的dr值, 98%的ee值得到具有连续五个手性中心的螺环氧吲哚衍生物32.
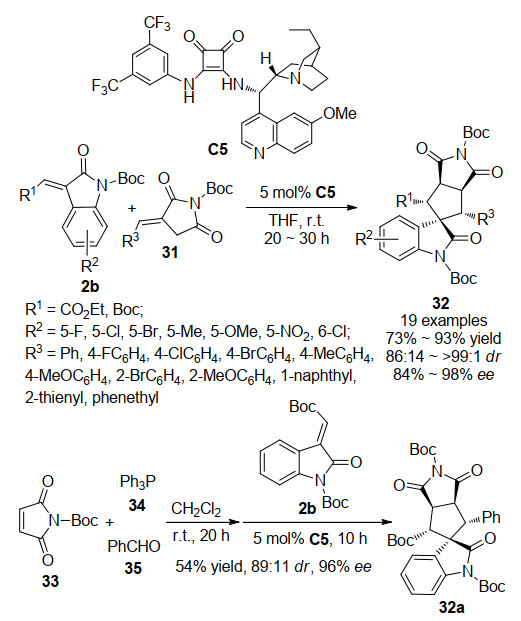
通过条件优化, 确定以方酰胺C5为催化剂, 四氢呋喃为溶剂, 5 mol%催化剂用量, 室温环境为最优反应条件, 反应的收率可提高到83%, 非对应选择性提高到86:14.进一步考察反应的底物适应性发现, 该反应的底物适应性广泛, 能以63%~93%的收率, 86:14~>99:1的dr值, 84%~98%的ee值得到目标产物.此外, 我们还尝试在该催化体系下以马来酰亚胺为起始底物一锅法合成螺环氧吲哚衍生物32, 不仅反应的总收率能达到54%, 立体选择性也能够很好地保持.
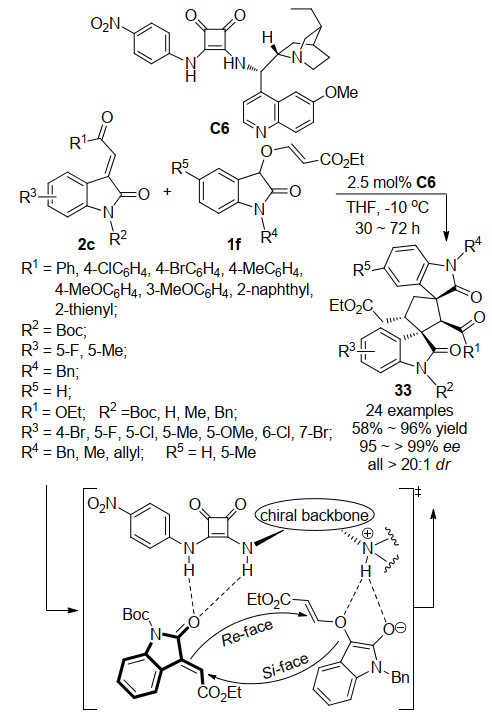
同年, 本课题组[24]通过3-羟基氧吲哚与丙炔酸乙酯在N-甲基吗啡啉做催化剂条件下反应, 成功开发了一种新型的氧吲哚启动串联反应试剂1f, 并利用方酰胺催化剂C6, 催化其与3-亚甲基氧吲哚2c发生不对称Michael/Michael串联反应, 成功实现了螺环双氧吲哚骨架化合物33的高立体选择性合成.该反应策略不仅反应活性高, 仅需2.5 mol%的催化剂用量, 且底物适应范围广, 收率为58%~96%, 对映选择性为95%~>99%, 非对映选择性均可达到>20:1.根据反应产物的立体构型, 我们推测了一个合理的反应过渡态来解释该反应的历程(Scheme 9).另外, 在催化剂筛选过程中, 尝试了硫脲催化剂催化反应, 虽然能够取得与方酰胺催化反应相当的反应收率, 但是对映选择性却大大下降, 仅能得到36%的ee值.可能是由于方酰胺结构较硫脲更具有刚性, 在此反应中, 与反应受体形成氢键时, 更易控制反应受体的朝向, 从而获得更为出色的对映选择性.
2020年, 林文伟课题组[25]以3-苯乙酰基香豆素34为启动串联反应试剂, 以方酰胺C1为催化剂, 二氯甲烷为溶剂, 30 ℃条件下开发了与3-亚甲基氧吲哚2的不对称Michael/Michael串联反应, 高产率、高立体选择性地构建了具有连续五个立体手性中心, 三个连续季碳手性中心的螺环氧吲哚35.该策略底物适应性广泛, 作者尝试了三种类型的3-亚甲基氧吲哚, 当取代基R3为酯基和苯甲酰基时, 都能获得出色的收率以及立体选择性; 当取代基R3为苯基时, 反应的对映选择性有所下降, 可获得61%~90%的ee值(Eq. 1).
|
|
(1) |
|
|
(2) |
2019年, 洪伯诚课题组[26]成功报道了一例硝基氧吲哚1g与硝基烯烃酯36的不对称Michael/Michael串联反应(Eq. 2).作者利用双功能方酰胺C7的催化体系, 成功克服了硝基氧吲哚在Michael加成反应中的自身烷基化问题, 导向合成具有四个连续手性中心的Michael/ Michael串联反应产物, 然而该体系由于无法很好的控制第一步硝基氧吲哚Michael加成的进攻方向, 而产生两种非对映异构体37和37', 非对映选择性为70:30~91:9, 主要非对映异构体的对映选择性为92%~97%.另外, 作者还考察了底物硝基烯烃酯的顺反异构体对反应的影响, 在该催化体系下分别尝试了(E)-36与(Z)-36底物, 发现底物36的顺反异构体对反应的立体选择性没有影响.
不对称Michael/lactonization串联反应是目前构建手性螺环氧吲哚内酯的最有效方法之一. 2015年, 袁伟成课题组[27]以3-羟基氧吲哚1h为氧吲哚启动串联反应试剂, 乙腈为溶剂, 利用方酰胺催化剂C8, 通过与α, β-不饱和酰基膦酸酯38的不对称Michael/内酯化串联反应合成手性螺环氧吲哚内酯39 (Scheme 10a).反应收率为82%~98%, 立体选择性为81:19 dr, 71%~95% ee.当反应底物为大空间位阻的β-二取代酰基膦酸酯时, 收率明显下降为33%而立体选择性能够保持(89% ee).当取代基R3为苯环及呋喃环时, 反应的立体选择性有所下降, 分别为81:19 dr, 71% ee和92:8 dr, 78% ee.受上述成果的启发, 作者在该催化体系下, 又尝试了3-氨基氧吲哚1c与α, β-不饱和酰基膦酸酯38发生不对称Michael/内酰胺化串联反应合成螺环氧吲哚内酰胺40.然而反应的收率仅有11%, 将溶剂换为二氯甲烷后, 反应能够顺利地进行, 并以31%~65%的收率, 92:8~>99:1 dr, 85%~97% ee获得相应的螺环氧吲哚内酰胺产物.该反应的产率相对较低, 可能是由于反应第二步的内酰胺化相对困难所致.根据反应产物的立体化学结构, 作者还提出了一个可能的反应过渡态, 如Scheme 10a所示.
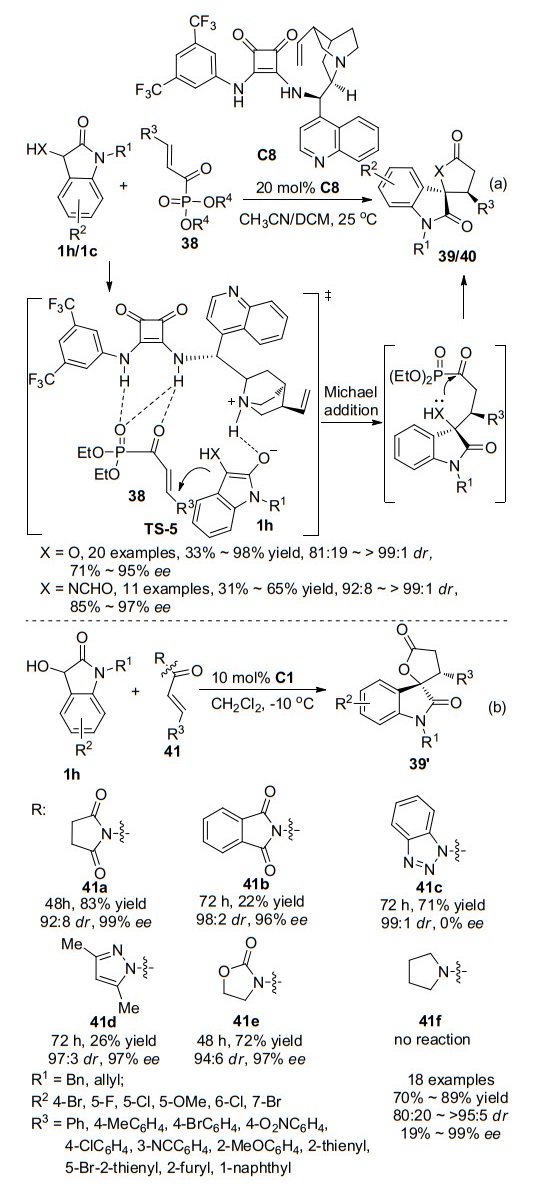
随后, 2018年, 本课题组[28]跟进报道了一例利用方酰胺C1催化3-羟基氧吲哚1h与α, β-不饱和N-酰化琥珀酰亚胺41a的不对称Michael/内酯化串联反应(Scheme 10b).首先我们对离去基团进行了筛选, 包括琥珀酰亚胺、邻苯二甲酰亚胺、苯并三唑、二甲基吡唑、恶唑烷酮以及吡咯, 得到α, β-不饱和N-酰化琥珀酰亚胺的反应效果最佳, 反应时间48 h, 收率可达83%, 立体选择性达到92:8 dr, 99% ee.在确定最优反应底物类型后, 对催化剂进行筛选, 发现硫脲与方酰胺在控制反应的非对映体选择性上有较大的差异.硫脲主要控制生成另一对非对映异构体(19:81 dr), 且对映选择性有明显的下降(69% ee).随后对其他反应条件进行优化, 并进行底物拓展实验.相较袁伟成的工作, 该策略的对映选择性有明显的提高, 普遍可达到 > 96% ee, 仅当取代基R3为邻位取代的邻甲氧基苯和呋喃环时, 反应的对映选择性明显下降, 分别下降为19%和39%.
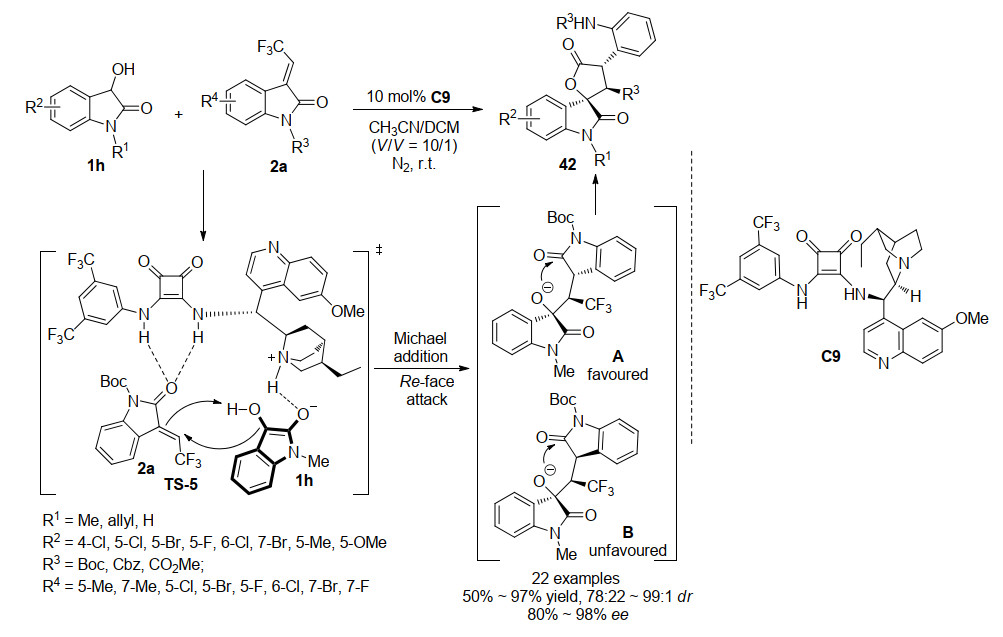
2019年, 邓卫平课题组[29]同样以3-羟基氧吲哚1h为底物, 在氮气保护下, 以二氯甲烷/乙腈(V:V=10:1)混合溶液为溶剂, 利用双功能方酰胺C9, 催化其与3-三氟乙烯基氧吲哚2a, 通过不对称Michael/内酯化串联反应高产率、高立体选择性地获得含三氟甲基的螺环氧吲哚内酯化合物42.该报道是首例通过内酰胺C—N键的断裂来合成螺环氧吲哚内酯化合物, 并提供了一种合成含三氟甲基的螺环氧吲哚内酯化合物的新方法.作者根据产物的立体化学, 提出了一个可能的反应过渡态, 与袁伟成课题组提出的催化过渡态基本相同, 双功能方酰胺的叔胺部分活化3-羟基氧吲哚启动串联反应试剂, 双N—H与反应受体形成氢键, 从而催化3-羟基氧吲哚从Si面向3-三氟乙烯基氧吲哚的Re面进攻, 控制反应的立体选择性, 产生反应中间体A, 随后进行羟基氧进攻内酰胺, C—N键断裂形成C—O键, 产生螺环氧吲哚内酯化合物(Scheme 11).
2015年, 孙兴文课题组[30]开发了一种构建五元螺环氧吲哚的新方法, 该方法基于双功能奎宁衍生方酰胺C10催化的酮亚胺中间体的Michael/Mannich串联反应.作者首先将乙酰丙酮43与亚硝基苯44投入到方酰胺催化体系中, 反应完全后直接加入3-亚甲基氧吲哚2b, 使其与上一步反应生成的具有多个反应位点的酮亚胺进行Michael/Mannich串联反应, 以中等至优秀的收率(31%~94%), 优秀的立体选择性(4:1~>20:1 dr, 74%~>99% ee), 一锅法合成含有三个连续手性中心, 两个连续季碳手性中心的螺环氧吲哚化合物45.该一锅法反应策略很容易进行放大反应, 且仅需在2 mol%的催化剂用量下即可实现高效的合成, 无论是以氯仿还是甲苯作溶剂, 都能达到理想的反应效果(Scheme 12).值得注意的是, 为了考察该反应的反应机理, 作者还进行了N保护基的控制实验, 当Boc基团换作苄基或无保护基时, 反应无法进行, 当用苄氧羰基(Cbz)保护时, 反应效果基本可以保持, 这个结果说明催化剂与保护基Boc或Cbz有氢键作用.根据控制实验的结果以及产物的立体化学结构, 作者提出了可能的反应机理, 如Scheme 13所示.
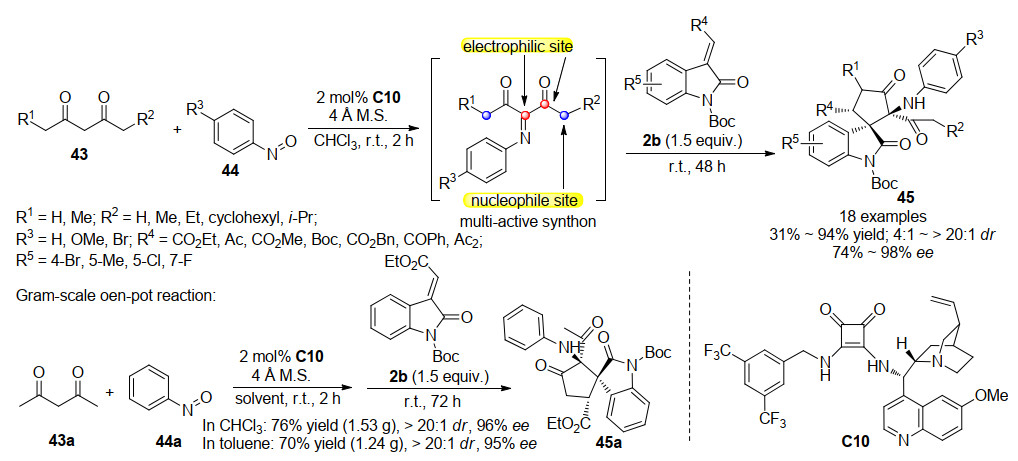
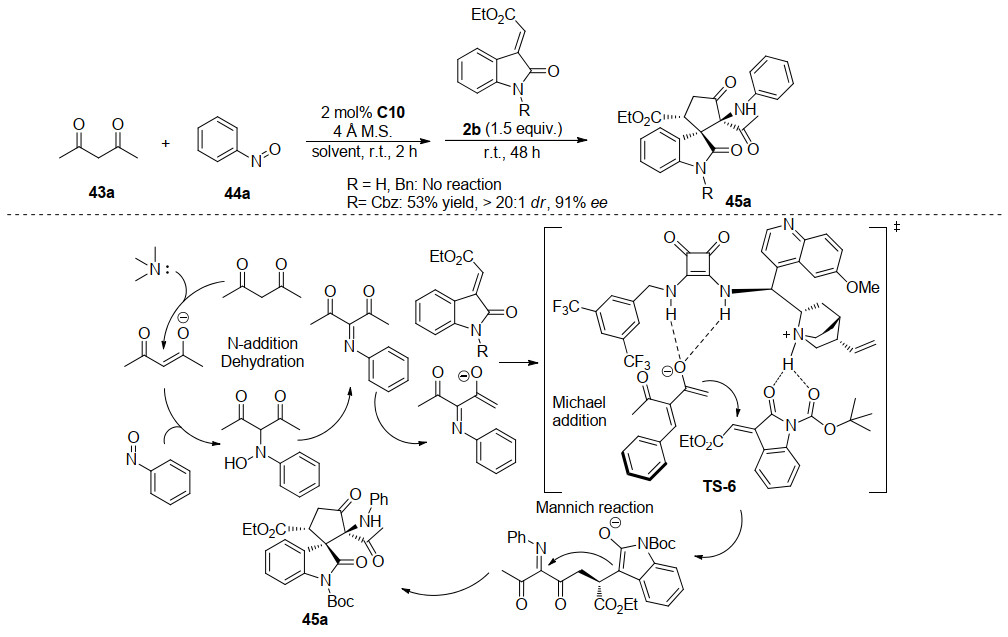
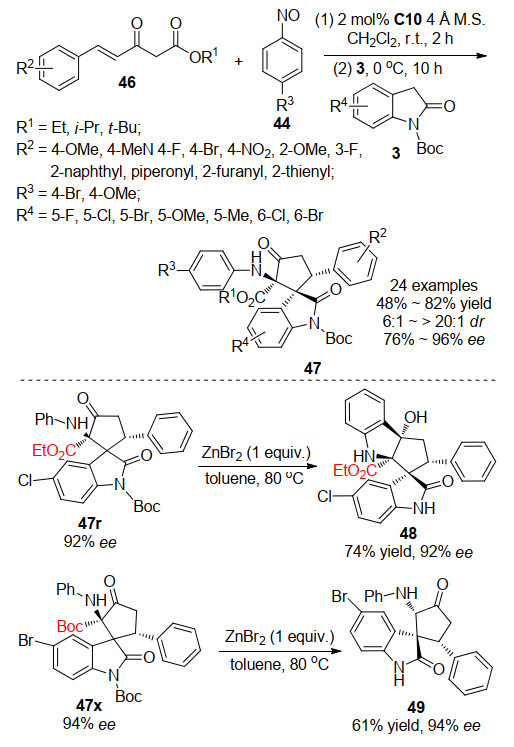
随后, 2018年, 孙兴文课题组[31]基于此研究报道, 成功发展了一例基于双功能方酰胺催化的三组分一锅法合成螺环氧吲哚衍生物.作者利用α, β-不饱和酮酸酯46, 可以与亚硝基芳烃44反应形成含有两个亲电位点的C4合成子, 这种C4合成子可以进一步与由双功能方酰胺C10催化剂促进的2-吲哚酮3反应.如Scheme 14所示, 通过不对称Michael/Mannich串联过程进行的[4+1]环化反应, 生成了带有三个连续立体中心的α-氨基-β-酮酸酯单元的螺环戊烷氧吲哚47.该策略通过三组分一锅法以48%~82%的收率, 6:1~>20:1的dr, 76%~96%的ee获得新型的螺环戊烷氧吲哚47.此外, 所获得的光学纯螺环戊烷氧吲哚可以轻松地进行进一步的衍生化反应, 从而获得其他重要的骨架化合物.作者尝试利用不同的Lewis酸促进手性螺环戊烷氧吲哚发生分子内的Friedel-Crafts反应, 令人高兴的是, 在ZnBr2存在下取代基R为乙酯基的螺环戊烷氧吲哚能够平稳地环化, 同时Boc基团也被除去, 以74%的收率获得了环戊[b]吲哚衍生物48, 而没有对映体纯度的降低.有趣的是, 当取代基R为Boc基团时, 在相同条件下, 螺环戊烷氧吲哚并不能发生Friedel-Crafts环化反应, 而是发生了叔丁基的去除和随后的脱羧反应, 脱羧最有可能通过与相邻的酮羰基的六元环过渡态进行, 并且所得的烯醇形式异构化为酮产物49.
2016年, Kesavan课题组[32]开发了一种由L-脯氨酸衍生的新型双功能方酰胺有机催化剂, 并利用该类催化剂成功实现了α, β-不饱和酮酸酯衍生的氧吲哚2d与吡唑啉酮50的不对称Michael/半缩酮化串联反应, 高收率(80%~95%)、高对映选择性(83%~ > 99% ee)地合成了呋喃基螺环氧吲哚51 (Scheme 15a).用氟原子取代羟基通常可以提高药物的安全性和有效性, 因此作者利用氟化剂二乙基氨基三氟化硫(DAST)探索了半缩醛产物51a的氟化反应.将化合物51a用DAST在二氯甲烷中于0 ℃处理10 d, 以64%的收率得到具有良好非对映选择性和出色对映选择性的氟化产物52.
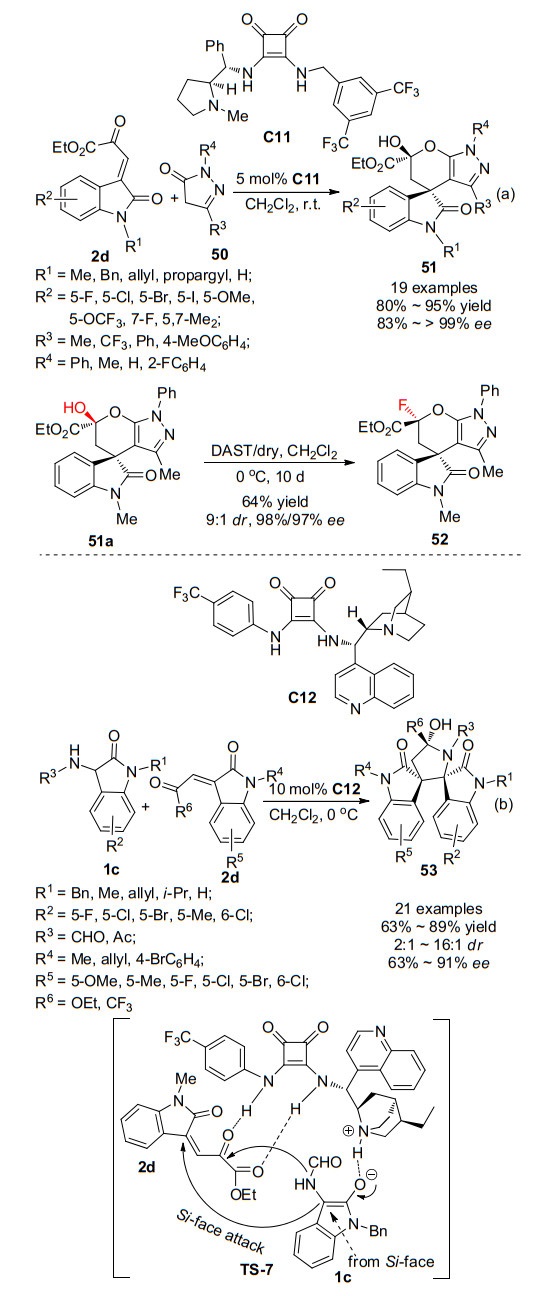
2018年, 本课题组[33]发展了一种构建吡咯烷基螺环双氧吲哚骨架的新方法(Scheme 15b), 采用3-氨基氧吲哚1c、α, β-不饱和酮酸酯衍生的氧吲哚2d为原料, 在手性方酰胺C12催化条件下, 利用方酰胺催化剂C12的叔胺基团活化启动串联反应试剂1c, 催化剂的N—H键与α, β-不饱和酮酸酯2d的双羰基形成氢键, 促进两底物之间发生对映选择性的Michael/氮杂半缩酮化串联反应, 得到吡咯烷基螺环双氧吲哚衍生物53.
2015年, 本课题组[34]成功开发了一种新型串联反应试剂对甲苯磺酰胺基甲基烯酮54, 并将其应用于3, 3'-吡咯烷基螺环氧吲哚衍生物的合成中.该方法以3-亚甲基氧吲哚2b/2c和氨基甲基烯酮54为底物, 以手性方酰胺C5为催化剂, 通过催化不对称aza-Michael/Michael串联反应, 高收率(72%~99%)地合成了一系列具有优异的非对映选择性和对映选择性的光学纯的新型3, 3'-吡咯烷基螺环氧吲哚衍生物55和56(高达>99:1 dr, >99% ee).当我们尝试利用硫脲作为催化剂催化反应时, 发现反应几乎不发生, 此现象进一步说明, 方酰胺片段的两个N—H基团具有更大的间距, 从而较硫脲具有更强的酸性, 能够对特定底物, 如该反应的两个底物进行良好的识别, 高活性、高立体选择性地催化反应.另外, 本文也提出了可以合理解释方酰胺的催化过程的串联反应机理(Scheme 16).
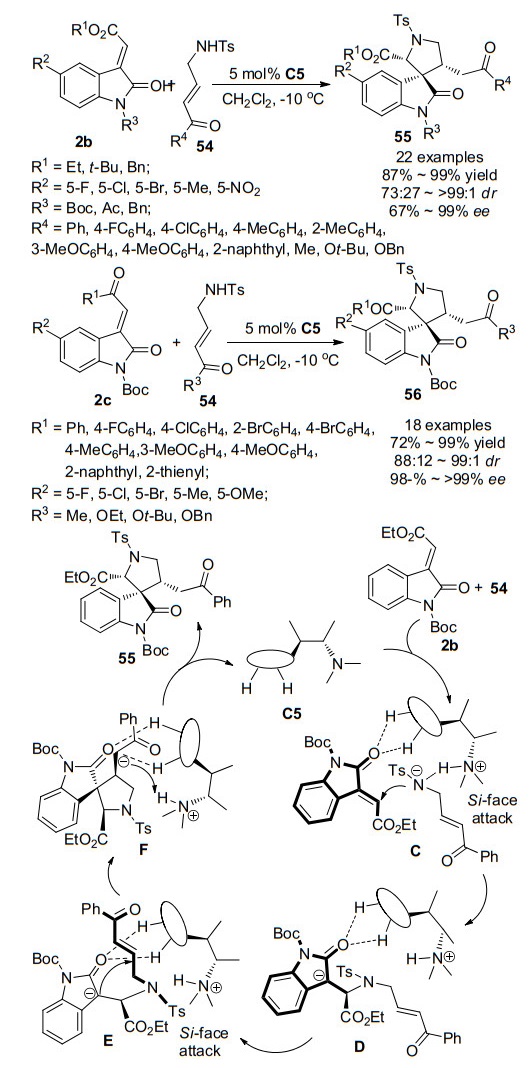
基于该反应策略, 近期孙兴文题组[35]实现了螺环氧吲哚并哌啶酮58的不对称合成.通过方酰胺C10催化3-亚甲基氧吲哚2b与N-苄氧基丙烯酰胺57, 高效发生不对称aza-Michael/Michael串联反应(Eq. 3).该反应可以取得23%~99%的收率及58%~ > 99%的对映选择性.研究表明, 当底物3-亚甲基氧吲哚C-4位有取代基时, 由于空间效应, 导致反应的对映选择性降低, 而产率几乎不受影响; 当底物N-苄氧基丙烯酰胺的α和β位有位阻基团时, 会导致产量下降, 而对映选择性不受影响.
|
|
(3) |
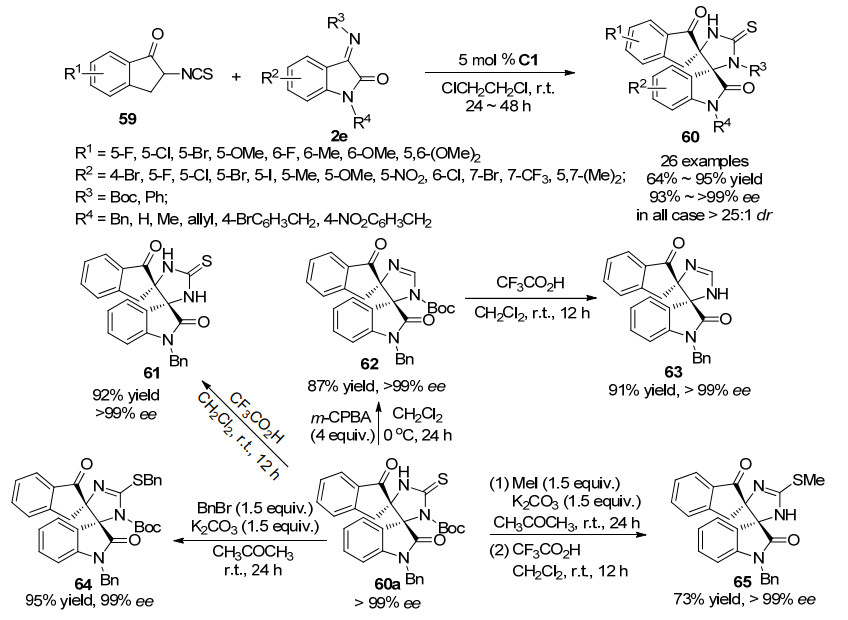
异硫氰酸酯基团已经成为有机催化不对称串联反应构建许多重要含氮杂环的重要合成砌块, 因此, 我们设想将异硫氰酸酯引入到1-茚酮的C-2位, 合成的2-异氰酸酯茚酮也可以用作串联反应试剂, 参与螺环氧吲哚衍生物的构建.基于这一设想, 2018年, 本课题组[36]成功合成了2-异氰酸酯茚酮59, 并将其应用于螺环氧吲哚衍生物的合成中.该方法以2-异氰酸酯茚酮59和3-亚胺氧吲哚2e为底物, 以手性方酰胺C1为催化剂, 通过催化不对称Michael/环化串联反应路径, 合成了一系列光学纯的新型螺环氧吲哚衍生物60 (>25:1 dr, 97%~>99% ee).当氧吲哚C-4位有取代基时, 由于空间位阻效应, 反应无法进行.此外, 我们还尝试了多种衍生化反应, 以良好至优秀的产率(73%~95%), 出色的对映选择性(99%~>99% ee)得到了多种不同的新型螺环氧吲哚(Scheme 17).
甲亚胺与各种烯烃的1, 3-偶极环加成反应是构建手性吡咯烷骨架的主要方法[37], 近年来, 利用该策略以氧吲哚有起始原料构建吡咯烷基螺环氧吲哚的报道越来越多.催化不对称1, 3-偶极环加成反应已经成为对映选择性合成吡咯烷基螺环氧吲哚的高效合成方法.
2015年, 赵洪武课题组[38]建立了首个由靛红衍生的甲亚胺基化物与马来酰亚胺的催化不对称1, 3-偶极环化串联反应.在大多数情况下, 通过10 mol%手性方酰胺C1催化, 采用靛红2f、苄胺66及马来酰亚胺4为底物, 二氯甲烷为溶剂, 硬脂酸、0.4 nm分子筛为添加剂, 室温下, 所得到手性吡咯烷基螺环氧吲哚化合物67的产率中等(高达89%), 非对映和对映选择性良好(最高>20:1 dr, >99% ee).研究表明, 当采用硫脲为催化剂催化该反应时, 只能获得一对消旋体产物.另外, 马来酰亚胺的保护基对反应的对映选择性影响巨大, 当无保护基或烷基保护基换作具有芳香性的苯基或苄基时, 反应的对映选择性降至12%~65% (Eq. 4).
|
|
(4) |
同年, 王锐等[39]利用本课题组所开发的新型氧吲哚串联反应试剂3-三氟乙基亚胺氧吲哚2g[40], 与硝基烯烃68在方酰胺C1催化条件下, 进行不对称1, 3-偶极环化串联反应(Scheme 18a).该反应在温和条件下平稳进行, 以高收率和优异的对映和非对映选择性获得了一系列含三氟甲基的吡咯烷基螺环氧吲哚化合物69.该研究表明, 3-三氟乙基亚胺氧吲哚是在碱催化作用下产生具有反应活性的甲亚胺叶立德, 并与缺电子烯烃发生1, 3-偶极环加成反应.
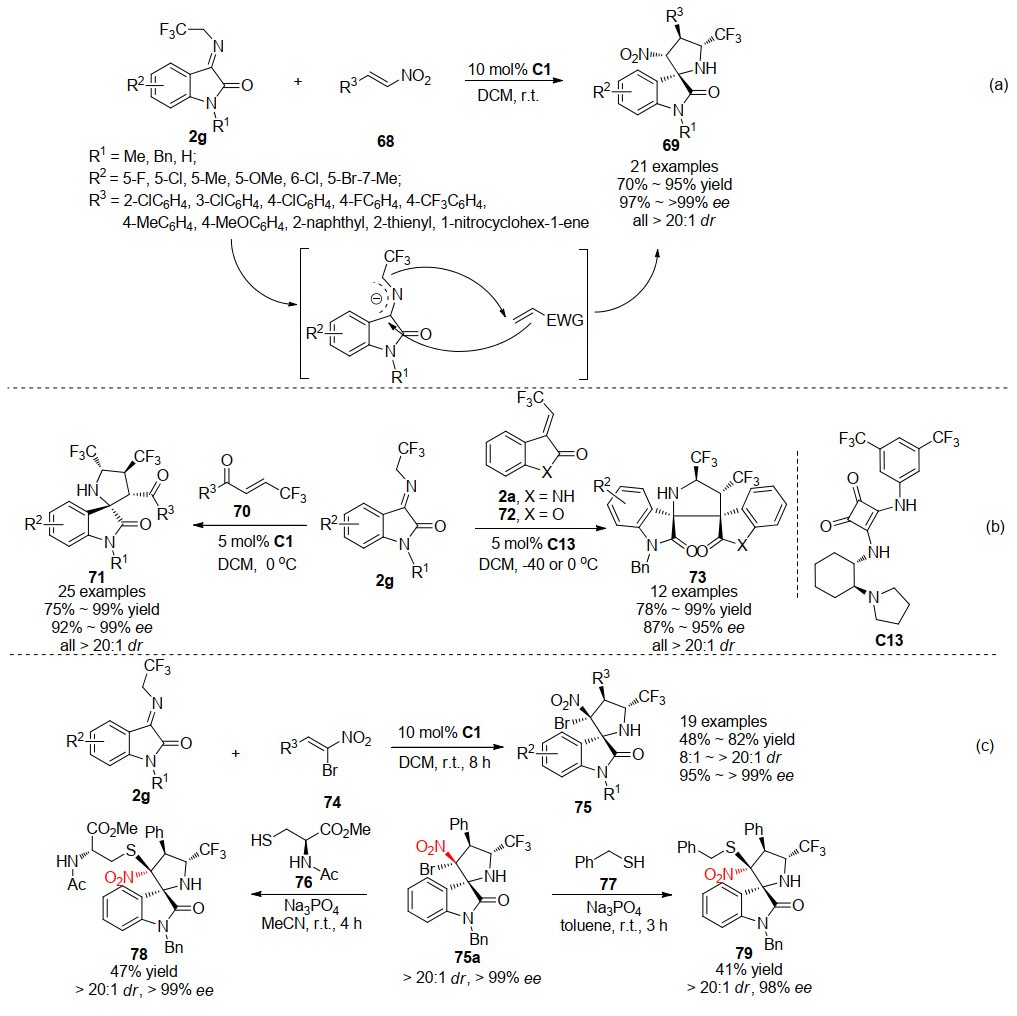
基于此项研究, 2018年, 袁伟成课题组[41]利用β-三氟甲基烯酮70与3-三氟乙基亚胺氧吲哚2g反应, 通过对映选择性环化反应得到了带有两个三氟甲基的相应螺环氧吲哚产物71.该反应也由金鸡纳生物碱衍生的方酰胺C1催化, 但剂量比王锐工作组使用的低(Scheme 18b).另外, 袁伟成课题组还利用环外烯烃底物, 如3-三氟乙烯基氧吲哚2a及3-三氟乙烯基苯并呋喃酮72为底物, 在方酰胺催化剂C13催化下, 与3-三氟乙基亚胺氧吲哚2g进行不对称1, 3-偶极环化串联反应, 高收率、高立体选择性地获得了带有连续两个三氟甲基的双螺环氧吲哚73.随后, 韩波课题组[42]也报道了一例3-三氟乙基亚胺氧吲哚2g与溴代硝基烯烃74的不对称1, 3-偶极环化串联反应, 同样利用方酰胺催化剂C1作为催化剂, 得到了一系列高立体选择性的含三氟甲基的吡咯烷基螺环氧吲哚75.作者在尝试硫脲催化反应时发现, 相较于方酰胺催化剂, 硫脲催化反应的立体选择性有明显的降低(18:1 dr, 53% ee vs.>20:1 dr, >99% ee).考虑到溴化物的独特结构, 作者利用含巯基的亲核试剂, 如N-乙酰基-L-半胱氨酸甲酯、苄硫醇处理产物75a, 通过SN2过程, 以中等产率得到了构型反转的产物78和79 (Scheme 18c).
为了对连续螺环结构的含三氟甲基螺环氧吲哚骨架化合物进行合成, 大量的研究者在金鸡纳生物碱衍生的方酰胺有机催化剂的活化下, 将3-三氟乙基亚胺氧吲哚转化为含三氟甲基的甲亚胺叶立德, 并催化其与不同种类的环外烯烃进行1, 3-偶极环化串联反应.
2017年, 鲁桂课题组[43]利用3-亚甲基氧吲哚2b与3-三氟乙基亚胺氧吲哚2g反应, 报道了一例选择性环外1, 3-偶极环化串联反应(Scheme 19a).在奎宁衍生的方酰胺C14催化作用下, 以高度立体选择性的方式有效地构建了一系列潜在的具有重要生物学意义的含三氟甲基的吡咯烷基螺环双氧吲哚产物80 (84%~99%的产率, 高达>20:1 dr和>99% ee). 2018年, 本课题组[44]利用方酰胺C2催化剂, 催化不饱和罗丹宁15与3-三氟乙基亚胺氧吲哚2g进行环外不对称1, 3-偶极环化串联反应(Scheme 19b).随后, 2019年蒋先兴[45]、阎文锦[46]课题组利用相似的策略, 分别采用2, 3-二羰基吡咯烷27、不饱和吡唑啉酮20为底物, 通过手性方酰胺C15及C16促进与3-三氟乙基亚胺氧吲哚2g的不对称1, 3-偶极环化串联反应.值得注意的是, 蒋先兴通过控制催化剂及添加剂苯甲酸的用量, 来控制反应的非对映选择性, 合成具有光学纯的两种含三氟甲基的双螺环氧吲哚非对映异构体82和82' (Scheme 19c).

2016年, 王锐课题组[47]利用2-氧代丙二酸二乙酯与三氟乙胺盐酸盐缩合, 合成了高反应活性的三氟乙胺衍生的酮亚胺, 受CF3和酮亚胺基团的影响, 亚甲基中氢原子的酸度得到了增强, 提供了进行1, 3-偶极环加成反应的可能性.随后他们成功通过方酰胺催化剂C16催化三氟乙胺衍生的酮亚胺84进行翻转反应, 再与3-亚甲基氧吲哚2h进行不对称1, 3-偶极环加成串联反应, 得到2'-CF3-吡咯烷基螺环氧吲哚87, 而非5'-CF3-吡咯烷基螺环氧吲哚86.该策略仅需1 mol%催化剂, 即可实现含三氟甲基螺环氧吲哚高效高立体选择性合成(Scheme 20a).
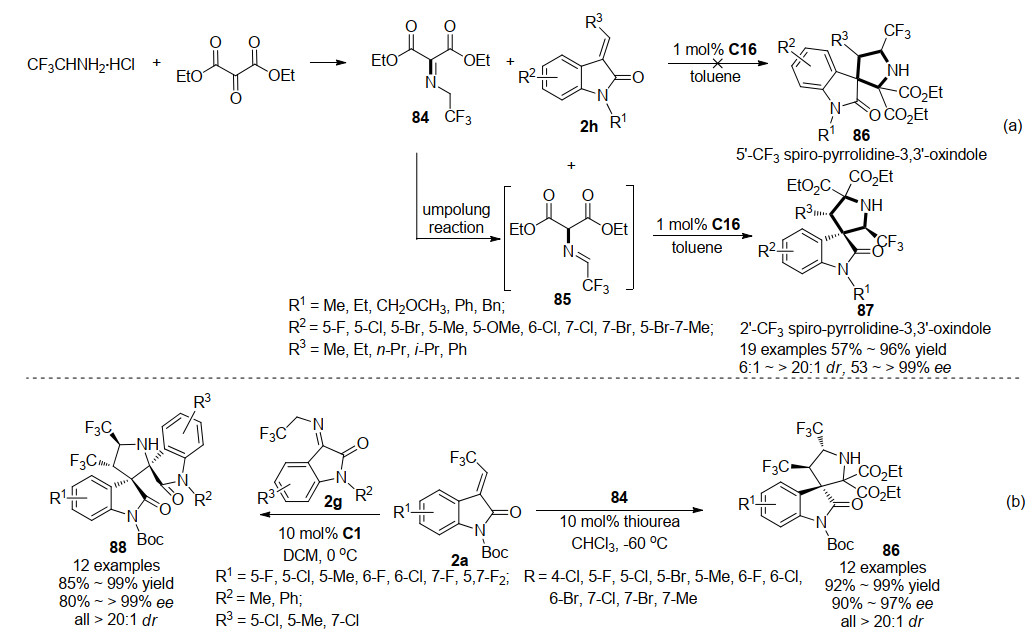
2018年, 鲁桂课题组[48]也报道了一例三氟乙胺衍生的酮亚胺84与3-三氟乙烯基氧吲哚2a的不对称1, 3-偶极环加成反应(Scheme 20b), 该反应与王锐所报道的不同, 未发生三氟乙胺衍生的酮亚胺的翻转反应, 直接通过硫脲催化不对称1, 3-偶极串联反应, 高收率(92%~99%), 高立体选择性(>20:1 dr, 90%~97% ee)地得到了含双三氟甲基的吡咯烷基螺环氧吲哚86.另外, 鲁桂课题组还尝试了方酰胺C1催化的3-三氟乙基亚胺氧吲哚2g与3-三氟乙烯基氧吲哚2a的不对称1, 3-偶极环加成反应, 同样能以优秀的产率和立体选择性获得含双三氟甲基的吡咯烷基螺环双氧吲哚化合物88.
2016年, 韩政良课题组[49]报道了一例方酰胺催化的3-亚烷基氧吲哚2i与靛红2f的不对称插烯aldol/环化串联反应, 将这类乙烯基亲核试剂3-亚烷基氧吲哚应用于Aldol反应中.作者利用方酰胺C1和C17为催化剂, 二氯甲烷为溶剂, 室温条件下高产率(62%~99%)、高对映选择性(87%~99%/87%~99% ee)地构建了一组具有潜在生物活性的骨架分子, 螺环氧吲哚并二氢吡喃酮对映异构体(Scheme 21a).作者还进一步尝试了3-亚芳基氧吲哚的不对称插烯aldol/环化串联反应, 无论是顺势还是反式结构, 都能获得高收率及高对映选择性.根据产物绝对构型和前人报告的机理, 韩政良课题组提出了该反应的催化机理, 3-亚烷基氧吲哚2i被催化剂C1质子化生成顺式烯醇酸酯G, 然后通过Si面进攻靛红2f得到醇盐中间体H.中间体H质子化后便得到产物89, 并使催化剂C1再生.

2017年, 韩政良课题组[50]又报道了一例以环烷烃取代的3-亚甲基氧吲哚2j与靛红2f的不对称插烯aldol/环化串联反应(Scheme 21b), 同样通过方酰胺催化剂C1催化, 可以顺利合成大量具有连续季碳和叔碳立体中心的螺环氧吲哚内酯化合物90, 并获得中等至良好的收率(62%~99%), 以及中等至优秀的立体选择性(7:1~>20:1 dr, 42%~99% ee).为了对比方酰胺与硫脲对该反应的催化效果, 作者在以上两项工作中都尝试了硫脲催化剂, 结果表明, 当底物为3-乙烯基氧吲哚2i或环状3-亚烷基氧吲哚2j时, 方酰胺的催化效果明显更出色, 而当底物为非环状3-亚烷基氧吲哚2k时, 方酰胺无法催化反应, 硫脲催化剂可以获得70%~99%的收率, 6:1~>20:1的dr值以及73%~>99%的ee值(Scheme 21c).
2016年, 杜欣课题组[51]首次报道了手性4-噻唑烷酮并螺环氧吲哚化合物的合成方法.该方法基于3-亚胺氧吲哚2e与2, 5-二羟基-1, 4-二噻烷92的不对称[3+2]环化反应形成螺环氧吲哚并酮亚胺化合物93, 再进一步利用氯铬酸吡啶盐(PCC)氧化得到4-噻唑烷酮并螺环氧吲哚94 (Scheme 22a).作者利用方酰胺C1为催化剂, 二氯甲烷为溶剂, 以中等总产率(51%~65%)、出色对映选择性(86%~98% ee)获得目标产物.随后, 作者又使用间氯过氧苯甲酸(m-CPBA)进一步处理产物, 通过控制间氯过氧苯甲酸的投放比例来控制氧化进程, 进而得到不同的氧化产物95及96.
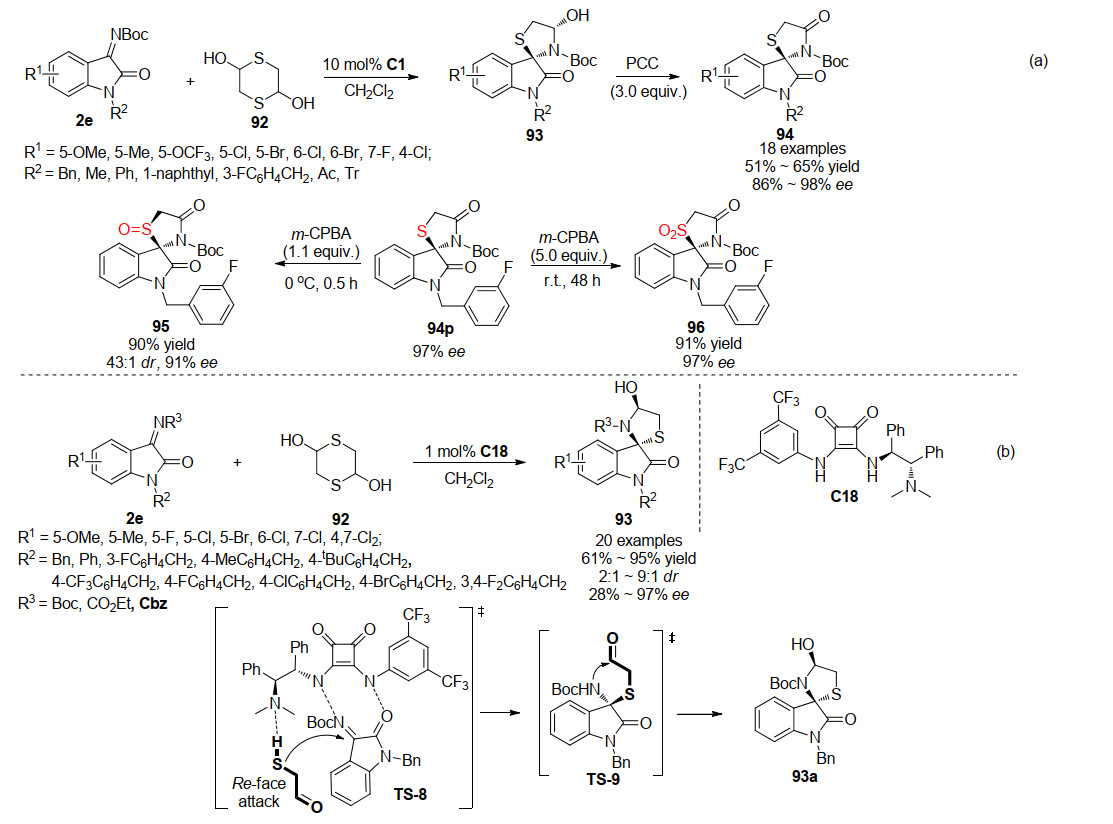
同年, 李鑫课题组[52]背靠背地报道了一例合成4-噻唑烷酮并螺环氧吲哚化合物的不对称方法, 同样是以3-亚胺氧吲哚2e与2, 5-二羟基-1, 4-二噻烷92为底物, 利用方酰胺催化剂C18催化不对称[3+2]环化反应获得螺环氧吲哚并酮亚胺化合物93.此工作所需催化剂用量相较杜欣课题组的报道更少, 仅需1 mol%.另外, 李鑫课题组还对此反应提出了一个可能的反应机理, 如Scheme 22b所示.
催化不对称Diels-Alder环加成反应已成为快速构建复杂手性环状分子的有效方法, 然而关于方酰胺催化的不对称Diels-Alde串联反应构建螺环氧吲哚的报道却非常有限. 2015年, 鲁桂课题组[53]成功利用方酰胺催化剂C16, 催化3-亚甲基氧吲哚2b与2-亚苄基吲哚97进行不对称Diels-Alder串联反应, 快速高效地构建了许多高度官能化的咔唑螺环氧吲哚骨架化合物98 (Eq. 5).该策略具有广泛的底物适应性, 可获得48%~90%的产率, 4:1~>20:1的dr以及4%~99%的ee.当取代基R5为氰基时, 反应的立体选择性急剧下降(4:1 dr, 4% ee); 当取代基R2为苯基时, 反应几乎不发生.
|
|
(5) |
2015年, 孙兴文课题组[54]报道了一例方酰胺催化的三组分(1, 3-二酮化合物99、硝基烯烃68及3-亚甲基氧吲哚2b)一锅法连续多步不对称Michael/Michael/aldol串联反应(Scheme 23a).起初, 孙兴文课题组以无保护的3-亚甲基氧吲哚为底物尝试反应, 不幸的是, 反应24 h后没有获得所需的螺环己烷氧吲哚, 只以几乎定量的收率分离出1, 3-二酮和硝基烯烃的迈克尔加合物.随后, 测试了3-亚甲基氧吲哚N原子上的不同保护基. N-甲基或N-苄基保护的底物在该串联反应中表现出很低的反应活性, N-乙酰基保护的底物能以15%的产率、1:1的dr及>99%的ee得到产物, 而N-Boc保护的底物能以25%的收率、>20:1的dr及>99%的ee得到产物.因此, 作者采用N-Boc保护的3-亚甲基氧吲哚为底物进行反应条件的优化.为了进一步提高反应的产率, 作者采用分步反应的方式, 将1, 3-二酮化合物99与硝基烯烃68在方酰胺催化条件下反应1 h, 随后加入3-亚甲基氧吲哚进行一锅法串联反应21 h, 分步一锅法反应大大提高了反应的收率.最终确定以方酰胺C10为催化剂, 二氯甲烷为溶剂, N-Boc保护的3-亚甲基氧吲哚为底物, 室温下分步一锅法为最优的反应条件.对反应产物的研究发现, 该反应历经不对称Michael/Michael/aldol串联反应, 以中等至良好的产率(18%~89%)、出色的立体选择性(>20:1 dr, >99% ee)得到环己基螺环氧吲哚100而非Michael/Michael/Henry串联反应得到环己基螺环氧吲哚101.
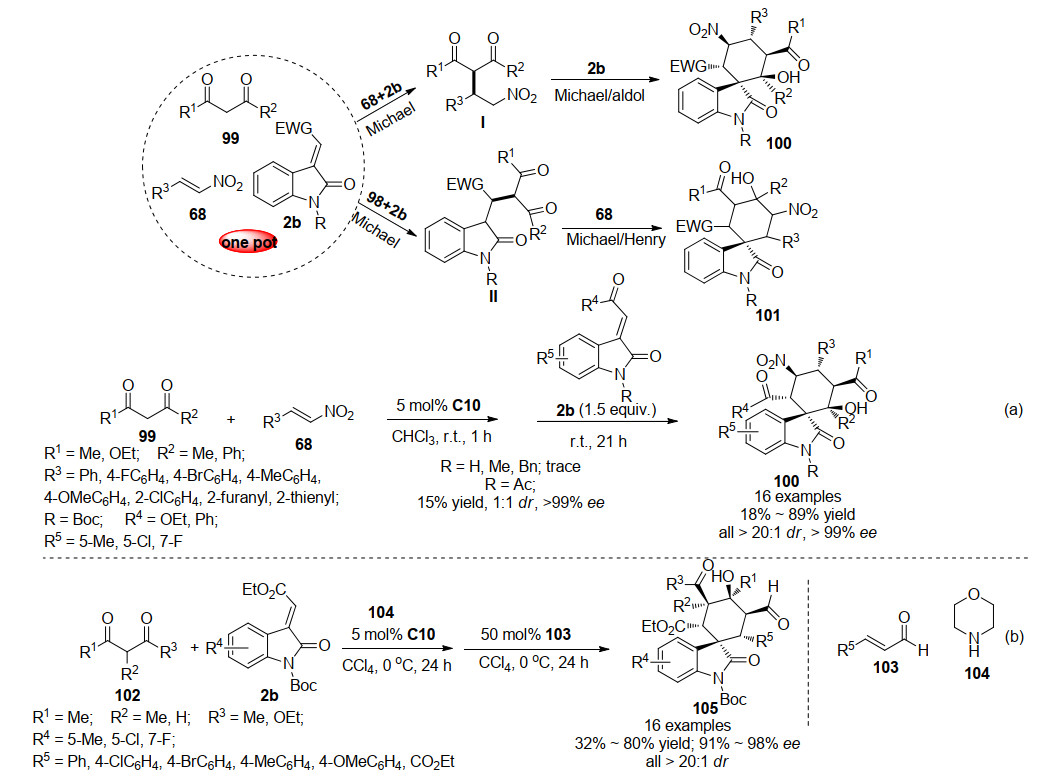
随后, 2016年孙兴文课题组[55]利用同样的反应策略, 以1, 3-二酮化合物102、3-亚甲基氧吲哚2b及肉桂醛103为底物, 方酰胺C10及吗啉104为催化剂, 分步一锅法不对称Michael/Michael/aldol串联反应合成了全取代环己基螺环氧吲哚105.与之前报道不同的是, 该方法首先将1, 3-二酮化合物102与3-亚甲基氧吲哚2b在方酰胺催化条件下进行第一步反应, 随后加入肉桂醛及吗啉催化剂进行下一步串联反应(Scheme 23b).
2019年, 阳华课题组[56]开发了一种新的高效催化策略, 利用手性方酰胺的有机催化体系, 建立了3-羟基氧吲哚1h和不带保护基的3-亚甲基氧吲哚2b的一锅法分步不对称Michael/内脂化/内酰胺化串联反应(Scheme 24).首先在方酰胺C17氩气保护条件下进行不对称Michael内脂化/反应, 随后在对甲苯磺酸加热到80 ℃条件下进一步发生内酰胺化反应, 以高收率、高对映选择性及中等至良好的非对映选择性获得了具有三个连续手性中心的新型螺环氧吲哚并喹啉酮化合物106.根据实验结果和主要异构体的绝对构型, 作者初步提出了可能的涉及催化剂双重活化的反应机理.另外, 在该催化模式下, 作者还尝试了3-氨基氧吲哚1c的反应效果, 取得84%的收率, 93:7的dr以及71%的ee, 反应的对映选择性有明显的下降.
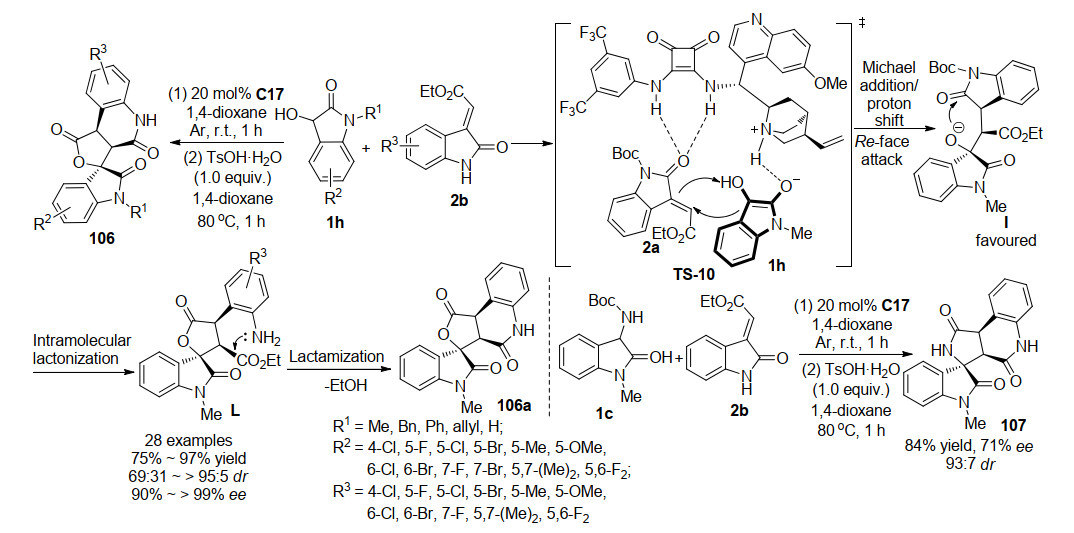
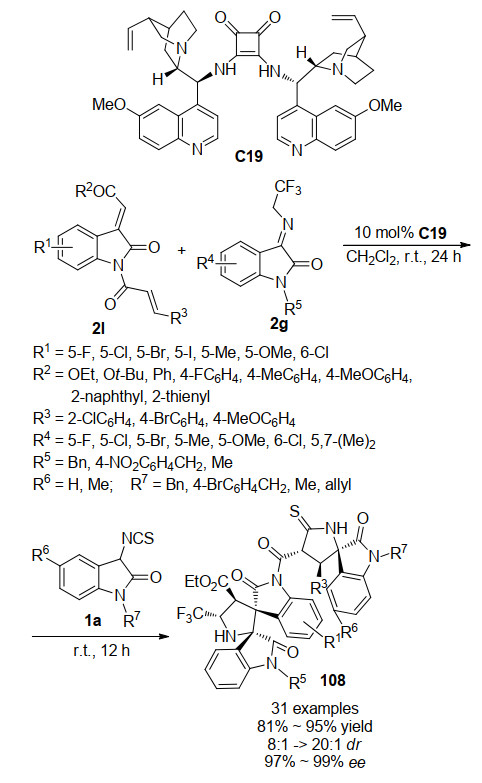
2019年, 本课题组[57]也开发了一例方酰胺催化的三组分一锅法分步串联反应(Scheme 25), 设计了3-亚甲基氧吲哚与肉桂酰氯反应, 合成了具有多个亲电反应位点的新型氧吲哚串联反应试剂2l, 并利用对环外烯烃具有高反应活性的3-三氟乙基亚胺氧吲哚2g与其发生第一步区域选择性的不对称Michael/Mannich串联反应, 随后一锅法加入第三组分3-异硫氰酸酯氧吲哚1a进行不对称Michael/环化串联反应, 得到具有七个手心中心的多螺环杨吲哚化合物108.该反应具有广泛的底物适应性, 能够以很好的反应活性及立体选择性得到目标产物.
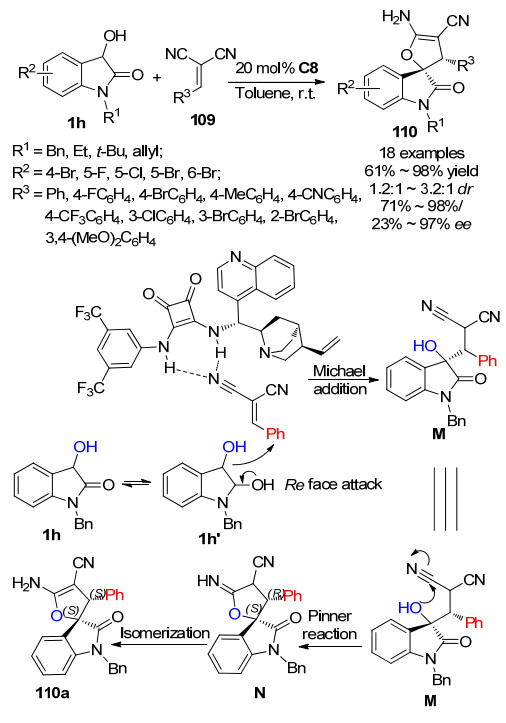
2019年, Pan课题组[58]报道了3-羟基氧吲哚1h与亚苄基丙二腈109的不对称Michael/Pinner/isomerization串联反应, 合成了二氢呋喃基螺环氧吲哚110.通过方酰胺C8催化剂催化, 该串联反应能以良好的产率(61%~98%)及对映选择性(71%~98%)获得目标产物.然而, 该策略的不足之处是, 反应的非对映选择性无法良好地控制, 仅能以1.2:1~3.2:1的dr获得一对非对映异构体, 且对非主要非对映异构体的对映选择性较差, 取得23%~97%的ee.为了更好地理解产物的反应途径和立体化学, 作者提出了一个合理的反应机理.如Scheme 26所示, 方酰胺催化剂C8的N—H部分与亚苄基丙二腈109的氰基结合形成氢键, 因此亚苄基丙二腈的Si面被催化剂C8封闭.另一方面, 催化剂的奎宁丁活化3-羟基氧吲哚1h, 通过互变异构形成其烯醇式1h', 并从Re面与亚苄基丙二腈发生Michael加成反应, 生成中间体M, 随后进行Pinner反应得到中间体N.最后, 中间体N异构化生成了产物110a.
双功能方酰胺催化的不对称串联反应已被认为是构建螺环氧吲哚骨架化合物最强力、最有效的策略之一.在过去的五年中, 我们目睹了该领域的重大发展.在这篇综述中, 我们着重介绍了多个课题组通过不同的方酰胺催化串联反应策略对螺环氧吲哚结构化合物进行对映选择性合成的最新进展, 包括我们小组的最新进展.然而尽管近年来在该领域中新的不对称催化方法发展迅速, 但是在起始材料和催化剂用量及回收再利用方面仍有改进的空间.尤其是在开发易于获得的底物、不对称合成大分子多元螺环氧吲哚以及构建轴手性吲哚骨架方面仍然需要努力.因此, 突破这些挑战性问题, 开发出简单、实用的合成方法, 以有效地构建多种螺环氧吲哚, 特别是大元环稠合的螺环氧吲哚, 仍是化学家们所面临的挑战.同时进一步发展手性方酰胺的不对称催化反应策略, 也将促进该类催化剂在手性药物开发和复杂手性分子合成等方面的应用.
(a) Ding, Y.; Tian, Z.; Zhu, N. Chin. J. Org. Chem. 2010, 30, 1156(in Chinese).
(丁研, 田喆, 朱能, 有机化学, 2010, 30, 1156.)
(b) Rios, R. Chem. Soc. Rev. 2012, 41, 1060.
(c) Carreira, E. M.; Fessard, T. C. Chem. Rev. 2014, 114, 8257.
(d) Ding, A.; Meazza, M.; Guo, H.; Yang, J. W.; Rios, R. Chem. Soc. Rev. 2018, 47, 5946.
(a) Ball-Jones, N. R.; Badillo, J. J.; Franz, A. K. Org. Biomol. Chem. 2012, 10, 5165.
(b) Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, Ⅲ, C. F. ACS Catal. 2014, 4, 743.
(c) Pavlovska, T. L.; Redkin, R. G.; Lipson, V. V.; Atamanuk, D. V. Mol. Diversity 2016, 20, 299.
(d) Yu, B.; Cai, Z.; Wang, S.; Liu, H. Chin. J. Org. Chem. 2017, 37, 1952(in Chinese).
(余斌, 蔡祖恽, 王帅, 刘宏民, 有机化学, 2017, 37, 1952.)
(a) Vine, K. L.; Matesic L.; Locke, J. M.; Ranson, M.; Skropeta, D. Anti-Cancer Agents Med. Chem. 2009, 9, 397.
(b) Arun, Y.; Bhaskar, G.; Balachandran, C.; Ignacimuthu, S.; Perumal, P. T. Bioorg. Med. Chem. Lett. 2013, 23, 1839.
(c) Das, D.; Banerjee, R.; Mitra, A. J. Chem. Pharm. Res. 2014, 6, 108.
(d) Bian, Z.; Marvin, C. C.; Pet-tersson, M.; Martin, S. F. J. Am. Chem. Soc. 2014, 136, 14184.
(e) Kathirvelan, D.; Haribabu, J.; Reddy, B. S. R.; Balachandran, C.; Duraipandiyan, V. Bioorg. Med. Chem. Lett. 2015, 25, 389.
(a) Hong L, Wang R. Adv. Synth. Catal. 2013, 355, 1023.
(b) Mei, G.-J.; Shi, F. Chem. Commun. 2018, 54, 6607.
(a) Grondal, C.; Jeanty, M.; Enders, D. Nat. Chem. 2010, 2, 167.
(b) Ying, A.; Wu, C.; Fu, Y.; Ren, S.; Liang, H. Chin. J. Org. Chem. 2012, 32, 1587(in Chinese).
(应安国, 武承林, 付永前, 任世斌, 梁华定, 有机化学, 2012, 32, 1587.)
(c) Chanda, T.; Zhao, J. C.-G. Adv. Synth. Catal. 2018, 360, 2.
Malerich, J. P.; Hagihara, K.; Rawal, V. H. J. Am. Chem. Soc. 2008, 130, 14416. doi: 10.1021/ja805693p
(a) Storer, R. I.; Aciro, C.; Jones, L. H. Chem. Soc. Rev. 2011, 40, 2330.
(b) Rouf, A.; Tanyeli, C. Curr. Org. Chem. 2016, 20, 2996.
(c) Han, X.; Zhou, H.-B.; Dong, C. Chem. Rec. 2016, 16, 897.
Chauhan, P.; Mahajan, S.; Kaya, U.; Hack, D.; Enders, D. Adv. Synth. Catal. 2015, 357, 253. doi: 10.1002/adsc.201401003
Chen, W.-B.; Wu, Z.-J.; Hu, J.; Cun, L.-F.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2011, 13, 2472. doi: 10.1021/ol200724q
(a) Han, W.-Y.; Li, S.-W.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. Chem.-Eur. J. 2013, 19, 5551.
(b) Chen, Q.; Liang, J.; Wang, S.; Wang, D.; Wang, R. Chem. Commun. 2013, 49, 1657.
(c) Tan, F.; Lu, L.-Q.; Yang, Q.-Q.; Guo, W.; Bian, Q.; Chen, J.-R.; Xiao, W.-J. Chem.-Eur. J. 2014, 20, 3415.
(d) Wang, L.-Q.; Yang, D.-X.; Li, D.; Liu, X.-H.; Zhao, Q.; Zhu, R.-R.; Zhang, B.-Z.; Wang, R. Org. Lett. 2015, 17, 4260.
Liu, L.; Zhao, B.-L.; Du, D.-M. Eur. J. Org. Chem. 2016, 2016, 4711.
Du, D.; Jiang, Y.; Xu, Q.; Li, X.-G.; Shi, M. ChemistryOpen 2016, 5, 311, doi: 10.1002/open.201600034
Kayal, S.; Mukherjee, S.; Org. Biomol. Chem. 2016, 14, 10175. doi: 10.1039/C6OB02187E
Lin, Y.; Liu, L.; Du, D.-M. Org. Chem. Front. 2017, 4, 1229. doi: 10.1039/C6QO00852F
Lin, N.; Long, X.-W.; Chen, Q.; Zhu, W.-R.; Wang, B.-C.; Chen, K.-B.; Jiang, C.-W.; Weng, J.; Lu, G. Tetrahedron 2018, 74, 3734. doi: 10.1016/j.tet.2018.05.052
Song, Y.-X.; Du, D.-M. Synthesis 2018, 50, 1535. doi: 10.1055/s-0036-1591527
Zhu, W.-R.; Chen, Q.; Lin, N.; Chen, K.-B.; Zhang, Z.-W.; Fang, G.; Weng, J.; Lu, G. Org. Chem. Front. 2018, 5, 1375. doi: 10.1039/C8QO00044A
(a) Zhao, B.-L.; Lin, Y.; Du, D.-M. Adv. Synth. Catal. 2019, 361, 3387.
(b) Lin, Y.; Zhao, B.-L.; Du, D.-M. J. Org. Chem. 2019, 84, 10209.
Chen, J.; Huang, B.-Q.; Wang, Z.-Q.; Zhang, X.-J.; Yan, M. Org. Lett. 2019, 21, 9742. doi: 10.1021/acs.orglett.9b03911
Li, J.-H.; Feng, T.-F.; Du, D.-M. J. Org. Chem. 2015, 80, 11369. doi: 10.1021/acs.joc.5b01940
Song, Y.-X.; Du, D.-M. Org. Biomol. Chem. 2019, 17, 5375. doi: 10.1039/C9OB00998A
Wen, J.-B.; Du, D.-M. Org. Biomol. Chem. 2020, 18, 1647. doi: 10.1039/C9OB02663K
Zhao, B.-L.; Du, D.-M. Chem. Commun. 2016, 52, 6162. doi: 10.1039/C6CC00705H
Zhao, B.-L.; Du, D.-M. Adv. Synth. Catal. 2016, 358, 3992. doi: 10.1002/adsc.201600782
Vagh, S. S.; Karanam, P.; Liao, C.-C.; Lin, T.-H.; Liou, Y.-C.; Edukondalu, A.; Chen, Y.-R.; Lin, W. Adv. Synth. Catal. 2020, 362, 1. doi: 10.1002/adsc.201901597
Chaudhari, P. D.; Hong, B.-C.; Wen, C.-L.; Lee, G.-H. ACS Omega 2019, 4, 655. doi: 10.1021/acsomega.8b03049
Chen, L.; Wu, Z.-J.; Zhang, M.-L.; Yue, D.-F.; Zhang, X.-M.; Xu, X.-Y.; Yuan, W.-C. J. Org. Chem. 2015, 80, 12668. doi: 10.1021/acs.joc.5b02253
Ming, S.; Zhao, B.-L.; Du, D.-M. Org. Biomol. Chem. 2017, 15, 6205. doi: 10.1039/C7OB01307H
Yang, Z.-T.; Zhao, J.; Yang, W.-L.; Deng, W.-P. Org. Lett. 2019, 21, 1015. doi: 10.1021/acs.orglett.8b04039
Sun, Q.-S.; Zhu, H.; Chen, Y.-J.; Yang, X.-D.; Sun, X.-W.; Lin, G.-Q. Angew. Chem., Int. Ed. 2015, 54, 13253. doi: 10.1002/anie.201506206
Wang, C.-C.; Huang, J.; Li, X.-H.; Kramer, S.; Lin, G.-Q.; Sun, X.-W. Org. Lett. 2018, 20, 2888. doi: 10.1021/acs.orglett.8b00927
Kumarswamyreddy, N.; Kesavan, V. Org. Lett. 2016, 18, 1354. doi: 10.1021/acs.orglett.6b00287
Lin, Y.; Zhao, B.-L.; Du, D.-M. J. Org. Chem. 2018, 83, 7741. doi: 10.1021/acs.joc.8b00632
Zhao, B.-L.; Du, D.-M. Asian J. Org. Chem. 2015, 4, 1120. doi: 10.1002/ajoc.201500306
Tang, Q.-G.; Cai, S.-L.; Wang, C.-C.; Lin, G.-Q.; Sun, X.-W. Org. Lett. 2020, 22, 3351. doi: 10.1021/acs.orglett.0c00779
Zhao, B.-L.; Du, D.-M. Org. Lett. 2018, 20, 3797. doi: 10.1021/acs.orglett.8b01389
(a) Moyano, A.; Rios, R. Chem. Rev. 2011, 111, 4703.
(b) Kaur, A.; Singh, B.; Vyas, B.; Silakari, O. Eur. J. Med. Chem. 2014, 79, 282.
(c) Adrio, J.; Carretero, J. C. Chem. Commun. 2014, 50, 12434.
Zhao, H.-W.; Yang, Z.; Meng, W.; Tian, T.; Li, B.; Song, X.-Q.; Chen, X.-Q.; Pang, H.-L. Adv. Synth. Catal. 2015, 357, 2492. doi: 10.1002/adsc.201500162
Sun, Q.-T.; Li, X.-Y.; Su, J.-H.; Zhao, L.; Ma, M.-X.; Zhu, Y.-Y.; Zhao, Y.-Y.; Zhu, R.-R.; Yan, W.-J.; Wang, K.-R.; Wang, R. Adv. Synth. Catal. 2015, 357, 3187. doi: 10.1002/adsc.201500416
Ma, M.-X.; Zhu, Y.-Y.; Sun, Q.-T.; Li, X.-Y.; Su, J.-H.; Zhao, L.; Zhao, Y.-Y.; Qiu, S.; Yan, W.-J.; Wang, K.-Y.; Wang, R. Chem. Commun. 2015, 51, 8789. doi: 10.1039/C4CC10216A
You, Y.; Lu, W.-Y.; Wang, Z.-H.; Chen, Y.-Z.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. Org. Lett. 2018, 20, 4453. doi: 10.1021/acs.orglett.8b01730
Chen, F.-Y.; Xiang, L.; Zhan, G.; Liu, H.; Kang, B.; Zhang, S.-C.; Peng, C.; Han, B. Tetrahedron Lett. 2020, 61, 151806. doi: 10.1016/j.tetlet.2020.151806
Huang, W.-J.; Chen, Q.; Lin, N.; Long, X.-W.; Pan, W.-G.; Xiong, Y.-S.; Weng, J.; Lu, G. Org. Chem. Front. 2017, 4, 472. doi: 10.1039/C6QO00723F
Song, Y.-X.; Du, D.-M. J. Org. Chem. 2018, 83, 9278. doi: 10.1021/acs.joc.8b01245
Zhao, X.; Xiong, J.; An, J.; Yu, J.; Zhu, L.; Feng, X.; Jiang, X.; Org. Chem. Front. 2019, 6, 1989. doi: 10.1039/C9QO00452A
Wang, C.; Wen, D.; Chen, H.; Deng, Y.; Liu, X.; Liu, X.; Wang, L.; Gao, F.; Guo, Y.; Sun, M.; Wang, K.; Yan, W. Org. Biomol. Chem. 2019, 17, 5514. doi: 10.1039/C9OB00720B
Su, J.; Ma, Z.; Li, X.; Lin, L.; Shen, Z.; Yang, P.; Li, Y.; Wang, H.; Yan, W.; Wang, K.; Wang, R. Adv. Synth. Catal. 2016, 358, 3777. doi: 10.1002/adsc.201600688
Zhu, W.-R.; Zhang, Z.-W.; Huang, W.-H.; Lin, N.; Chen, Q.; Chen, K.-B.; Wang, B.-C.; Weng, J.; Lu, G. Synthesis 2019, 51, 1969. doi: 10.1055/s-0037-1612089
Han, J.-L.; Chang, C.-H. Chem. Commun. 2016, 52, 2322. doi: 10.1039/C5CC08883F
Han, J.-L.; Tsai, Y.-D.; Chang, C.-H. Adv. Synth. Catal. 2017, 359, 4043. doi: 10.1002/adsc.201701104
Cheng, P.; Guo, W.; Chen, P.; Liu, Y.; Du, X.; Li, C. Chem. Commun. 2016, 52, 3418. doi: 10.1039/C5CC10292H
Feng, B.-X.; Yang, J.-D.; Li, J.; Li, X. Tetrahedron Lett. 2016, 57, 3457. doi: 10.1016/j.tetlet.2016.06.084
Huang, L.-J.; Weng, J.; Wang, S.; Lu, G. Adv. Synth. Catal. 2015, 357, 993. doi: 10.1002/adsc.201400780
Sun, Q.-S.; Chen, X.-Y.; Zhu, H.; Lin, H.; Sun, X.-W.; Lin, G.-Q. Org. Chem. Front. 2015, 2, 110. doi: 10.1039/C4QO00299G
Sun, Q.-S.; Lin, H.; Sun, X.; Sun, X.-W. Tetrahedron Lett. 2016, 57, 5673. doi: 10.1016/j.tetlet.2016.11.019
Ren, J.-W.; Zheng, L.; Ye, Z.-P.; Deng, Z.-X.; Xie, Z.-Z.; Xiao, J.-A.; Zhu, F.-W.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Org. Lett. 2019, 21, 2166. doi: 10.1021/acs.orglett.9b00477
Zhao, B.-L.; Du, D.-M. Adv. Synth. Catal. 2019, 361, 3412. doi: 10.1002/adsc.201900218
Balha, M.; Mondal, B.; Pan, S. C. Org. Biomol. Chem. 2019, 17, 6557. doi: 10.1039/C9OB01199D

图 1 含有螺环氧吲哚骨架的生物活性分子
Figure 1 Biologically active molecules with spirooxindole skeleton

图 2 三类用于有机催化不对称合成螺环氧吲哚化合物的起始原料
Figure 2 Three types of starting materials for the organocatalytic asymmetric synthesis of spirooxindoles.

图式 1 3-异硫氰酸酯氧吲哚与马来酰亚胺的Michael/环化串联反应及其机理
Scheme 1 Michael/cyclization cascade reaction of 3-isothio- cyanato oxindoles with maleimides and its mechanism

图式 2 3-异硫氰酸酯氧吲哚参与的Michael/环化串联反应
Scheme 2 Michael/cyclization cascade reactions involving 3-isothiocyanato oxindoles

图式 3 3-异硫氰酸酯氧吲哚与环外烯烃的Michael/环化串联反应
Scheme 3 Michael/cyclization cascade reactions of 3-isothiocyanato oxindoles with exocyclic olefins

图式 4 3-丙二腈氧吲哚与烯烃的Michael/环化串联反应及机理
Scheme 4 Michael/cyclization cascade reactions of 2-(1-methyl-2-oxoindolin-3-yl)malononitriles with olefins and its mechanism

图式 5 3-氨基氧吲哚与乙烯基磺酰氟的Michael/环化串联反应
Scheme 5 Michael/cyclization cascade reactions of 3-amino- oxindoles with ethylene sulfonyl fluorides

图式 6 3-氯氧吲哚参与的Michael/烷基化串联反应
Scheme 6 Michael/alkylation cascade reactions involving 3-chlorooxindoles

图式 7 方酰胺催化3-氯氧吲哚的不对称Michael/烷基化串联反应机理
Scheme 7 Mechanism of squaramide-catalyzed asymmetric Michael/alkylation cascade reaction of 3-chlorooxyindoles

图式 8 3-亚甲基氧吲哚与α-亚烷基琥珀酰亚胺的Michael/ Michael串联反应
Scheme 8 Michael/Michael cascade reaction of 3-methylene- indolinones with α-alkylidene succinimides

图式 9 3-亚甲基氧吲哚与3-取代氧吲哚的Michael/Michael串联反应
Scheme 9 Michael/Michael cascade reaction of 3-methylenein- dolinones with 3-substituted oxindoles

图式 10 3-羟基氧吲哚参与的Michael/lactonization串联反应
Scheme 10 Michael/lactonization cascade reactions involving 3-hydroxyoxindoles

图式 11 3-羟基氧吲哚与3-三氟乙烯基氧吲哚的Michael/内酯化串联反应
Scheme 11 Michael/lactonization cascade reaction of 3-hydroxyoxindoles with 3-trifluoroethylidene oxindoles

图式 12 3-亚甲基氧吲哚参与的三组分一锅法Michael/Mannich串联反应
Scheme 12 One-pot three-component Michael/Mannich cascade reaction involving 3-methyleneindolinones

图式 13 三组分一锅法Michael/Mannich串联反应机理
Scheme 13 Mechanism of squaramide-catalyzed one-pot three-component Michael/Mannich cascade reaction

图式 14 2-吲哚酮参与的三组分一锅法Michael/Mannich串联反应
Scheme 14 One-pot three-component Michael/Mannich cascade reaction involving oxindoles

图式 15 α, β-不饱和酮酸酯衍生的氧吲哚参与的Michael/半缩酮化串联反应
Scheme 15 Michael/hemiketalization cascade reactions involving isatin-derived β, γ-unsaturated α-keto esters

图式 16 3-亚甲基氧吲哚与对甲苯磺酰胺基氨基甲基烯酮或烯酯的aza-Michael/Michael串联反应及机理
Scheme 16 Aza-Michael/Michael cascade reactions of 3-me- thyleneindolinones with tosylaminomethyl enones or enoates and its mechanism

图式 17 3-亚胺氧吲哚与2-异氰酸酯茚酮的Mannich/环化串联反应及衍生化反应
Scheme 17 Mannich/cyclization cascade reaction of isatinimines with 2-isothiocyanato-1-indanones and further transformations

图式 18 3-三氟乙基亚胺氧吲哚与缺电子烯烃的1, 3-偶极环化串联反应
Scheme 18 1, 3-偶极 cyclization cascade reactions of N-2, 2, 2-trifluoroethylisatin ketamines with electron-deficient olefins

图式 19 3-三氟乙基亚胺氧吲哚与环外烯烃的1, 3-偶极环化串联反应
Scheme 19 1, 3-偶极 cyclization cascade reactions of N-2, 2, 2-trifluoroethylisatin ketamines with exocyclic olefins

图式 20 3-亚甲基氧吲哚参与的1, 3-偶极环化串联反应
Scheme 20 1, 3-偶极 cyclization cascade reactions involving 3-methyleneindolinones

图式 21 3-亚烷基氧吲哚与靛红的插烯aldol环化串联反应
Scheme 21 Vinylogous aldol/cyclization cascade reactions of 3-alkylidene oxindoles with isatins

图式 22 3-亚胺氧吲哚与2, 5-二羟基-1, 4-二噻烷的[3+2]环化串联反应
Scheme 22 [3+2] annulation reactions of isatinimines with 1, 4-dithiane-2, 5-diols

图式 23 方酰胺催化的Michael/Michael/aldol顺序串联反应
Scheme 23 Squaramide-catalyzed Michael/Michael/aldol cascade sequence

图式 24 3-羟基氧吲哚与3-亚甲基氧吲哚的Michael/内酯化/内酰胺化顺序串联反应
Scheme 24 Michael/lactonization/lactamization sequence of 3-hydroxyoxindoles with 3-methyleneindolinones

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





 下载:
下载:

