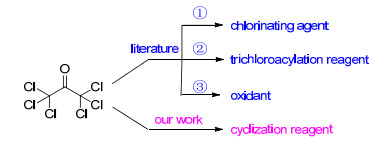
图式 1.
本工作思路来源和方法提出
Scheme 1.
Ideas and methods of this work

六氯丙酮简单易得, 反应后易于分离, 常用作氯离子的提供试剂, 用于烯胺的氯化[1]以及糖半缩醛的无溶剂氯化[2], 或者用作三氯甲酰化试剂, 在中性条件下制备三氯乙酰胺和三氯乙酸酯[3], 也可以和二甲基亚砜(DMSO)一起用作氧化剂, 如用于卞醇的氧化[4].
联苯并噻唑作为一类二齿配体, 能与一系列的无机金属离子进行配位[5].此外, 与一系列含氮类杂环化合物一样[6], 它也具有一定的生物活性[7].以联苯并噻唑作为重复结构单元的聚苯并噻唑, 不仅是一类具有光电活性的红色荧光材料[8], 还是一种耐高温耐化学氧化和尺寸稳定性的高性能材料[9].联苯并噻唑的制备一般通过使用2位取代的苯并噻唑和一分子的邻氨基苯硫醇进行反应, 比如使用2-甲酰氯苯并噻唑[10]和2-醛基苯并噻唑[11], 但是这两种底物的获取均不方便, 也可以通过草酸和邻氨基苯硫醇进行缩合[12].对于苯并噻唑类聚合物聚合物的制备, 实验室常用二元酸和2, 5-二氨基对苯二硫醇进行缩聚, 这种制备方法要求较高的反应温度和强酸性的反应溶剂.另外, 也有报道通过苯撑二噻唑的金属催化偶联进行制备[13].
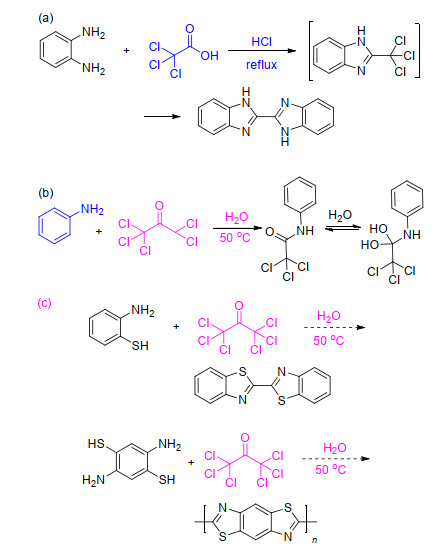
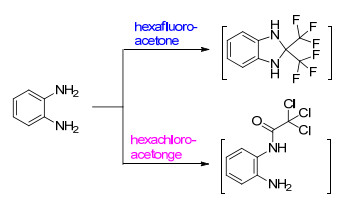
研究发现, 传统的合成方法中(Scheme 1a), 有一个活性很大的中间体2-三氯甲基苯并咪唑, 其对联苯并咪唑的构建起到关键作用[14].因此, 我们将从改变中间体的合成途径角度, 在避免高温和强酸性条件下制备联苯并噻唑.而根据文献报道的六氯丙酮的反应[15](Scheme 1b), 六氯丙酮可以作为一种制备2-三氯甲基联苯并噻唑的温和试剂.
如Scheme 1c所示, 尝试将六氯丙酮与邻氨基苯硫醇反应, 以提供一种新的绿色合成联苯并噻唑的方法, 并利用其能连接杂环的特点, 与2, 5-二氨基对苯硫醇反应来制备聚苯撑二噻唑.该方法的提出除了有效避免了强酸和高温带来的环境问题, 还提出了一种不同于常规报道的六氯丙酮的用途, 该法中六氯丙酮提供了两个碳源分别成环(Scheme 2).
从六氯丙酮和邻氨基苯硫醇出发, 对合成联苯并噻唑的反应条件进行了探索.首先根据化学计量比确定了反应底物的投量比为2.5:1(物质的量之比), 接着从绿色化学角度出发, 尝试以水作为反应溶剂, 设定反应温度为50 ℃, 最后对反应时间进行优化.我们发现, 产物的沉淀发生在反应开始后的40 min左右, 于是对不同反应时间下的产率进行了计算, 结果如表 1所示.随着反应时间的增加, 产率逐步上升, 反应2 h后产率上升为65%(表 1, Entry 3), 之后延长反应时间, 产率基本不再上升.因此, 基于水为溶剂、反应温度为50 ℃下的联苯并噻唑合成的最佳反应时间为2 h.对联苯并噻唑进行了单晶结构的表征,结果表明联苯并噻唑的两个苯并噻唑以头对头连接的方式形成共平面结构, C14H8N2S2的晶体学数据以CCDC编号1887631存放在剑桥晶体学数据中心.
 下载:
导出CSV
下载:
导出CSV
| Entry | Reaction timea/min | Yield/% |
| 1 | 60 | 20 |
| 2 | 90 | 43 |
| 3 | 120 | 65 |
| 4 | 150 | 64 |
| 5 | 180 | 67 |
| a The optimal reaction time of bibenzothiazole based on hexachloroacetone, n(HCA):n(2-aminobenzenethiol)=2.5:1, water as solvent under 50 ℃. | ||
接着在优化后的条件下分别用邻苯二胺、邻氨基苯酚和六氯丙酮进行反应, 成功制备了联苯并咪唑, 产率为75%(表 2, Entry 2).对其单晶结构进行表征, 单晶结果表明其有两个苯并咪唑头对头连接的共平面结构, C14H10N2的晶体学数据已以CCDC编号1959008的形式存放在剑桥晶体学数据中心.用同样的方法尝试合成联苯并噁唑, 没有得到产物(表 2, Entry 3).由表 2 Entries 1和2对比可知, 联苯并噻唑的产率(65%)低于联苯并咪唑(75%), 这是因为巯基的亲核性弱于氨基.这也是Philips反应中, 相对于邻苯二胺, 邻氨基苯硫醇必须在酸性更强的条件下才能和二元酸反应的原因[16].此外, 从表 2 Entry 3可知, 六氯丙酮和邻氨基苯酚不反应, 这一结果和一些醛基与二元芳香胺进行缩合时, 只能有效缩合邻氨基苯硫醇和邻苯二胺的文献报道一致[17], 即苯并噁唑一般需要催化剂参与反应才能进行[18].这可能与苯并噁唑啉稳定性有关, 后者若没有合适的催化剂催化脱氢, 很容易重新分解成起始原料[19].
下载:
导出CSV
| Entry | Substrate | Product | Yield/% |
| 1a |  |
 |
65 |
| 2a |  |
 |
75 |
| 3a |  |
— | — |
| 4b |  |
 |
— |
| 5a |  |
 |
— |
| a All the reactions conducted under the optimized conditions. b All the reagents were prepared by experiments according to literature reports. The reactions were carried out under the optimized conditions. | |||
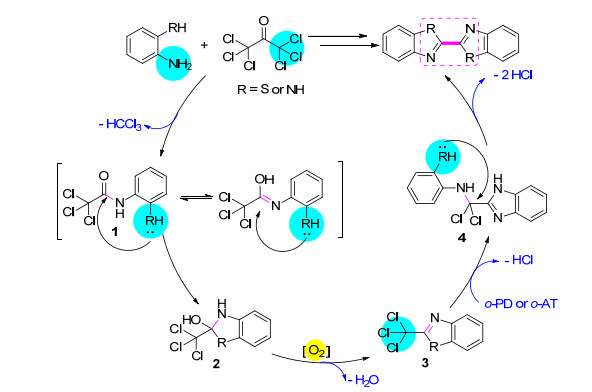
此外, 对不同取代基取代的邻苯二胺也进行了实验, 4-甲基邻苯二胺与六氯丙酮反应3 h得到了产率68%的5, 5'-二甲基联苯并咪唑(表 2, Entry 6), 4-氯苯-1, 2-二胺在延长反应时间至5 h后也未出现明显的反应.该方法不适用于吸电子基取代的的邻苯二胺, 且取代基的空间位阻对该反应有较明显的影响.为了更好地理解反应过程, 进一步做了两个探索实验:在氮气保护护下, 进行六氯丙酮和邻氨基苯硫醇反应, 以达到隔氧的效果并观察反应结果; 在反应结束后的滤液中, 用硝酸银进行氯化银沉淀实验.隔氧实验结果显示不反应, 表明氧气参与该反应进程, 而氯化银沉淀实验证实了体系中游离氯离子的存在.综上所述, 我们提出邻氨基苯硫醇和六氯丙酮如Scheme 3所示的反应机理:首先邻氨基苯硫醇的氨基亲核进攻六氯丙酮的羰基, 一分子氯仿离去, 生成结构1; 进而分子内巯基进攻, 生成结构2, 后者在氧气氧化下经分子内脱水, 生成结构3; 接着体系中另一分子的邻氨基苯硫醇脱除一分子氯化氢生成结构4; 最后一分子氯化氢离去后生成联苯并噻唑.这一机理同样用于联苯并咪唑体系.而氧气氛实验结果表明, 在氧气氛下, 产率并没有提高, 说明作为氧化剂的氧气在该反应过程中仅需微量即可.
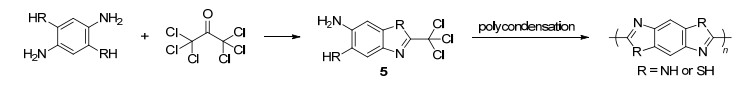
非常有趣的是, 在六氯丙酮的成环反应中, 它可以同时引入两个能进行杂环缩合的碳原子, 生成的两个杂环以头对头的形式以共价键相连.基于此, 用2, 5-二氨基对苯硫醇和均苯四胺代替邻苯二胺和邻氨基苯硫醇进行了反应, 成功制备了淡黄色的固体粉末聚苯撑二噻唑(表 2, Entry 4)和墨绿色的固体粉末聚苯撑二咪唑(表 2, Entry 5), 并且这两种聚合物产物均不溶于实验室常见的有机溶剂, 甚至难溶于浓硫酸.其中, 聚苯撑二噻唑的红外结果表明1462, 1428, 1309 cm-1处的吸收峰为噻唑环的伸缩振动峰, 925 cm-1处的吸收峰为苯环的吸收峰, 765 cm-1处的吸收峰为环的面外弯, 曲振动峰.聚苯撑二咪唑的红外特征吸收峰表明3475, 3367 cm-1处的是N—H与水分子或者链间氢键, 1612 cm-1处的是C=N伸缩振动峰.同样, 我们认为聚苯并唑类的合成可能也是通过Scheme 3所示的途径进行的, 而结构5是最重要的聚合中间体(Scheme 4).因此以2, 4, 5-三氨基苯硫醇作原料, 可以主链上同时含有苯并咪唑环和苯并噻唑环的聚合物.
由于六氟丙酮与六氯丙酮具有相似结构的骨架结构, 因此, 我们将六氟丙酮代替六氯丙酮与邻苯二胺进行反应, 在同样条件反应下2 h后, 没有白色沉淀生成.但将整个反应体系静置72 h后, 体系中突然生成大量的橘红色针状晶体, 最后确认其为邻苯二胺的氧化产物2, 3-二氨基酚嗪.这可能是由于三氟甲基不是易离去基团, 亲核进攻的反应位点在羰基上面, 这一点可以从相关文献报道得到佐证[20].如Scheme 5所示, 当六氟丙酮和邻苯二胺反应时, 生成的2, 2-二三氟甲基苯并咪唑作为中间体, 一定程度上延缓了邻苯二胺被氧气的氧化, 而六氯丙酮则是发生的氯仿消除反应.
阐明了六氯丙酮作为一个一步引入两个成环位点来构建联苯并唑类的试剂.运用六氯丙酮, 高效地合成了两个小分子(联苯并噻唑和联苯并咪唑)以及两种聚合物(聚苯撑二噻唑和聚苯撑二咪唑).使用六氯丙酮的成环, 提供了一种绿色合成路线, 所有这些杂环的合成只需水作溶剂, 反应温度低(50 ℃), 易分离(简单过滤), 纯度高.这不仅对传统的小分子杂环的合成具有重要意义, 而且在材料科学领域具有潜在应用价值, 特别是在制备高性能纤维聚合物中的应用, 六氯丙酮成环反应是一个非常不错的选择.相同条件下, 六氟丙酮虽然与六氯丙酮具有相似的分子骨架, 但无法实现六氯丙酮相同的功能.后续相关聚合物的制备正在进行中.
用Bruker AdvancedII (400 MHz)光谱仪(1H为600 MHz, 13C为101 MHz)记录1H NMR和13C NMR光谱.红外表征用溴化钾压片后进行固体红外的测量.使用石墨单色Mo Kα辐射在Nonius Kappa CCD衍射仪上收集晶体学数据.数据针对洛伦兹效应和极化效应进行了校正, 但并未针对吸收进行校正.通过直接方法(SHELSHEL XS)解析结构, 并通过针对F2的全矩阵最小二乘技术(SHELXL-97)进行精炼.溶剂水使用前蒸馏处理.除非另有说明, 否则所有物质和试剂均购买于九鼎化学试剂公司且使用前未经处理. 2, 5-二氨基对苯硫醇[21]和均苯四胺[22]根据文献报道进行合成.
在50 mL反应瓶中装入8 mL蒸馏水, 置于50 ℃的油浴搅拌器中.将邻氨基苯硫醇置于室温至呈液态, 用注射器称取2-氨基苯硫醇(2.5 mmol, 0.318 g)后将其转移至反应容器, 另用注射器称取六氯丙酮(1 mmol, 0.267 g)并逐滴将其滴加进反应装置.然后继续搅拌2 h.冷却后过滤得到淡黄色沉淀.沉淀用二氯甲烷洗涤3次, 然后在50 ℃的真空烘箱中干燥12 h, 得0.1744 g淡黄色固体a, 产率65%. m.p. 306 ℃ (Lit.[23] 306 ℃); 1H NMR (D2SO4, 600 MHz) δ: 8.46 (t, J=7.8 Hz, 1H), 8.53 (t, J=7.9 Hz, 1H), 8.81 (d, J=8.5 Hz, 1H), 8.85 (d, J=8.5 Hz, 1H); 13C NMR (D2SO4, 151 MHz) δ: 120.72 (s), 125.60 (s), 133.50 (s), 134.62 (s), 142.53 (s), 154.96~154.83 (m).
用化合物a的饱和N, N-二甲基甲酰胺(DMF)溶液置于冰箱养单晶.
在50 mL反应瓶中装入8 mL蒸馏水, 置于50 ℃的油浴搅拌器中.称取邻苯二胺(2.5 mmol, 0.273 g)并将其转移至反应瓶, 另用注射器称取六氯丙酮(1 mmol, 0.267 g)并逐滴将其滴加进反应瓶, 然后继续搅拌2 h.反应液首先由无色变成绿色, 然后有橘黄色固体析出, 并慢慢变淡后成白色, 最后沉淀出白色粉末团块.过滤, 再用乙醇洗涤3次, 在50 ℃的真空烘箱中干燥12 h, 得0.1756 g白色固体b, 产率75%. m.p. 469~470 ℃(Lit.[24] m.p.>300 ℃); 1H NMR (DMSO-d6, 400 MHz) δ: 7.28 (d, J=8.3 Hz, 4H), 7.56 (s, 2H), 7.75 (s, 2H), 13.56 (s, 2H); 13C NMR (DMSO-d6, 101 MHz) δ: 112.52 (s), 119.64 (s), 122.70 (s), 124.07 (s), 135.29 (s), 144.12 (d, J=31.7 Hz), 144.46~144.33 (m).核磁表征结果与文献[25]报道一致.
将化合物b的饱和乙酸乙酯溶液置于冰箱养单晶.
在50 mL反应瓶中装入8 mL蒸馏水, 置于50 ℃的油浴搅拌器中.称取4-甲基邻苯二胺(2.5 mmol, 0.301 g)并将其转移至反应瓶, 另用注射器称取六氯丙酮(1 mmol, 0.267 g)并逐滴将其滴加进反应瓶.然后继续搅拌3 h.反应液由褐色慢慢沉淀出白色粉末.过滤, 再用丙酮洗涤3次, 在50 ℃的真空烘箱中干燥12 h, 得0.178 g淡褐色固体c, 产率68%. m.p. 355 ℃; 1H NMR (DMSO, 400 MHz) δ: 2.45 (s, 6H), 7.11 (dd, J=15.9, 8.1 Hz, 2H), 7.69~7.27 (m, 4H), 13.35 (s, 2H); 13C NMR (101 MHz, DMSO) δ: 21.84 (s), 112.04 (d, J=12.6 Hz), 119.14 (s), 124.27 (s), 125.51 (s), 131.66 (s), 133.38 (d, J=8.1 Hz), 135.55 (s), 142.16 (s), 143.93 (s), 144.28 (s).核磁表征结果与文献[26]报道一致.
将邻氨基苯硫醇和六氯丙酮反应结束后的混合液过滤, 收集反应溶液于锥形瓶中, 配置硝酸银的饱和乙醇溶液, 然后逐滴将其加进瓶中并手动摇晃, 观察到白色沉淀的生成.
在50 mL反应管中装入8 mL蒸馏水, 塞上橡胶塞, 加入氮气保护装置后用油泵置换体系中残留的氧气, 置换时间约5 min, 置换结束后拧紧反应管的旋钮, 并持续通入氮气, 转移至50 ℃的油浴搅拌器中.将邻氨基苯硫醇置于室温至呈液态, 用注射器称取2-氨基苯硫醇(2.5 mmol, 0.318 g)后将其转移至反应容器, 另用注射器称取六氯丙酮(1 mmol, 0.267 g)并逐滴将其滴加进反应装置, 搅拌2 h后观察反应体系, 无淡黄色沉淀产生.
在50 mL反应瓶中装入8 mL蒸馏水, 塞上橡胶塞, 插入氧气球后用油泵置换体系中残留的氧气, 置换时间约5 min, 置换结束后持续通入氧气, 转移至50 ℃的油浴搅拌器中.将邻氨基苯硫醇置于室温至呈液态, 用注射器称取2-氨基苯硫醇(2.5 mmol, 0.318 g)后将其转移至反应容器, 另用注射器称取六氯丙酮(1 mmol, 0.267 g)并逐滴将其滴加进反应装置.然后继续搅拌2 h.冷却后过滤得到淡黄色沉淀.用二氯甲烷洗涤3次, 然后在50 ℃的真空烘箱中干燥12 h, 得0.1675 g淡黄色固体a, 产率62.4%.
称取制备的2, 6-二胺基苯并双噻唑(5 mmol, 1.115 g)在强碱下进行开环, 得到2, 5-二巯基对苯二胺[20], 用注射器迅速将其转移入50 mL反应瓶中, 瓶中加入10 mL水, 将称取的六氯丙酮(0.5 mmol, 0.06 g)迅速加入反应装置, 然后转移至50 ℃油浴搅拌器中反应12 h.得到淡黄色浑浊液.过滤得到固体, 用DMF冲洗两遍, 用水冲洗两遍, 再用乙醇冲洗两遍, 固体在真空干燥箱50 ℃下过夜干燥, 得到40 mg淡黄色固体粉末d, 产率49%.粉末d不溶于实验室常见的有机溶剂, 难溶于浓硫酸. IR (KBr) ν: 1462, 1428, 1309, 925, 765 cm-1.红外结果与文献报道的一致[27].
称取制备的1, 5-二硝基-2, 4-二氨基苯(5 mmol, 0.990 g)溶于乙醇, 并投入适量钯碳, 加入氢气包进行加氢还原, 反应48 h后得到均苯四胺.将称取的六氯丙酮(0.5 mmol, 0.06 g)用注射器将其加入还原装置, 然后转移至50 ℃油浴搅拌器中反应10 min, 继而立马去除加氢装置使体系敞口, 继续反应12 h.过滤并用乙醇洗涤三次, 干燥后得到43 mg墨绿色固体粉末e, 产率65%.粉末e不溶于实验室常见的有机溶剂, 难溶于浓硫酸.红外结果与文献报道的一致[28].
在50 mL反应瓶中装入8 mL蒸馏水, 置于50 ℃的油浴搅拌器中.称取邻苯二胺(2.5 mmol, 0.273 g)并将其加入反应瓶, 另用注射器称取六氯丙酮(1 mmol, 0.335 g)并逐滴将其滴加进反应装置.反应2 h后, 没有相应的沉淀产物析出.反应溶液在室温下静置72 h后, 析出大量橙色针状产物, 过滤, 在50 ℃的真空烘箱中干燥48 h, 最后得到0.3889 g黄褐色固体f, 产率17%. m.p. 289~290 ℃; 1H NMR (DMSO, 400 MHz) δ: 6.25 (s, 2H), 6.92 (s, 1H), 7.55 (s, 1H), 7.90 (s, 1H); 13C NMR (DMSO, 101 MHz) δ: 102.72, 126.89, 128.34, 140.58, 144.39~144.19.核磁表征结果与文献[29]报道一致.
辅助材料(Supporting Information) 联苯并噻唑、联苯并咪唑、5, 5'-二甲基联苯并咪唑、2, 3-二氨基酚嗪氢谱的核磁共振, 联苯并噻唑单晶结构、联苯并咪唑单晶结构, 聚苯撑二噻唑红外光谱、聚苯并二咪唑红外光谱的原始谱图..这些材料可以免费从本刊网站(http://siocjournal.cn/)上下载.
Maynard, G. D. e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001, Chapter Hexachloroacetone 1.
Traboni, S.; Liccardo, F.; Bedini, E.; Giordano, M.; Iadonisi, A. Tetrahedron Lett. 2017, 58, 1762. doi: 10.1016/j.tetlet.2017.03.068
Marcos, C. R.; Carlos A. M.; Evandro L. D.; Cesar Z. Liebigs Ann. 1997, 925.
Sheikhi, E.; Adib, M.; Akherati Karajabad, M.; Rezaei, N. ChemistrySelect 2019, 4, 13455. doi: 10.1002/slct.201903790
(a) Omaka, N. O.; Ekennia, A. C.; Njoku, N. N.; Onwudiwe, D. C. Appl. Organomet. Chem. 2018, 32.
(b) Richardson, C.; Richard, K. F. B.; Steel, P. J. Aust. J. Chem. 2008, 61, 183.
张志国, 郑丹, 麻娜娜, 毕晶晶, 有机化学, 2017, 37, 1824.Hamdi, A. E.; Helmut, G. A. Chin. J. Org. Chem. 2017, 37, 1824(in Chinese).
Dar, A. A.; Shadab, M.; Khan, S.; Ali, N.; Khan, A. T. J. Org. Chem. 2016, 81, 3149. doi: 10.1021/acs.joc.6b00113
(a) Osaheni, J. A.; Jenekhe, S. A. Macromolecules 1993, 26, 4726.
(b) Conboy, G.; Taylor, R. G. D.; Findlay, N. J.; Kanibolotsky, A. L.; Inigo, A. R.; Ghosh, S. S.; Ebenhoch, B.; Krishnan Jagadamma, L.; Thalluri, G. K. V. V.; Sajjad, M. T.; Samuel, I. D. W.; Skabara, P. J. Mater. Chem. C 2017, 5, 11927.
(c) Martin, S. J.; Bradley, D. D. C.; Osaheni, J. A.; Jenekhe, S. A. Mol. Cryst. Liq. Cryst. 2006, 256, 583.
Hattori, T.; Akita, H.; Kakimoto, M.; Imai, Y. J. Polym. Sci. 1992, 30, 197.
(a) Omaka, O. N.; Ekennia, A. C.; Njoku, N. N.; Onwudiwe, D. C. Appl. Organomet. Chem. 2018, 32.
(b) Dar, A. A.; Shadab; Khan, S.; Ali, N.; Khan, A. T. J. Org. Chem. 2016, 81, 3149.
Zhang, C.; Xu, J.; Zhao, X. Y.; Kang, C. M. J. Chem. Res. 2017, 9, 537.
Addison, A. W.; Rao, T. N.; Wahlgren, C. G. J. Heterocycl. Chem. 1983, 20, 1481.
(a) Huang, Q.; Qin, X.; Li, B.; Lan, J.; Guo, Q.; You, J. Chem. Commun. 2014, 50, 13739.
(b) Elagab, H. A.; Alt, H. G. Turk. J. Chem.2016, 40, 667.
(a) Preston, P. N. Chem. Rev.1974, 74, 279.
(b) Holan, G.; Samuel, E. L.; Ennis, B. C.; Hinde, R. W. J. Chem. Soc., Perkin. 1 1967, 20.
(c) Rezende, M. C.; Dall'Oglio, E. L.; Zucco, C. Synth. Commun. 2001, 31, 607.
(a) Panetta, C. A.; Casanova, T. G. J. Org. Chem. 1970, 35, 2423.
(b) Zucco, C.; Lima, C. F.; Rezende, M. C.; Vianna, J. F.; Nome, F. J. Org. Chem. 1987, 52, 5356.
(c) Rezende, M. C.; Dall'Oglio, E. L.; Zucco, C. Tetrahedron Lett. 1996, 37, 5265.
Addison, A. W.; Rao, T. N.; Wahlgren, C. G. J. Heterocycl. Chem. 1983, 20, 1481.
Samanta, S.; Das, S.; Biswas, P. J. Org. Chem. 2013, 78, 11184. doi: 10.1021/jo401445j
(a) Farahi, M.; Karami, B.; Azari, M. C. R. Chim. 2013, 16, 1029.
(b) Mazlumi, M.; Shirini, F.; Goli-Jolodar, O.; Seddighi, M. New J. Chem. 2018, 42, 5742.
(a) Freedlander, R. S.; Bryson, T. A.; Dunlap, R. B.; Schulman, E. M.; Lewis, C. A. J. Org. Chem. 1981, 46, 3519.
(b) Tauer, E.; Grellmann, K. H. J. Am. Chem. Soc.1981, 4252.
Spengler, J.; Böttcher, C.; Albericio, F.; Burger, K. Chem. Rev. 2006, 106, 4728. doi: 10.1021/cr0509962
Conboy, G.; Taylor, R. G. D.; Findlay, N. J.; Kanibolotsky, A. L.; Inigo, A. R.; Ghosh, S. S.; Ebenhoch, B.; Krishnan Jagadamma, L.; Thalluri, G. K. V. V.; Sajjad, M. T.; Samuel, I. D. W.; Skabara, P. J. J. Mater. Chem. C 2017, 5, 11927. doi: 10.1039/C7TC03959J
Boyer, J. I.; Buriks, R. S. Org. Synth. 1960, 40, 96. doi: 10.15227/orgsyn.040.0096
Rai, C.; Braunwarth, J. B. J. Org. Chem. 2002, 26, 3434.
Rezende, M. C.; Dall'Oglio, E. L.; Zucco, C. Tetrahedron Lett. 1996, 37, 5265. doi: 10.1016/0040-4039(96)01115-X
Omaka, O. N.; Ekennia, A. C.; Njoku, N. N.; Onwudiwe, D. C. Appl. Organomet. Chem. 2018, 32, 2.
Rezende, M. C.; Marques, C. A.; Dall'Oglio, E. L.; Zucco, C. Liebigs Ann. 1997, 928.
John, A.; Osaheni; Samson, A.; Jenekhe Chem. Mater. 1992, 4, 1286.
Osaheni, J. A.; Jenekhe, S. A. Macromolecules 1995, 28, 1173.
He, D.; Wu, Y.; Xu, B.-Q. Eur. Polym. J. 2007, 43, 3705.

图式 5 六氟丙酮与六氯丙酮的不同反应路径
Scheme 5 Different reaction paths of hexafluoroacetone and hexachloroacetone
表 1 六氯丙酮和邻氨基苯硫醇反应的时间优化
Table 1. Optimization of the reaction time of hexachloroacetone and 2-aminobenzenethiol
| Entry | Reaction timea/min | Yield/% |
| 1 | 60 | 20 |
| 2 | 90 | 43 |
| 3 | 120 | 65 |
| 4 | 150 | 64 |
| 5 | 180 | 67 |
| a The optimal reaction time of bibenzothiazole based on hexachloroacetone, n(HCA):n(2-aminobenzenethiol)=2.5:1, water as solvent under 50 ℃. | ||
 下载: 导出CSV
下载: 导出CSV
表 2 六氯丙酮成环反应的底物拓展
Table 2. Substrate expansion of hexachloroacetone cyclization reaction
| Entry | Substrate | Product | Yield/% |
| 1a | |
|
65 |
| 2a | |
|
75 |
| 3a | |
— | — |
| 4b | |
|
— |
| 5a | |
|
— |
| a All the reactions conducted under the optimized conditions. b All the reagents were prepared by experiments according to literature reports. The reactions were carried out under the optimized conditions. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们