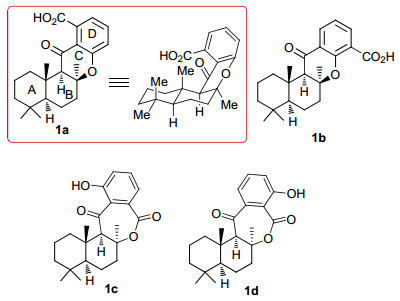
图 1.
15-oxopuupehenoic acid的四种可能结构
Figure 1.
Four possible structures of 15-oxopuupehenoic acid

5-脂氧合酶(5-LOX或5-LO)是人体中由ALOX5基因编码的酶.它是脂氧合酶家族的主要成员, 在体内的主要作用是将人体必需的脂肪酸(EFAs)代谢为炎症介质白三烯, 是许多疾病中药物抑制的靶标.例如5-LO是冠心病(CAD)中药物干预的目标, 它在脑细胞中表达并且可以参与神经病理过程, 一些患有5-LO变异等位基因的人患CAD的风险较高.利用小分子化合物来抑制和调节体内5-LO的活性和功能表达已成为治疗相关疾病的重要手段.
海洋海绵富含结构独特的杂萜类二级代谢物, 这些天然杂萜大多具有良好的生物活性, 例如抗病毒、抗疟疾、抗癌和抗真菌等[1], 许多可作为新药研发的先导化合物. 15-oxopuupehenoic acid是Crews小组[2]2009年从巴布新几内亚的特种海绵Hyrtios sp.中分离得到的一个新杂倍半萜, 活性研究发现它具有较好的5-脂氧合酶抑制活性.分离作者由波谱数据分析其可能的结构为1a~1d (图 1), 进而通过二维1H-13C相关(gHMBC correlations)核磁谱图分析判定其结构为1a.
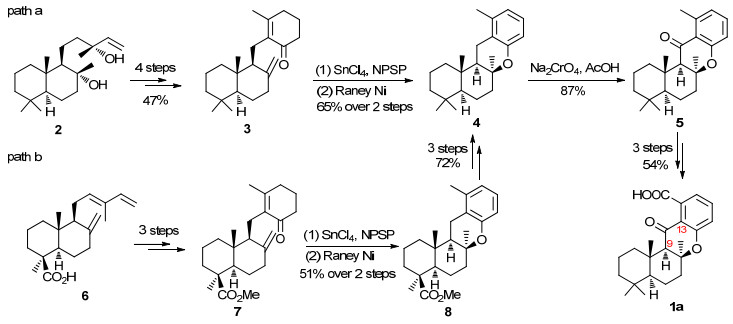
从骨架结构来看, 1a和1b均为6/6/6/6四环二氢色满酮, ABC三环的稠合方式为trans-anti-cis, 二者的差异在于D环上取代羧基的位置不同. 2014, Alvarez- Manzaneda和Chahboun小组[3]利用四氯化锡促进的硒试剂参与的环化反应为关键反应完成了化合物1a的全合成(Scheme 1).他们首先从手性香紫苏醇2出发, 经过6步反应制得四环化合物4, 再经4步官能团能转化即得到1a (Scheme 1, path a).根据1a核磁谱中观察到的Me-13和H-9的NOE效应, 他们确定合成化合物的相对构型符合目标分子的立体化学要求; 再结合所用手性原料的绝对构型信息, 他们判定所合成的化合物就是Crews等[2]推测的天然产物15-oxopuupehenoic acid可能的结构1a.但是化学合成得到的1a的谱图数据无法与分离作者报道的相吻合, 比旋光值也相差很大.于是他们又以手性湿地松酸6为初始原料, 再次合成了环化产物4 (Scheme 1, path b), 确认了化合物4结构的正确性.据此他们认为天然15-oxopuupehenoic acid的真实结构可能不是1a.
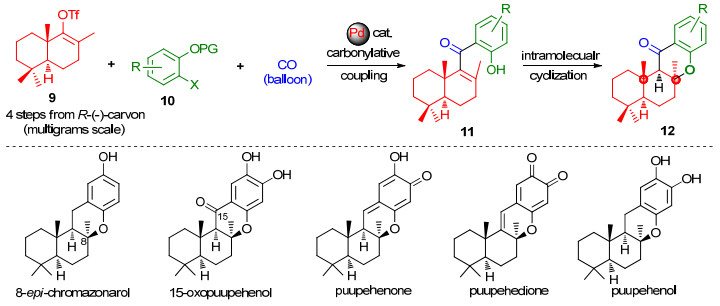
我们小组[4]近期从廉价易得的R-(-)-香芹酮出发, 利用汇聚的偶联环化策略, 以钯催化的羰化Stille偶联反应和水合肼促进的环化反应为关键反应构建二氢色满酮骨架, 完成了海绵杂倍半萜8-epi-chromazonarol的首次不对称全合成(Scheme 2).为更好地说明这一策略在该类天然产物合成中的通用性, 我们又进一步改进了偶联/环化反应, 以羰化Suzuki偶联反应[5-9]和KOH促进的环化反应为关键反应, 以8步(最长线性路线), 18%的总收率完成了(-)-15-oxopuupehenol的不对称全合成.我们还从相同的关键中间体出发, 通过简单的仿生转化, 完成了其天然类似物puupehenone的全合成和puupehedione、puupehenol的形式合成(Scheme 2)[10].在完成这些工作后, 我们开始了对15-oxopuupehenoic acid的合成研究. Crews等[2]最初提出其四种可能的结构中, 根据我们发展的合成方法可以快捷的得到化合物1a和1b.由于1a已经被证实不是天然15-oxopuupe- henoic acid的真实结构[3], 我们拟合成1b, 然后通过比较所得1b与天然产物相关波谱数据的异同以验证1b是否为其真实结构.
我们采用的具体合成路线如Scheme 3所示.首先, 从廉价易得的R-(-)-香芹酮出发, 经过两次烷基化反应、酸促进的碳正离子环化反应及加氢还原反应得到双环酮15[4, 10-11].在双(三甲基硅烷基)氨基钾(KHMDS)为碱的条件下, 化合物15的羰基转化成烯基三氟甲磺酸酯9.这些反应都可以以多克级的规模平稳进行, 从而保证了可以得到足量的双环偶联片段9.在我们之前报道的Suzuki羰化偶联反应条件下[10], 芳基硼酸16, CO (101 kPa, balloon)和双环片段9发生羰化偶联反应, 得到了合成所需的α, β-不饱和芳基酮17.这步反应目前可以克级规模进行, 收率为40%;我们尝试了进一步优化反应条件, 如增大钯催化剂的用量和升高反应温度等, 偶联反应的收率都没有明显提高.随后我们使用BBr3脱除甲醚保护基以89%的收率得到环化前体酚18, 再经KOH促进的分子内环化反应[11]顺利得到了四环化合物19.这步环化反应的收率为80%, 可放大反应规模至500毫克级别.四环化合物19已经具有目标分子1b的基本骨架和所有的碳原子, 只需要将苄位的甲基转化为羧基就可以得到1b.依照Alvarez-Manzaneda和Chahboun小组[3]所采用逐级氧化的方法, 化合物19经过苄位自由基溴代、氧化成醛, Pinnick氧化三步反应顺利转化为目标产物1b.通过对1b一维NOE实验中观察到的C13-Me (δ 1.37)和C9-H (δ 2.13)的NOE效应分析, 并结合所用手性香芹酮起始原料的绝对构型, 我们判定合成化合物1b的立体构型是正确的.
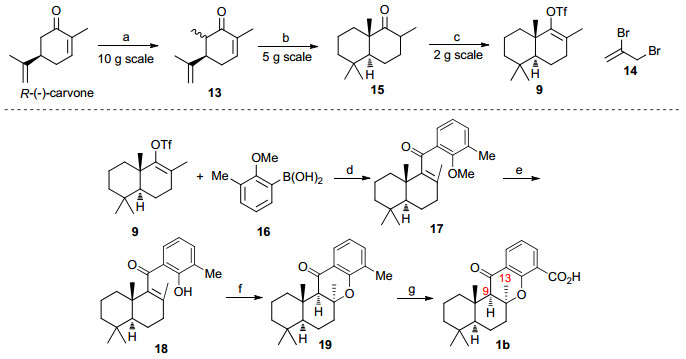
Reagents and conditions: (a) LDA, CH3I, THF, -25 ℃, 89%; (b) (1) LDA, 14, -20 ℃, THF, 12 h, then TFA, r.t., 72 h; (2) Pd/C, H2 (3.03 MPa), MeOH, r.t., 40% yield in 2 steps; (c) KHMDS, PhNTf2, -78 ℃, THF, 90%; (d) PdCl2, PPh3, K2CO3, CO (101 kPa), DMF, 80 ℃, 40%; (e) BBr3, CH2Cl2, 0 ℃, 89%; (f) KOH, DEG, 125 ℃, 80%; (g) (1) NBS, (BzO)2, 80 ℃, CCl4; (2) Me3NO, DMSO, CH2Cl2; (3) NaClO2, isoprene, 5% NaH2PO4, t-BuOH, r.t., 65% in 3 steps
但令人遗憾的是, 如表 1和2所示, 化合物1b的波谱数据和Crews等[2]提供的天然15-oxopuupehenoic acid的相关数据也不相符.二者的比旋光值相差也较大, 1b的比旋光值为
 下载:
导出CSV
下载:
导出CSV
| No. | Synthetic 1ba, c | 15-oxopuupehenoic acidb, c | Δd |
| 1 ax | 1.21 (m) | 1.21 (m) | 0 |
| 1 eq | 1.69 (m) | 1.62 (m) | 0.07 |
| 2 | 1.75 (d, J=6.2 Hz) | 1.66 (m) | 0.09 |
| 3 ax | 1.25 (m) | 1.18 (m) | 0.07 |
| 3 eq | 1.75 (d, J=6.2 Hz) | 1.42 (m) | 0.33 |
| 5 | 0.97 (d, J=6.4 Hz) | 0.91 (dd, J=11.5, 2.5 Hz) | 0.06 |
| 6 | 1.44(s) | 1.44 (m) | 0 |
| 7 ax | 1.25 (m) | 1.57 (m) | -0.32 |
| 7 eq | 2.38 (dd, J=11.1, 2.3 Hz) | 2.26 (m) | 0.12 |
| 9 | 2.13 (s) | 2.04 (s) | 0.09 |
| 11 | 0.94 (s) | 0.92 (s) | 0.02 |
| 12 | 0.84 (s) | 0.84 (s) | 0 |
| 13 | 1.37 (s) | 1.22 (s) | 0.15 |
| 14 | 0.86 (s) | 0.88 (s) | -0.02 |
| 18 | 7.18 (t, J=7.7 Hz) | 7.01 (dd, J=8, 1.5 Hz) | 0.17 |
| 19 | 8.36 (dd, J=7.4, 1.4 Hz) | 7.48 (dd, J=8, 7.5 Hz) | 0.88 |
| 20 | 8.13 (d, J=7.7, 1.5 Hz) | 7.27 (dd, J=7.5, 1.5 Hz) | 0.86 |
| a 1H NMR data (400 MHz). b 1H NMR data (600 MHz). c The solvent signal of CHCl3 (δ 7.26) was used as reference. d Chemical shift difference between synthetic product 1b and natural product; for the convenience of calculation, the middle value of chemical shift is taken for multiple peaks. | |||
下载:
导出CSV
| Number | Synthetic 1ba, c | 15-oxopuupehenoic acidb, c | Δd |
| 1 | 39.9 | 40.0 | -0.1 |
| 2 | 18.0 | 18.1 | -0.1 |
| 3 | 41.5 | 41.4 | 0.1 |
| 4 | 33.4 | 33.4 | 0 |
| 5 | 54.1 | 54.2 | -0.1 |
| 6 | 18.2 | 18.3 | -0.1 |
| 7 | 39.8 | 39.5 | 0.3 |
| 8 | 84.5 | 80.4 | 4.1 |
| 9 | 64.7 | 64.9 | -0.2 |
| 10 | 38.7 | 38.7 | 0 |
| 11 | 33.7 | 33.7 | 0 |
| 12 | 21.9 | 21.9 | 0 |
| 13 | 26.8 | 26.4 | 0.4 |
| 14 | 15.4 | 15.4 | 0 |
| 15 | 192.4 | 196.2 | -3.8 |
| 16 | 118.5 | 119.2 | -0.7 |
| 17 | 158.1 | 161.0 | -2.9 |
| 18 | 121.8 | 121.3 | 0.5 |
| 19 | 132.2 | 135.3 | -3.1 |
| 20 | 123.3 | 122.2 | 1.1 |
| 21 | 139.4 | 132.6 | 6.8 |
| 22 | 164.9 | 171.9 | -7.0 |
| a 13C NMR data (101 MHz). b 13C NMR data (150 MHz). c The solvent signal of CHCl3 (δ 77.0) was used as reference. d Chemical shift difference between synthetic product 1b and natural product. | |||
以R-(-)-香芹酮为起始原料, 通过偶联-环化的汇聚式合成策略, 以Suzuki羰化偶联反应和KOH促进的分子内环化反应为关键反应构建了其独特的苯并-γ-吡喃酮骨架, 完成了15-oxopuupehenoic acid可能结构1b的不对称全合成.然而化合物1b的谱图数据和天然15-oxopuupehenoic acid也不相符, 这一结果提示分离化学家需要对15-oxopuupehenoic acid的结构进行进一步的修正.
1H NMR和13C NMR由Virian BB 300M, Bruker 400M测定, 若没有特殊说明, 相对位移均以TMS (1H NMR: δ 0.0)或者CDCl3 (1H NMR: δ 7.26, 13C NMR: δ 77.0)为标准; 旋光由Perkin-Elmer 241MC型自动旋光仪测定; 高分辨质谱由Bruker Daltonics APEXII测定.二氯甲烷用氢化钙回流2 h以上后收集使用, N, N-二甲基甲酰胺(DMF)和二甲基亚砜(DMSO)溶液经过重蒸或者购买的超干溶液.使用的药品均为化学纯或分析纯.
将底物双环烯基三氟甲基磺酸酯(9, 340 mg, 1 mmol)和2-甲氧基-3-甲基苯硼酸(16, 183 mg, 1.1 mmol)置于200 mL圆底烧瓶中, 然后依次加入碳酸钾(414 mg, 3 mmol)、三苯基膦(79 mg, 0.6 mmol)和氯化钯(51 mg, 0.3 mmol), 加入20 mL DMF溶解反应混合物.用一氧化碳气体(气袋或气球)置换三次, 在室温条件下搅拌2 h左右, 然后在80 ℃下反应过夜.反应混合物冷却至室温, 抽滤以将体系中的固体过滤掉, 加入100 mL乙醚, 先用水洗涤(15 mL×3), 再用饱和食盐水洗涤.有机相用无水硫酸钠干燥, 过滤后旋干溶剂, 硅胶柱色谱分离纯化[V(石油醚):V(乙酸乙酯)=200:1]得到136 mg无色油状液体化合物17, 产率40%.
氩气保护下, 将底物17 (70 mg, 0.21 mmol)溶于2 mL干燥的二氯甲烷中.在0 ℃下, 缓慢滴加三溴化硼(0.022 mL, 0.23 mmol), 然后在该温度下搅拌反应2 h. TLC检测体系中无反应原料后加水淬灭.乙酸乙酯萃取(15 mL×3), 合并有机相并用15 mL饱和食盐水洗涤后, 无水硫酸钠干燥, 过滤, 旋除溶剂, 硅胶柱色谱分离纯化[V(石油醚):V(乙酸乙酯)=100:1]得到60 mg无色油状液体化合物18, 产率89%.
称量底物18 (55 mg, 0.17 mmol)于圆底烧瓶中, 加入一缩二乙二醇溶液溶解底物, 然后加入氢氧化钾(48 mg, 0.85 mmol)的水溶液(1 mL), 在125 ℃下反应24 h.薄层色谱(TLC)检测体系中无反应原料后冷却至室温, 乙酸乙酯萃取(15 mL×3).合并有机相, 依次用1 mol/L HCl溶液和饱和食盐水洗涤后, 无水硫酸钠干燥.过滤后旋除溶剂, 硅胶柱色谱分离纯化[V(石油醚):V(乙酸乙酯)=100:1]得到44 mg白色固体化合物19, 产率80%. m.p. 126~130 ℃ (CH2Cl2);
称量N-溴代琥珀酰亚胺(NBS) (20 mg, 0.11 mg), 过氧化苯甲酰(2 mg, 0.009 mmol)于圆底烧瓶中, 置换氩气, 加入化合物19 (30 mg, 0.09 mmol)的CCl4 (5 mL)溶液.在80 ℃下反应14 h, TLC检测体系中无反应原料.冷却至室温, 旋除溶剂, 硅胶柱分离纯化[V(石油醚):V(乙酸乙酯)=100:1]得到32 mg无色油状液体.
将上述无色油状液体溶解在CH2Cl2和DMSO (1.6 mL/1.6 mL)混合溶液中, 在0 ℃下加入三甲基氮氧化物.在室温下搅拌反应过夜, TLC检测体系中无反应原料后旋除二氯甲烷, 加入10 mL水, 乙酸乙酯萃取(10 mL×3).合并有机相, 用饱和食盐水洗涤, 无水硫酸钠干燥, 过滤后旋除溶剂, 硅胶柱色谱层析分离纯化[V(石油醚):V(乙酸乙酯)=100:1]得到18 mg无色油状液体.
氩气保护下, 于圆底烧瓶中, 将上述无色油状液体溶于2 mL干燥的叔丁醇中, 在0 ℃下加入异戊二烯(0.23 mL, 2 mmol), 将10 mg亚氯酸钠溶解在2 mL质量分数为5%的磷酸二氢钠的水溶液中后加入反应体系内.在室温下反应14 h, TLC检测体系中无反应原料, 加入饱和碳酸氢钠溶液淬灭反应.旋除叔丁醇, 加入15 mL水, 乙酸乙酯萃取(10 mL×3).合并有机相, 并用饱和食盐水洗涤, 无水硫酸钠干燥, 过滤后旋除溶剂, 硅胶柱色谱分离纯化[V(石油醚):V(乙酸乙酯)=3:1]得到18 mg无色油状液体化合物1b, 三步总产率为65%.
辅助材料(Supporting Information) 17, 18, 19和1b的1H NMR和13C NMR谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(a) Kohmoto, S.; McConnell, O. J.; Wright, A.; Koehn, F.; Thompson, W.; Lui, M.; Snader, K. M. J. Nat. Prod. 1987, 50, 336.
(b) Sova, V.; Fedoreev, S. A. Khim. Prir. Soedin. 1990, 497.
(c) Longley, R. E.; McConnel, O. J.; Essich, E.; Harmody, D. J. Nat. Prod. 1993, 56, 915.
(d) El Sayed, K. A.; Dunbar, D. C.; Perry, T. L.; Wilkins, S. P.; Hamann, M. T.; Greenplate, J. T.; Wideman, M. A. J. Agric. Food Chem. 1997, 45, 2735.
(e) Faulkner, D. J. Nat. Prod. Rep. 1998, 15, 113 and references cited therein.
(f) ourguet-Kondracki, M. L.; Lacombe, F.; Guyot, M. J. Nat. Prod. 1999, 62, 1304.
(g) Popov, A. M.; Stekhova, S. I.; Utkina, N. K.; Rebachuk, N. M. Pharm. Chem. J. 1999, 33, 71.
(h) El Sayed, K. A.; Bartyzel, P.; Shen, X.; Perry, T. L.; Zjawiony, J. K.; Hamann, M. T. Tetrahedron 2000, 56, 949.
(i) Takamatsu, S.; Hodges, T. W.; Rajbhandari, I.; Gerwick, H.; Hamann, M. T.; Nagle, D. G. J. Nat. Prod. 2003, 66, 605.
(j) Pina, I. C.; Sanders, M. L.; Crews, P. J. J. Nat. Prod. 2003, 66, 2.
(k) Hamann, M. T. Curr. Pharm. Des. 2003, 9, 879.
(l) Graus, G. A.; Nguyen, T.; Bae, J.; Hostetter, J.; Steadham, E. Tetrahedron 2004, 60, 4223.
Robinson, S. J.; Hoobler, E. K.; Riener, M.; Loveridge, S. T.; Tenney, K.; Valeriote, F. A.; Holman, T. R.; Crews, P. J. Nat. Prod. 2009, 72, 1857. doi: 10.1021/np900465e
Boulifa, E.; Fernández, A.; Alvarez, E.; Alvarez-Manzaneda, R.; Mansour, A. I.; Chahboun, R.; Alvarez-Manzaneda, E. J. Org. Chem. 2014, 79, 10689. doi: 10.1021/jo502048y
Liu, L.; Song, H.-Y.; Chen, P.; Yuan, Z.-Y.; Feng, S.-B.; Zhang, W.-W.; Fang, B.-W.; Xie, X.-G.; She, X.-G. Org. Chem. Front. 2018, 5, 3013. doi: 10.1039/C8QO00901E
Sumino, S.; Ui, T.; Ryu, I. Org. Chem. Front. 2015, 2, 1085. doi: 10.1039/C5QO00185D
Wang, Z.-J.; Wang, X.-Y.; Wang, X.; Liang, Z.-W.; Xu, X. Catal. Commun. 2017, 101, 10. doi: 10.1016/j.catcom.2017.06.005
Wójcik, P.; Sygellou, L.; Gniewek, A.; Skarżyńska, A.; Trzeciak, A. Chem. Cat. Chem. 2017, 9, 4397.
Gautam, P.; Bhanage, B. M. J. Org. Chem. 2015, 80, 7810. doi: 10.1021/acs.joc.5b01160
Ketike, T.; Velpula, V. R. K.; Madduluri, V. R.; Kamaraju, S. R. R.; Burri, D. R. ChemistrySelect 2018, 3, 7164. doi: 10.1002/slct.201801100
Song, H.-Y.; Liu, L.; Yang, M.-Y.; Wu, G.-M.; Chen, P.; Xie, X.-G.; She, X.-G. Org. Chem. Front. 2020, 7, 35. doi: 10.1039/C9QO01027K
Gesson, J. P.; Jacquesy, J. C.; Renoux, B. Tetrahedron 1989, 45, 5853. doi: 10.1016/S0040-4020(01)89112-2

图 1 15-oxopuupehenoic acid的四种可能结构
Figure 1 Four possible structures of 15-oxopuupehenoic acid

图式 1 Manzaneda and Chahboun小组对1a的全合成
Scheme 1 Manzaneda and Chahboun's total synthesis of 1a

图式 2 我们小组对8-epi-chromazonarol及其天然类似物的不对称全合成
Scheme 2 Our asymmetric total syntheses of 8-epi-chromazonarol and its natural analogues

图式 3 15-oxopuupehenoic acid可能结构1b的全合成
Scheme 3 Total synthesis of the possible structure 1b of 15-oxopuupehenoic acid
Reagents and conditions: (a) LDA, CH3I, THF, -25 ℃, 89%; (b) (1) LDA, 14, -20 ℃, THF, 12 h, then TFA, r.t., 72 h; (2) Pd/C, H2 (3.03 MPa), MeOH, r.t., 40% yield in 2 steps; (c) KHMDS, PhNTf2, -78 ℃, THF, 90%; (d) PdCl2, PPh3, K2CO3, CO (101 kPa), DMF, 80 ℃, 40%; (e) BBr3, CH2Cl2, 0 ℃, 89%; (f) KOH, DEG, 125 ℃, 80%; (g) (1) NBS, (BzO)2, 80 ℃, CCl4; (2) Me3NO, DMSO, CH2Cl2; (3) NaClO2, isoprene, 5% NaH2PO4, t-BuOH, r.t., 65% in 3 steps
表 1 合成产物1b与天然15-oxopuupehenoic acid1H NMR数据(δ)比较
Table 1. Comparison of the 1H NMR data (δ) of synthetic 1b and natural 15-oxopuupehenoic acid
| No. | Synthetic 1ba, c | 15-oxopuupehenoic acidb, c | Δd |
| 1 ax | 1.21 (m) | 1.21 (m) | 0 |
| 1 eq | 1.69 (m) | 1.62 (m) | 0.07 |
| 2 | 1.75 (d, J=6.2 Hz) | 1.66 (m) | 0.09 |
| 3 ax | 1.25 (m) | 1.18 (m) | 0.07 |
| 3 eq | 1.75 (d, J=6.2 Hz) | 1.42 (m) | 0.33 |
| 5 | 0.97 (d, J=6.4 Hz) | 0.91 (dd, J=11.5, 2.5 Hz) | 0.06 |
| 6 | 1.44(s) | 1.44 (m) | 0 |
| 7 ax | 1.25 (m) | 1.57 (m) | -0.32 |
| 7 eq | 2.38 (dd, J=11.1, 2.3 Hz) | 2.26 (m) | 0.12 |
| 9 | 2.13 (s) | 2.04 (s) | 0.09 |
| 11 | 0.94 (s) | 0.92 (s) | 0.02 |
| 12 | 0.84 (s) | 0.84 (s) | 0 |
| 13 | 1.37 (s) | 1.22 (s) | 0.15 |
| 14 | 0.86 (s) | 0.88 (s) | -0.02 |
| 18 | 7.18 (t, J=7.7 Hz) | 7.01 (dd, J=8, 1.5 Hz) | 0.17 |
| 19 | 8.36 (dd, J=7.4, 1.4 Hz) | 7.48 (dd, J=8, 7.5 Hz) | 0.88 |
| 20 | 8.13 (d, J=7.7, 1.5 Hz) | 7.27 (dd, J=7.5, 1.5 Hz) | 0.86 |
| a 1H NMR data (400 MHz). b 1H NMR data (600 MHz). c The solvent signal of CHCl3 (δ 7.26) was used as reference. d Chemical shift difference between synthetic product 1b and natural product; for the convenience of calculation, the middle value of chemical shift is taken for multiple peaks. | |||
 下载: 导出CSV
下载: 导出CSV
表 2 合成产物1b与天然15-oxopuupehenoic acid的13C NMR数据比较
Table 2. Comparison of 13C NMR data of synthetic 1b and natural 15-oxopuupehenoic acid
| Number | Synthetic 1ba, c | 15-oxopuupehenoic acidb, c | Δd |
| 1 | 39.9 | 40.0 | -0.1 |
| 2 | 18.0 | 18.1 | -0.1 |
| 3 | 41.5 | 41.4 | 0.1 |
| 4 | 33.4 | 33.4 | 0 |
| 5 | 54.1 | 54.2 | -0.1 |
| 6 | 18.2 | 18.3 | -0.1 |
| 7 | 39.8 | 39.5 | 0.3 |
| 8 | 84.5 | 80.4 | 4.1 |
| 9 | 64.7 | 64.9 | -0.2 |
| 10 | 38.7 | 38.7 | 0 |
| 11 | 33.7 | 33.7 | 0 |
| 12 | 21.9 | 21.9 | 0 |
| 13 | 26.8 | 26.4 | 0.4 |
| 14 | 15.4 | 15.4 | 0 |
| 15 | 192.4 | 196.2 | -3.8 |
| 16 | 118.5 | 119.2 | -0.7 |
| 17 | 158.1 | 161.0 | -2.9 |
| 18 | 121.8 | 121.3 | 0.5 |
| 19 | 132.2 | 135.3 | -3.1 |
| 20 | 123.3 | 122.2 | 1.1 |
| 21 | 139.4 | 132.6 | 6.8 |
| 22 | 164.9 | 171.9 | -7.0 |
| a 13C NMR data (101 MHz). b 13C NMR data (150 MHz). c The solvent signal of CHCl3 (δ 77.0) was used as reference. d Chemical shift difference between synthetic product 1b and natural product. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们