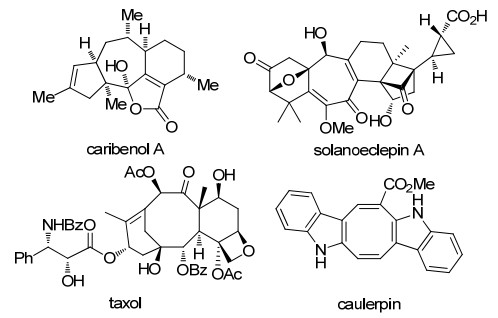
图 1.
含有中环骨架的代表性天然产物
Figure 1.
Representative natural products with medium ring skeletons

环状化合物在天然产物和生物活性化合物中普遍存在.其中3至6元环结构一般称为小环化合物, 7至9元环结构称为中环化合物, 而10元及以上的结构称为大环化合物.相比于小环和大环化合物的合成, 中环化合物的合成一直更具有挑战, 因为环化过程存在熵的不利影响以及过渡态的非键相互作用, 同时还需要克服中环化合物自身的跨环作用力.
然而, 中环结构在很多具有重要生物活性和药理活性化合物中广泛存在, 例如, Caribenol A是海洋生物中分离得到的天然产物, 具有抗肺结核及抗疟原虫活性, 紫杉醇(taxol)是该家族中最著名的天然产物(图 1), 目前是临床上最有效的抗癌药物之一, 因此发展高效的中环合成方法一直以来备受关注.
近年来, C—H键活化官能化反应得到了蓬勃的发展, 不仅大大拓宽了底物的适用范围, 而且有利于发展全新的合成途径, 大大提高了合成效率和原子经济性.因此, 如何利用C—H键活化官能化反应构建中环化合物也受到了越来越多的关注, 这样不仅可以使用更为价廉易得的原料, 而且可以获得全新的活性和选择性.然而, 相对于传统的使用高活性试剂的方法, 通过相对惰性C—H键活化的方法来构建中环面临更大的挑战, 需要考虑金属催化剂的种类、中间体的稳定与否、活性及选择性如何控制等多重挑战, 目前取得成功的例子还较为有限, 但是显示了较好的发展前景.
本文主要总结了近年来通过过渡金属催化的不饱和键对C—H键的插入反应来构建中环化合物的反应.此类反应中最为重要的策略是成环前如何稳定C—H键金属化中间体.因此, 按照稳定策略的不同, 将分为4个部分进行介绍, 即通过共振结构稳定金属中间体构建中环、通过杂原子配位稳定金属中间体构建中环、通过双齿配位稳定金属中间体构建中环以及通过大位阻卡宾配体稳定金属中间体构建中环.
过渡金属催化的烯醛环化是构建环状化合物的重要方法.早在1972年, Sakai小组[1]就首次报道了Rh催化的烯醛环化反应, 合成了环戊酮衍生物.但是, 由于酰基金属中间体不太稳定, 容易脱羰, 该类反应长期以来一直局限于5元环的合成.考虑到烯醛原料易于合成, 通过该反应进行中环化合物的合成将是一个理想的选择.但是由于中环的环化速率显著降低, 金属中间体更易于脱羰分解, 因此, 这类反应一直具有挑战性, 难以获得成功.
2000年, Shair小组[2]在底物中巧妙引入了烯基环丙烷结构, 成功地实现了8元环辛烯酮化合物的合成(Scheme 1).在乙烯氛围下使用20 mol% [Rh(dppe)]ClO4或[Rh(dppe)]OTf作为催化剂, Cl(CH2)2Cl作为溶剂, 在65 ℃下反应, 以54%~65%的收率得到8元环酮.反应的具体机理见Scheme 1.
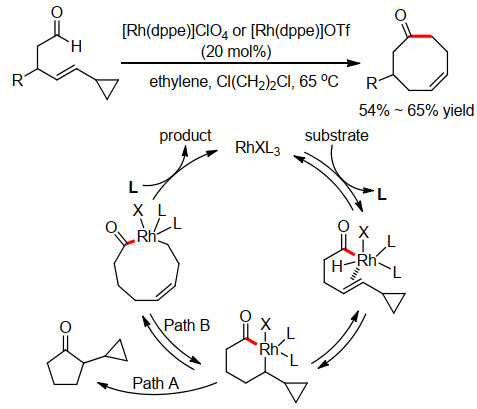
首先, Rh(Ⅰ)催化剂插入醛的C—H键, 生成酰基- Rh(Ⅲ)中间体, 随后发生分子内氢金属化生成6元金属铑环中间体, 该中间体可以直接还原消除生成环戊酮(Path A), 也可以导致相邻的3元环开环, 从而生成9元Rh环, 再还原消除生成环辛烯酮(Path B).反应中, 环丙烷的开环和酰基Rh异构化至关重要, 可以产生类似于烯丙基Rh的共振结构来获得较好的稳定作用, 从而有效避免关环前金属中间体的分解.
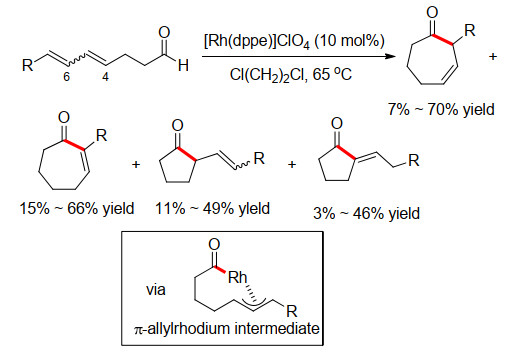
受此工作启发, 2002年, Mori小组[3]使用双烯底物进行类似的反应尝试, 成功发展了Rh(Ⅰ)-催化分子内氢酰化合成7元环化合物的方法(Scheme 2).该反应中额外存在的烯烃有助于生成烯丙基金属中间体, 有效稳定酰基Rh中间体, 避免其在关环前分解.值得注意的是, 双烯的取代基和烯烃构型对环化的选择性也有重要影响.在C(7)位有取代基时, 主要生成环庚烯酮衍生物, 而C(7)位无取代基时, 则优先生成环戊酮衍生物.此外, 当C(6)位烯烃为E式构型时, 环化反应主要生成环庚烯酮衍生物, 而相应的Z式烯烃则生成环戊酮衍生物.
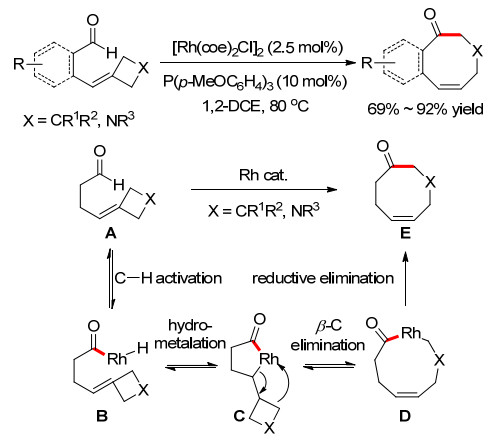
2010年, Aïssa小组[4]在底物中引入了张力较大的4元环, 以便通过β-C消除反应形成共振稳定的金属中间体, 完成了8元环化合物的合成(Scheme 3).在以往的文献报道中, 关于金属介导的吖丁啶开环的报道极为少见, 且仅描述了吖丁啶的消除反应.该反应为8元环化合物的合成提供了新的途径, 扩大了铑催化的分子内氢酰化的范围.
除了通过共振结构来稳定金属中间体外, 在底物上引入额外杂原子, 让其和金属配位形成环状螯合物, 也是一种有效的中间体稳定策略. 2002年, Bendorf小组[5]使用S原子作为螯合基团, 在铑催化下, 成功实现了烯烃或者炔烃的分子内氢酰化反应, 合成了一系列7/8元杂环酮化合物(Scheme 4). 2012年, 该小组[6]发现使用N原子代替S原子, 也可以顺利实现烯烃或者炔烃的分子内氢酰化反应.
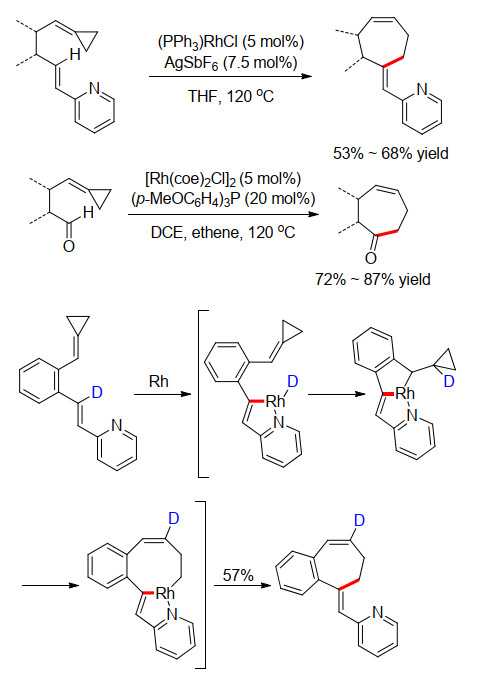
2007年, Fürstner小组[7]引入吡啶配位基, 实现了铑催化烯烃的C—H键活化, 再和亚烷基环丙烷发生环化反应, 合成了一系列7元环庚烯化合物(Scheme 5). E-型的环外烯烃的形成经历了吡啶氮原子导向的C—H键活化/氢金属化/环加成串联的反应机理.氘代实验证明了该机理的合理性, 可以检测到氘原子从最初的C—H(D)活化位点定量转移到新形成的双键上.此外, 作者还在此反应模式下考察了醛类底物的反应情况.发现当在乙烯氛围下进行反应时, 无需再引入导向基团, 反应依然可以很顺利地进行, 获得了一系列7元环庚烯酮类产物.因为额外的乙烯配位可以有效地稳定酰基Rh中间体, 避免其脱羰分解.
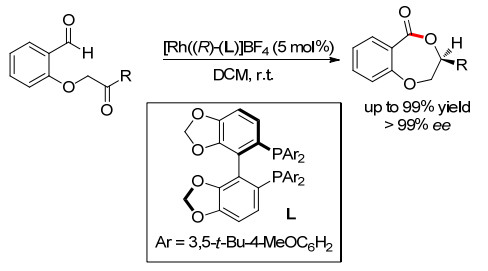
2008年, Dong小组[8]以氧原子作为配位原子, 以酮作为不饱和键, 顺利完成了醛C—H键的活化环化反应, 合成了一系列7元环内酯化合物, 为内酯的合成提供了一种新的原子经济性策略.此外, 手性膦配体的使用, 可以很好地控制反应的对映选择性, 最高可获得99%以上的ee值(Scheme 6).其中, 膦配体的碱性在促进氢酰化而非竞争性脱羰化中也起着关键的作用. 2009年, Dong小组对该反应的机理进行了探讨, 证明了O原子的存在对酰基金属中间体的稳定起着至关重要的作用[9].
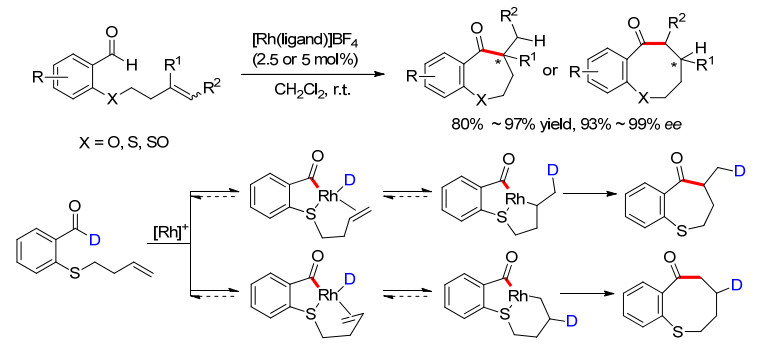
2009年, Dong小组[10]进一步将此反应体系拓展到与烯烃化合物的环化反应.发现在Rh金属和合适的手性膦配体条件下, 无论是使用氧、硫还是亚砜配位基, 分子内氢酰化反应都可以顺利地进行, 得到各类手性7-元酮或者8-元酮化合物(Scheme 7).杂原子的存在对反应至关重要, 不仅可以避免酰基Rh的脱羰, 有效地提高了反应的活性, 而且该配位作用增强了中间体的刚性, 便于反应立体选择性的控制.当这些原子被碳原子取代时, 反应没有活性, 很好地证明了这一点.此外, 作者还发现手性膦配体的结构对末端烯烃环化的exo和endo选择性有较大的影响.而对于内烯或者1, 1-二取代烯烃, 由于位阻作用, 则基本都是exo型环化.作者还进行了醛氢的同位素标记实验, 发现产物酮的α和β位没有出现同位素混杂, 只生成了β位氘代的产物, 从而说明还原消除不是反应的决速步.
随后, 在2011年, Dong小组[11]又实现了氮原子导向的酮的氢酰化反应.通过使用不同的手性双膦配体, 可以高的收率和高的对映选择性合成一系列7元以及8元环内酯(Scheme 8).作者比较了氮、氧和硫等不同的导向基团对反应的影响, 发现氮促进氢酰化的速度更快, 而且氮原子的螯合作用可以完全抑制竞争性脱羰反应.
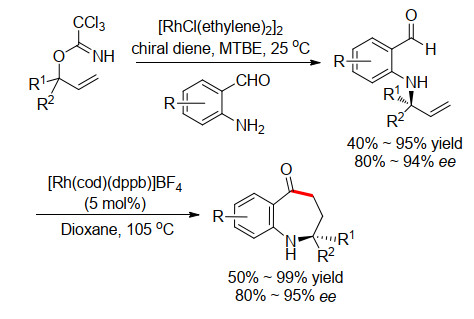
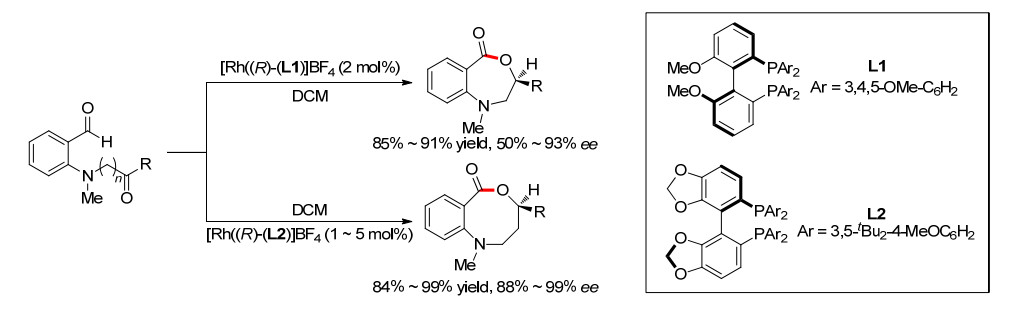
2014年, Nguyen小组[12]同样报道了N原子配位导向的分子内烯醛的氢酰化反应, 构建了7元中环(Scheme 9).作者首先通过手性双烯配体辅助的铑催化剂实现了手性的烯丙基胺化关环前体的合成, 获得了较高的收率和对映选择性.然后再在Rh催化剂的作用下, 发生分子内的烯烃氢酰化反应, 得到一系列7元氮杂环酮化合物, 而且环化过程较好地保留了关环前体的对映选择性.
虽然上述通过形成共振结构和引入杂原子配位的两类方法可以较好地稳定金属中间体, 抑制了酰基金属的脱羰反应, 大大提高了环化反应的活性, 但是这些方法中底物需要经过特殊的修饰, 导致产物的类型受到了明显的限制, 不利于该类环化方法的广泛应用.因此, 发展不依赖底物的修饰, 而是通过催化剂控制的方法受到了越来越多的重视.
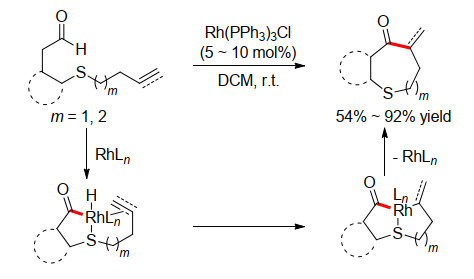
在1979年, Suggs小组[13]为醛基化合物设计了独特的氨基吡啶有机催化剂, 该试剂可以和醛现场生成3-甲基-2-胺基吡啶基的醛亚胺, 在和过渡金属作用后, 形成双齿配位的金属中间体, 可以有效消除脱羰副反应, 促进了分子间氢酰化反应.受此启发, Douglas小组[14]将此体系应用于烯醛的分子内氢酰化反应, 构建中环化合物.对于没有杂原子存在的全碳链接的底物, 反应依然可以很好地进行(Scheme 10).因为形成双齿配位的金属中间体不仅完全抑制了脱羰反应, 而且大大增强了过渡态的刚性, 从而显著提升了环化反应的活性.此外, 作者还对氨基吡啶结构进行了修饰, 试图通过引入手性基团来控制环化反应的立体选择性.但是, 由于引入的手性基团远离反应中心, 最终只获得最高31% ee值.
另一类催化剂控制策略就是近年来发展的大位阻卡宾-Ni催化体系.该体系可以直接实现较为惰性的芳基或杂芳基C—H键的活化, 代替早前主要局限于更为活泼的醛基C—H键, 从而有效避免了脱羰副反应.该金属中间体不仅具有较好的稳定性, 而且对不活泼双键也具有较好的反应活性.
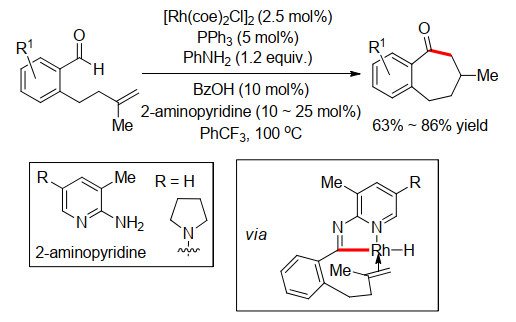
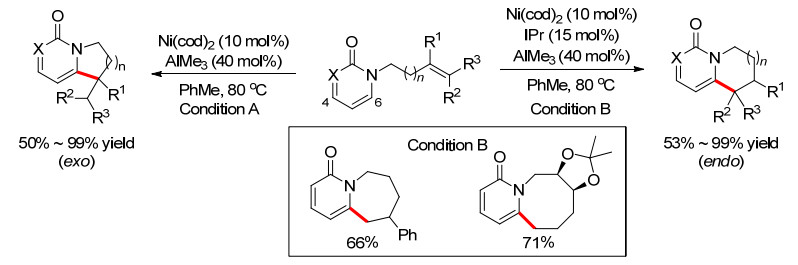
虽然早在2013年, Yoshikai和Cramer[15]在进行C—H键和烯烃的6元环化时, 就已经发现了7元中环的生成, 但还只是反应的副产物, 收率和选择性不高. 2015年, Cramer小组[16]利用卡宾-Ni催化体系终于实现了中环的选择性合成(Scheme 11).当在AlMe3作用下, 吡啶酮易于芳构化, 从而导致C(6)—H的酸性增强, 更有利于和Ni的氧化加成生成Ni—H物种, 这时候不需要添加卡宾配体就可以和烯烃很好地环化, 得到exo-型环化产物.然而当添加卡宾配体时, 可以切换反应的选择性, 生成endo-型环化产物.虽然大多数产物都是经典的5元和6元环, 但是通过改变烯烃的结构和长度, 作者首次实现了两例endo-型环化的7元和8元环产物.
2018年, Cramer小组[17]使用手性的卡宾配体, 实现了该环化反应的立体选择性控制(Scheme 12).同样, 多数底物都是6元环产物, 只有一例是7元环产物, 获得82%的收率和90%的ee值.
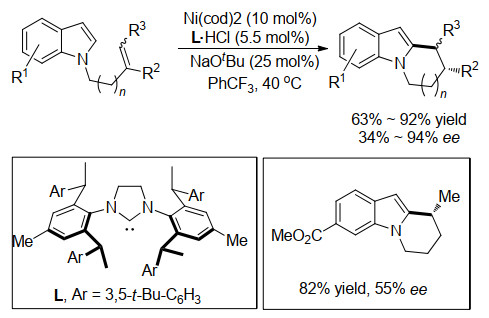
2019年, Cramer小组[18]将底物范围拓展到富电子的吲哚和吡咯类化合物.该类化合物的C—H键更富电子, 不易和Ni发生氧化加成反应.然而, 使用大位阻的卡宾配体依然可以很好地促进Ni对此类C—H键的氧化加成, 并顺利实现随后和烯烃的环化反应(Scheme 13).虽然对于各类小环产物的活性和选择性都较为理想, 但是对于仅有的一例7元中环的合成, 仅获得82%的收率和50%的ee值.
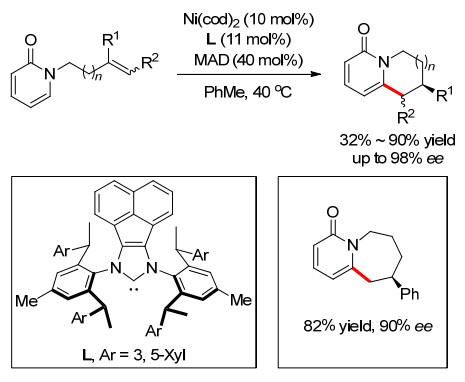
2019年, 施世良小组[19]使用Ni-卡宾催化实现了吡啶和烯烃的分子内环化反应.使用大体积的手性氮杂环卡宾配体可以实现吡啶3-位或者4-位对映选择性的C—H键活化.多数6元环化产物具有较好的收率和高的对映选择性, ee值最高可达99%以上.通过反应条件的进一步优化下, 他们还获得了一例7元环化产物, 但是只得到55%的收率和36%的ee值(Scheme 14).
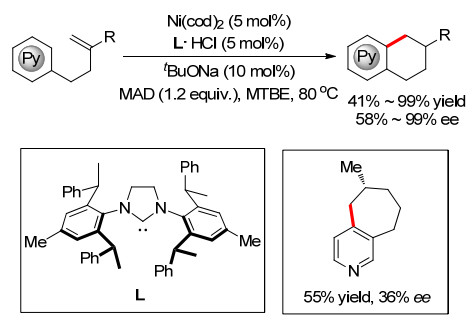
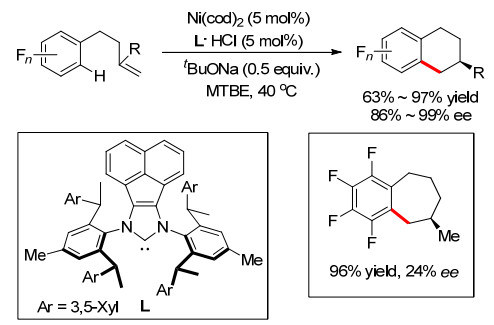
同年, 施世良小组[20]在Ni催化条件下, 使用手性氮杂环卡宾配体实现了多氟芳烃和烯烃的分子内不对称环化反应, 合成了多种手性多氟四氢萘衍生物, 并获得了较高的对映选择性(Scheme 15).通过优化反应条件, 他们还实现了一例7元环的合成, 收率达96%, 但是ee值只有24%.
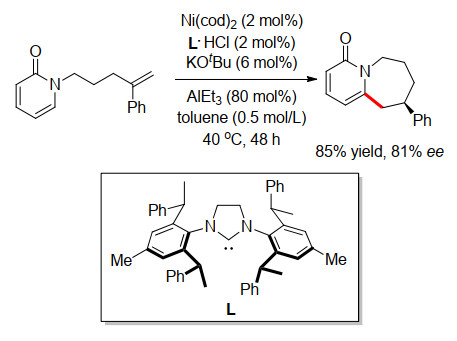
2020年, 施世良小组[21]利用吡啶分子内环化的类似催化体系, 又尝试了吡啶酮和嘧啶酮等杂环的分子内不对称环化反应.发现当将AlEt3用量由40 mol%增加至80 mol%, 可以获得一例7元环化产物, 收率为85%, ee值为81% (Scheme 16).
总结了近年来通过过渡金属催化π-不饱和化合物对C—H键插入反应构建中环化合物的研究进展.按照金属中间体稳定策略的不同, 合成方法主要分为四种类型.第一类通过形成共振结构来稳定金属中间体, 促进中环的构建.反应类型主要是过渡金属催化的醛和各类π-不饱和化合物的分子内氢酰化反应.但是, 这种合成策略具有很大的局限性, π-不饱和化合物主要局限于高度活化的烯基环丙烷、双烯和亚烷基环丁烷等底物.第二类是通过杂原子配位稳定的金属中间体, 促进中环的构建.底物中需要预先引入O、N、S或者亚砜基团参与和金属的配位, 稳定生成的金属中间体, 抑制脱羰反应, 并增加过渡态的刚性, 提高环化反应的活性.虽然该类方法可以把π-不饱和化合物拓展到更为价廉易得的简单烯烃, 但是这些杂原子的引入同样大大限制了中环产物的类型.第三种是在反应体系中引入有机催化剂, 让其和底物发生可逆的共价键作用后, 和金属发生C—H键活化, 形成双齿刚性金属中间体, 不仅消除了脱羰反应, 而且大大增加了过渡态的刚性, 有效提高了反应的活性.有机催化剂的使用避免了底物的额外修饰, 从而比前两种方法具有更大的底物范围和应用性.然而该方法依然主要针对较为活泼的醛基C—H键, 对C—H键的种类选择有很大的限制.第四种是使用大位阻的卡宾-Ni催化剂.该催化剂对各类C—H键活化都有较好的催化活性, 大大拓展了C—H键的底物种类.生成的金属中间体具有较好的稳定性和活性, 可以和各类不活泼烯烃较好地环化.但是由于配体结构有限, 过渡态的刚性也有限, 目前并不能实现很高的对映选择性, 并且多数产物还是以小环为主, 中环产物只是零星的出现, 活性受到了连接链和烯烃结构的限制.因此, 从底物适用范围来说, 第四类方法最为理想, 应该是未来发展的重点.但是在此方向上, 需要发展更为高效的卡宾配体, 进一步提升反应的活性和选择性.
Sakai, K.; Ide, J.; Oda, O.; Nakamura, N. Tetrahedron Lett. 1972, 1287.
Aloise, A. D.; Layton, M. E.; Shair, M. D. J. Am. Chem. Soc. 2000, 122, 12610. doi: 10.1021/ja0055920
Sato, Y.; Oonishi, Y.; Mori, M. Angew. Chem., Int. Ed. 2002, 41, 1218. doi: 10.1002/1521-3773(20020402)41:7<1218::AID-ANIE1218>3.0.CO;2-D
Crépin, D.; Dawick, J.; Aïssa, C. Angew. Chem., Int. Ed. 2010, 49, 620. doi: 10.1002/anie.200904527
Bendorf, H. D.; Colella, C. M.; Dixon, E. C.; Marchetti, M.; Matukonis, A. N.; Musselman, J. D.; Tiley, T. A. Tetrahedron Lett. 2002, 43, 7031. doi: 10.1016/S0040-4039(02)01552-6
Bendorf, H. D.; Ruhl, K. E.; Shurer, A. J.; Shaffer, J. B.; Duffin, T. O.; LaBarte, T. L.; Maddock, M. L.; Wheeler, O. W. Tetrahedron Lett. 2012, 53, 1275. doi: 10.1016/j.tetlet.2011.12.125
Aïssa, C.; Fürstner, A. J. Am. Chem. Soc. 2007, 129, 14836. doi: 10.1021/ja0746316
Shen, Z. M.; Khan, H. A.; Dong, V. M. J. Am. Chem. Soc. 2008, 130, 2916. doi: 10.1021/ja7109025
Shen, Z. M.; Dornan, P. K.; Khan, H. A.; Woo, T. K.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 1077. doi: 10.1021/ja806758m
Coulter, M. M.; Dornan, P. K.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 6932. doi: 10.1021/ja901915u
Khan, H. A.; Kou, K. G. M.; Dong, V. M. Chem. Sci. 2011, 2, 407. doi: 10.1039/C0SC00469C
Arnold, J. S.; Mwenda, E. T.; Nguyen, H. M. Angew. Chem., Int. Ed. 2014, 53, 3688. doi: 10.1002/anie.201310354
Suggs, J. W. J. Am. Chem. Soc. 1979, 101, 489. doi: 10.1021/ja00496a040
Beletskiy, E. V.; Sudheer, C.; Douglas, C. J. J. Org. Chem. 2012, 77, 5884. doi: 10.1021/jo300779q
(a) Ding, Z.; Yoshikai, N. Angew. Chem., Int. Ed. 2013, 52, 8574.
(b) Donets, P. A.; Cramer, N. J. Am. Chem. Soc. 2013, 135, 11772.
Donets, P. A.; Cramer, N. Angew. Chem., Int. Ed. 2015, 54, 633.
Diesel, J.; Finogenova, A. M.; Cramer, N. J. Am. Chem. Soc. 2018, 140, 4489. doi: 10.1021/jacs.8b01181
Diesel, J.; Grosheva, D.; Kodama, S.; Cramer, N. Angew. Chem., Int. Ed. 2019, 58, 11044. doi: 10.1002/anie.201904774
Zhang, W. B.; Yang, X. T.; Ma, J. B.; Su, Z. M.; Shi, S. L. J. Am. Chem. Soc. 2019, 141, 5628. doi: 10.1021/jacs.9b00931
Cai, Y.; Ye, X. D.; Liu, S.; Shi, S. L. Angew. Chem., Int. Ed. 2019, 58, 13433. doi: 10.1002/anie.201907387
Shen, D.; Zhang, W. B.; Li, Z. Y.; Shi, S. L.; Xu, Y. J. Adv. Synth. Catal. 2020, 362, 1125. doi: 10.1002/adsc.201901582

图 1 含有中环骨架的代表性天然产物
Figure 1 Representative natural products with medium ring skeletons

图式 1 铑催化氢酰化合成环辛烯酮
Scheme 1 Rh-catalyzed hydroacylation for the synthesis of cyclooctenones

图式 2 铑催化氢酰化合成环庚烯酮
Scheme 2 Rh-catalyzed hydroacylation for the synthesis of cycloheptenones

图式 9 铑催化的连续的烯丙基胺化和烯烃氢酰化
Scheme 9 Rh-catalyzed sequential allylic amination and alkenes hydroacylation

图式 10 协同催化烯烃分子内氢酰化
Scheme 10 Cooperative catalyzed alkenes intramolecular hydroacylation

图式 13 Ni催化对映选择性烯烃分子内氢芳基化
Scheme 13 Ni-catalyzed enantioselective alkenes intramolecular hydroarylation

图式 14 Ni催化对映选择性烯烃分子内氢芳基化
Scheme 14 Ni-catalyzed enantioselective alkenes intramolecular hydroarylation

图式 15 Ni催化对映选择性烯烃分子内氢芳基化
Scheme 15 Ni-catalyzed enantioselective alkenes intramolecular hydroarylation

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:








