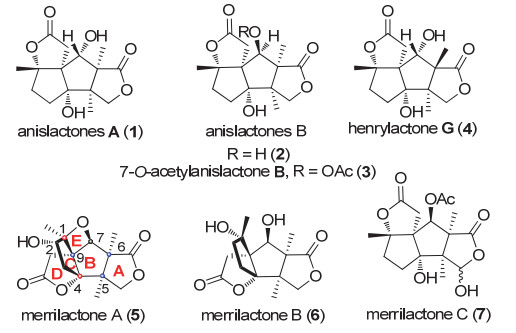
图 1.
几种Anislactone类倍半萜天然产物
Figure 1.
Examples of anislactone sesquiterpenes

八角又称八角茴香, 是被子植物门、双子叶植物纲、八角目、八角科、八角属的一种常绿乔木, 广泛分布于我国西南各地, 是一种“药食同源”的经济树种.八角的果实及枝叶中积累了大量且种类丰富的次级代谢产物, 其中主要包括挥发油、倍半萜类、黄酮类和木质素类等.除了作为日常餐饮的调味品以外, 医药上可用于治疗神经衰弱、消化不良等病症[1].
1989年Kouno研究小组[2]从白花八角(Illicium anisatum)中分离得到了anislactone型倍半萜anislactones A及anislactones B. 2000年日本Fukuyama小组从滇西八角(Illicium merrillianum)中分离得到了具有新颖骨架的倍半萜类天然产物merrilactone A-C以及7-O-acetyl- anislactone B.此系列天然产物表现出优异的神经营养活性, 特别是(-)-merrilactone A在0.1 mmol/L的低浓度下仍表现出良好的生理活性.更为重要的是(-)- merrilactone A是一种低分子量非肽类亲脂性化合物有望穿越血脑屏障[3-4], 这为开发治疗阿尔兹海默症的药物提供了新颖的先导化合物.后续研究中, 张雁冰课题组从红茴香中分离得到了新型的倍半萜henrylactone G[5], 此化合物系anislactones B的异构体.该类化合物具有较高的氧化态、拓扑多环结构及密集的手性中心特别是连续季碳手性中心, 这在合成上极具挑战性, 因此吸引了众多合成化学家的关注(图 1).
Merrilactone A分子由[5-5]二环碳环骨架、双γ-丁内酯结构单元及一个连接C1-C7的氧杂环丁烷桥构成, 分子中具有七个手性中心, 其中包括三个全碳手性季碳中心、两个四取代碳原子.这种复杂的笼状结构在全合成中极具挑战性, 因此吸引了多个国内外课题组的关注.迄今为止, 国内外已有七个课题组完成了merrilactone的全合成工作, 其中只有一个课题组完成了(±)-anislactone A的合成.本文按照课题组分类, 依照时间顺序对这些研究工作具体总结如下.
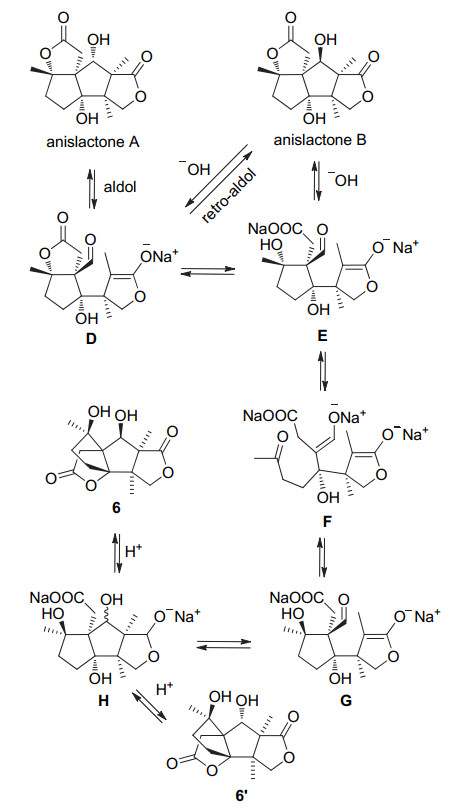
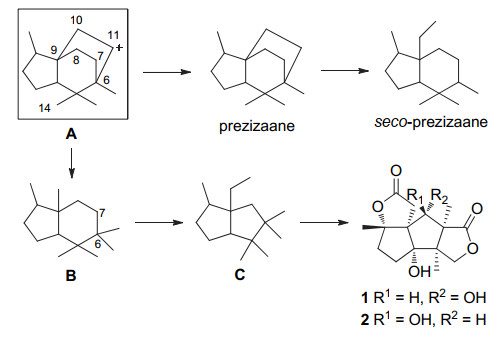
在分离出多个anislactone型倍半萜天然产物之后, Fukuyama小组[4]对该类天然产物的生源途径以及转化关系进行了推测(Scheme 1).基于前人的研究, 前体A在seco-prezizaane型倍半萜的生物合成中扮演了关键的角色, 同样在anislactone型倍半萜的生物合成中也是必经历的中间体.前体A中C-10与C-11之间碳碳键的断裂生成了二环中间体B, 接着C-6与C-7之间碳碳键断裂、缩环构建出anislactone型倍半萜的碳环骨架, 进而通过后续氧化生成了该类天然产物.
对于这类天然产物之间的转化关系, Fukuyama课题组[4]对anislactone B进行碱处理成功得到了天然产物anislactone A、中间体6及其对映体6' (Scheme 2).该作者提出在碱存在下, anislactone B通过retro-aldol/adol缩合反应转化为anislactone A.而在B、D环同时开环后, 再经过两次的retro-aldol/adol缩合反应, 则可转化为merrilactone潜在合成前体6及其对映体6'.这一研究对anislactone类天然产物的多样性合成提供了借鉴.
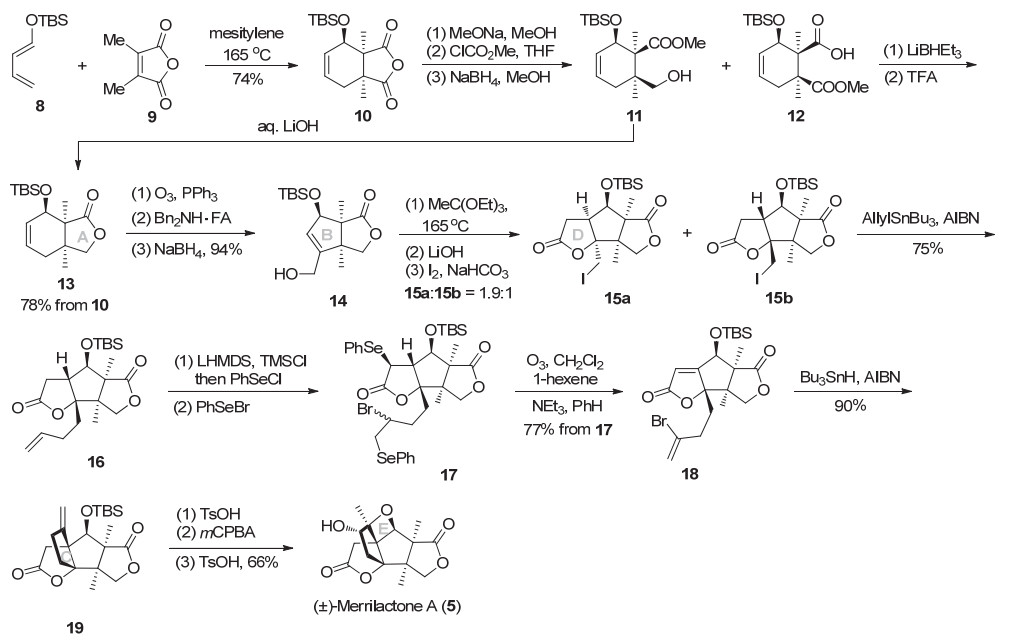
2002年, Danishefsky等[6]以Diels-Alder反应为关键反应实现连续季碳的构建, 经氧化开环、关环构筑B环, 碘代内酯化构建D环, 并利用自由基环化构建C环, 最后以homo-Payne重排完成氧杂环丁烷环E环的制备, 以20步10.7%总收率完成了(±)-merrilactone A的全合成.如Scheme 3所示, 双烯体8与二甲基顺丁烯二酸酐发生Diels-Alder环加成反应得到具有连续季碳中心的酸酐10.化合物10经过皂化开环、还原得到甲酯11及羧酸衍生物12, 前者在碱性条件下关环可得到内酯13, 后者经还原后在酸性条件下关环, 同样可转化为内酯13.之后, 环己烯臭氧化开环、二苄胺催化的分子内aldol缩合脱水、还原得到烯丙醇14.利用Johnson- Claisen重排、皂化、碘化环化得到三环中间体15a和15b (dr 1.9:1), 完成了ABD环的构建.碘代物15b经Keck烯丙基化得到烯烃16. LHMDS做碱, 在D环内酯α-位引入苯硒基, 然后双键溴硒化得二硒17.再经氧化、碱性条件脱硒得到溴代烯烃18.分子内自由基关环反应构建C环, 同时构建三个季碳, 得双酯19.该方法可以高效构建出ABCD四环中间体, 在后续的该天然产物的全合成中被广泛采用.在酸性条件下, 双酯19中的双键异构化到环内, 经mCPBA氧化双键得到环氧(α/β=3.5:1), 对甲苯磺酸催化下homo-Payne重排构建氧杂环丁烷E环, 完成(±)-merrilactone A (5)的全合成.值得指出的是, 之后其他课题组在merrilactone A的合成中, 都采用了最后两步双键环氧化/酸促进开环氧策略, 来完成E环的构建, 此方法为构建该天然产物E环的最高效策略.
Danishefsky小组完成了merrilactone A的消旋体的合成之后, 开展了该天然产物的不对称合成研究.在之前的合成路线中, 酸酐10进行水解等官能团转化构建γ-丁内酯13时几乎没有区域选择性; 而Johonson- Claisen重排构建反应的非对映选择性极差.为解决这些问题, 该课题组设计新的合成路线实现(-)-merrilac- tone A的不对称全合成[7].如Scheme 4所示, 多氯取代的环戊二烯20与甲基顺丁烯二酸酐21进行Diels-Alder反应专一性得到内型产物, 底物控制的立体选择性甲基化构建连续季碳得到二酯22.然后二酯经锂铝氢还原酯基、钠消除脱氯, 同时脱除甲基缩酮保护得到二醇23.缩酮保护二醇, 然后羰基与HWE反应延长碳链, 用镁还原不饱和酯, 再用酸后处理脱除缩酮保护得到甲酯24. mCPBA氧化24的双键, 同时伯醇分子内开环氧可得到二醇25.在这一步, 化合物24先用二甲基过氧化酮(DMDO)选择性环氧化, 手性钴催化剂催化伯醇开环氧可实现去对称化, 以86% ee值和86%产率得到光学活性的二醇25.
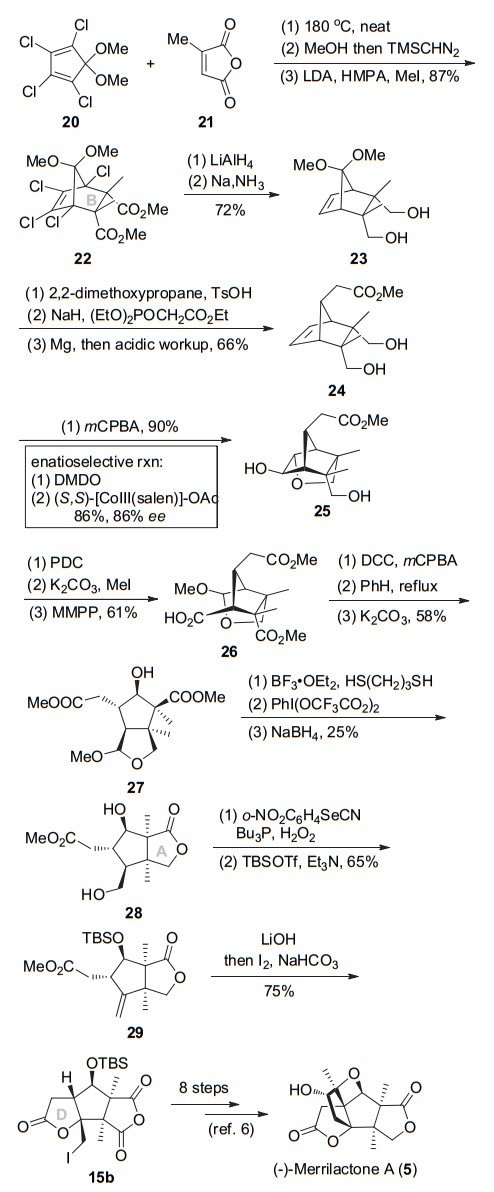
接下来重铬酸吡啶(PDC)同时氧化伯醇和仲醇、甲酯化、用单过氧邻苯二甲酸镁六水合物(MMPP)将酮进行Bayer-Villiger氧化, 氧化后得到的缩醛被醇解可得到羧酸26.羧酸26与mCPBA用DCC缩合、二酰基过氧加热发生类Bayer-Villiger氧化重排、然后碱性条件脱除酯基可得到脱羧氧化产物27.醇27在路易斯酸催化下打开缩醛环, 得到的伯醇和甲酯内酯化, 同时醛基和乙二硫醇生成硫缩醛, 然后用三氟醋酸碘苯脱除硫缩醛, NaBH4还原醛从而完成AB环的构建.内酯28伯羟基经硒化、消除、仲羟基叔丁基二甲基硅基(TBS)保护得到烯烃29.然后经水解甲酯、一锅的碘环化即可合成消旋路线中已知三环中间体15b, 从而完成(-)-merrilactone A的形式不对称对称全合成.
2003年, Inoue和Hirama教授[8]以分子内aldol缩合去对称化反应实现BC环的构建, 利用Fetizon氧化构建出A环, 最终以27步1.4%总收率完成了(±)-merrilac- tone A全合成.具体合成过程如Scheme 5所示, 首先, 商业化的原料二氯乙烯30与顺丁烯二酸酐9, 由光促进的[2+2]环化实现连续手性季碳的构建, 用锌粉消除脱氯及LiAlH4还原酸酐可得到二醇31.苄基保护羟基, 接着四氧化锇氧化双键, Swern氧化为二酮, 进而与烯丙基格氏试剂亲核加成得到两个烯丙基处于同面的异构体32.用Grubbs一代催化剂催化的烯烃复分解环化反应构建[4.2.0]二环中间体, 接着二醇用醋酸铅氧化开环, 得到环辛烯二酮33.经过条件优化, 以LHMDS为碱在-100 ℃下反应, 二酮经由分子内的aldol缩合去对称化反应可实现BC环的立体选择性构建(dr 3.1:1), 经过柱层析分离得到主要非对映异构体34.该去对称反应一步成功构建出BC二环中间体, 为合成该天然产物碳环骨架开辟出新的方法.化合物34经环氧化、1, 8-二氮杂二环十一碳-7-烯(DBU)促进的开环氧、2-碘酰基苯甲酸(IBX)氧化得到共轭烯二酮35.该底物叔醇经缩醛化、自由基环化引入C9位季碳, 得到二酮36 (乙氧基构型β/α=3.5:1), 在酸条件下可将α异构体完全转化为β异构体.接着将酮转化为烯醇硅醚, 进而与Eschenmoser试剂加成并氧化消除得到不饱和酮37.缩醛在三氟乙酸条件下脱除乙氧基, 然后甲磺酰活化消除得到烯醇醚.接下来用L-Selectride进行1, 4-还原, 用Comins试剂捕获烯醇中间体得烯醇三氟甲磺酸酯38.醋酸钯催化还原脱除OTf, DIABL-H还原羰基主要得到羟基朝上产物(β/α=6:1), 然后钠/液氨条件脱二苄基得到三元醇39.经烯醇醚水解、Fetizon氧化半缩醛同时区域选择性氧化1, 4-二醇为内酯得到四环中间体40.最后经DMDO氧化双键、酸性条件下环氧开环构建氧杂环丁烷完成了(±)-merrilactone A的全合成.
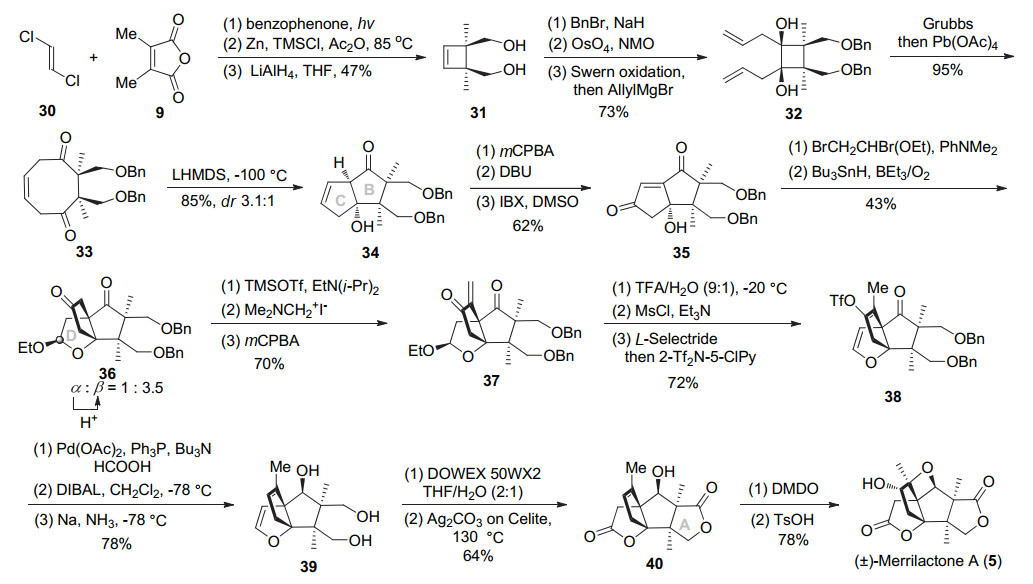
完成了merrilactone A消旋体的合成之后, 2006年Inoue小组[9]采用类似策略利用选择性Sharpless双羟化预先引入手性中心并经过相应的官能团转化构建出具有不同电性、位阻羟基保护基的中间体, 从而实现了(-)-merrilactone A的不对称全合成, 路线共31步总收率为1.1%.如Scheme 6所示, 二甲基顺丁烯二酸酐9经半还原、Wittig反应、羧基甲酯化得到不饱和酯41.末端双键区域及对映选择性的Sharpless双羟化得到的内酯经重结晶可将产物ee值从90%提高到>99%. Piv保护伯醇后与不饱和内酯和顺式二氯乙烯发生光促[2+2]环化, 锌粉消除脱氯, 然后锂铝氢还原内酯同时脱除伯醇的Piv保护得到三元醇42.缩酮保护邻二醇, 另外一个伯醇用苄基保护, 然后盐酸脱除缩酮保护, 四醋酸铅氧化切断邻二醇进而DIBAL-H还原醛得到醇43.为了提高后续分子内aldol缩合的区域选择性, 选用大位阻BTB (2, 6-bis-(trifluoromethyl)benzyl)保护伯羟基, 参照之前的合成路线经三步得到二醇44, 然后经过烯烃复分解反应及四醋酸铅氧化开环得到环辛烯二酮45.经过一系列条件优化发现用NaHMDS做碱, 可区域选择性拔掉与较小位阻Bn保护基相邻羰基的α氢, 然后分子内Adol缩合非对映选择性得到醇酮46 (dr 17:1), 从而实现了BC环的立体选择性构建.基本依照之前报道路线, 该化合物经过两步连续的氧化及引入溴代缩醛得到自由基环化前体47. AIBN/Bu3SnH作用下, 顺利发生分子内自由基环化构建C9位季碳手性中心(乙氧基构型β/α=4:1).参照该课题组之前的消旋路线[8], 光学纯三环化合物48经过10步转化可完成(-)-merrilactone A的不对称全合成. 2007年该课题组发展了手性氨基锂50诱导的环辛烯二酮49的去对称化反应, 以79%收率57% ee及dr 6:1得到醇酮ent-46, 通过重结晶可将ee值提高至99%.从醇酮ent-46出发, 采用之前一样的步骤, 再用14步完成了(+)-merrilactone A的全合成工作[10].
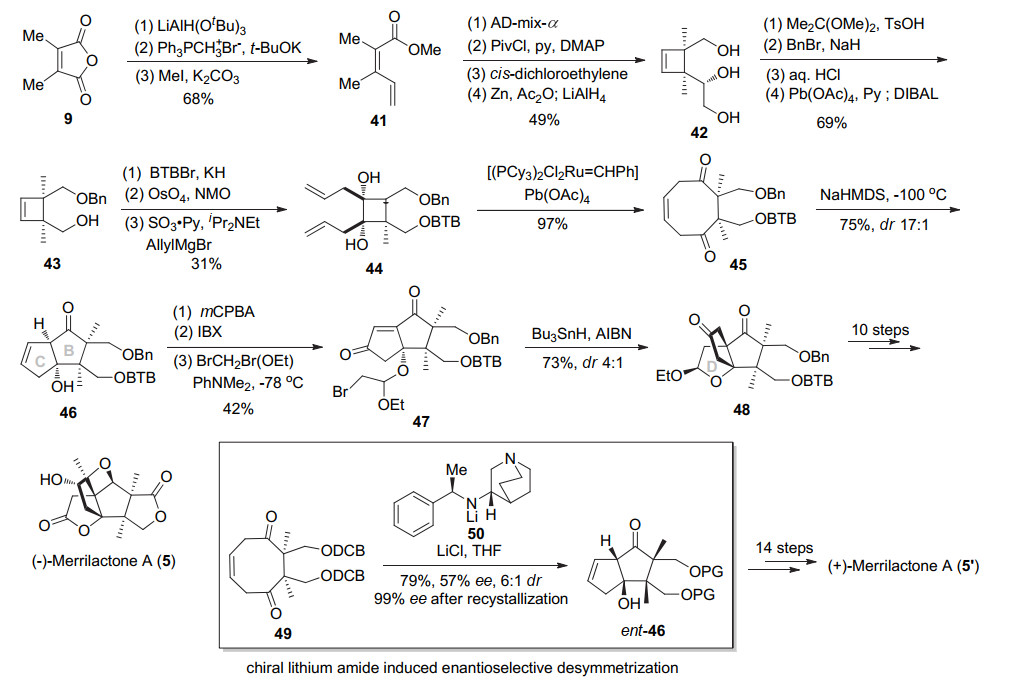
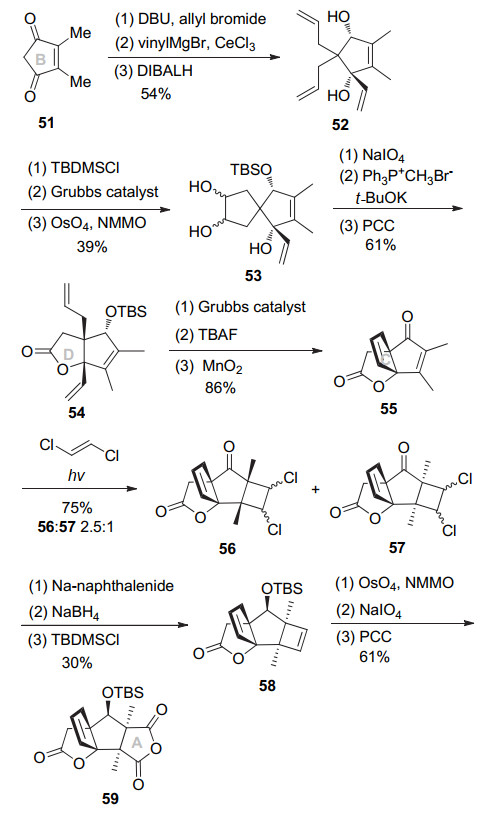
2005年, Mehta小组[11]报道了以RCM反应及光促[2+2]环加成反应构建merrilactone四环体系的合成路线.具体路线如Scheme 7所示, 环戊烯二酮51的α位双烯丙基化后, 使用低活性烯基铈试剂选择性和一个羰基亲核加成, 然后DIBAL-H还原另外一个羰基立体选择性得到顺式二醇52.用TBS选择性保护二级醇, 然后经由Grubbs试剂催化的烯烃复分解反应环化, 再经OsO4催化双羟化可得到三元醇53.该三元醇用NaIO4氧化断开、叔醇和同面的醛基半缩醛环化, 另外一个醛基发生Wittig反应, 后由氯铬酸吡啶盐(PCC)氧化得到三烯54, 完成D环的构建.该化合物由烯烃复分解环化构筑C环, 接下来四丁基氟化铵(TBAF)脱除TBS, MnO2氧化烯丙醇得到烯酮55.内酯55经与反式1, 2-二氯乙烯进行光促[2+2]环化反应, 主要得到甲基朝上环丁烷56和异构体57 (56:57=2.5:1).非主要异构体57经钠萘消除脱氯, 氧化断裂双键, 再用PCC氧化环化构建A环得到酸酐59, 从而完成了merrilactone A的四环骨架构建.
2005年, Mehta教授[12]采用之前报道的策略, 通过依次构建B-C-D-A-E环次序, 最终以27步0.28%总收率完成了(±)-merrilactone A的合成.具体如Scheme 8所示, 环戊烯二酮51在DBU作用下与甲醛经两次aldol反应、缩酮保护邻二羟基、选择性还原一个羰基得到醇酮60.接着酮与烯丙基铈试剂反应, 选择性氧化烯丙醇后脱除缩酮保护得到三醇61.选择性将叔醇和顺式伯醇用缩酮保护, 接着PDC氧化另外一个伯羟基成醛, 甲基化后再氧化为酮, 最后Wittig反应得到双烯62.用Grubbs催化剂进行烯烃复分解环化构建出C环, 得到二环产物63.接下来光促[2+2]环化反应构建A环的连续季碳中心得到多环丁烷64及65 (1:2).和之前的合成路线相比, 可能由于环缩酮的位阻比较大, 主要得到两个甲基朝下的异构体.钠萘条件下消除脱氯、DIBAL-H还原羰基得到单一β构型仲醇, 接着TBS保护羟基得到环丁烯66.酸性条件下环丁烯66脱除缩酮保护, 然后四丙基高钌酸铵(TPAP)氧化伯醇, 进而Wittig反应制备出甲基烯基醚67.接下来酸催化分子内环化得半缩醛, 进而PCC氧化得到内酯构建出D环.进一步臭氧断双键、选择性还原得到半缩醛68, 然后经PCC氧化构建A环, 然后TBAF脱除TBS保护基得到已知中间体40. DMDO氧化双键, 对甲苯磺酸催化开环氧构筑氧杂环丁烷从而完成(±)-merrilactone A的合成.该路线相较与其他路线整体选择性以及合成效率都偏低.
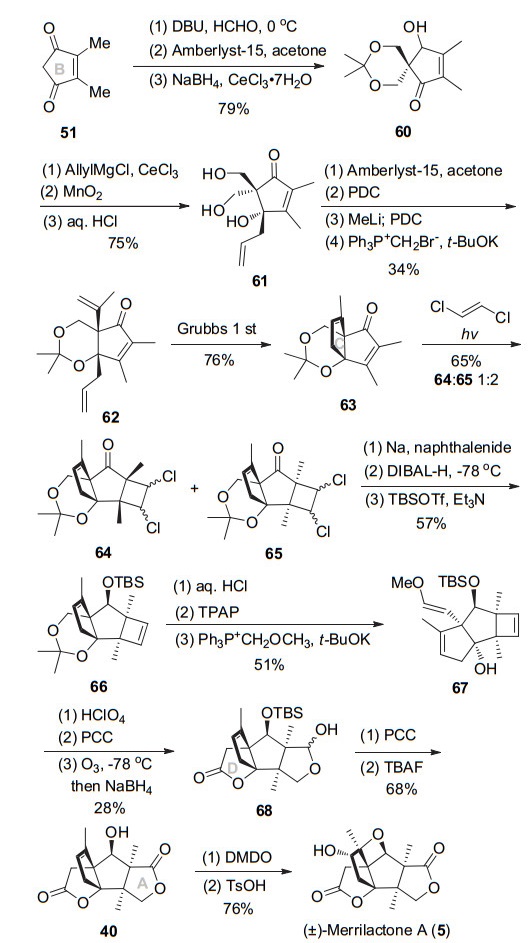
2006年, Frontier小组[13]应用铱催化Nazarov环化构建BD并环、自由基环化构筑C环、内酯环化构建A环、开环氧合成E环的次序完成merrilactone A消旋体的全合成, 该路线总长16步, 总收率19.6%, 为目前所报道的最高效合成路线之一.其具体路线如Scheme 9所示, 炔丙酸乙酯和炔醛69亲核加成, 接着与三丁基锡铜试剂对炔的Michael加成环化得到内酯, 然后将烯基锡在溴作用下转化为烯基溴得到内酯70.将不饱和内酯转化为硅氧基呋喃, 接下来用叔丁基锂和溴交换, 然后亲核进攻Weinreb酰胺得到呋喃酮72.三价铱73催化呋喃酮72的Nazarov环化, 可立体选择性得到单一非对映异构体74, 选择性构建B环及连续四取代碳手性中心.化合物74用AgNO3选择性脱除末端炔三甲基硅基(TMS), 然后用Bu3SnH/AIBN启动串联锡基自由基加成/环化反应实现手性季碳及C环的构建.利用Nazarov环化一步构建出BD二环以及自由基关环实现C环的连续性构建, 这一策略使用极大提高了合成的效率. TBAF脱除三异丙基硅基(TIPS)及TBS保护, 接着伯醇用氯甲酸乙酯酯化得到酮76.酮76在碱性条件下进行亲核内酯化, 得到内酯77及内酯开环产物, 此混合物不分离, 可通过酸催化内酯环反应完全转化为77.用NaH做碱, 可在内酯77的α位立体选择性甲基化, 接着NaBH4还原羰基得到四环中间体78.最终, 参照Danishefsky教授的方法, 酸促进双键移位、mCPBA环氧化、对甲苯磺酸促进的环氧开环, 完成了(±)-merrilactone A的全合成.
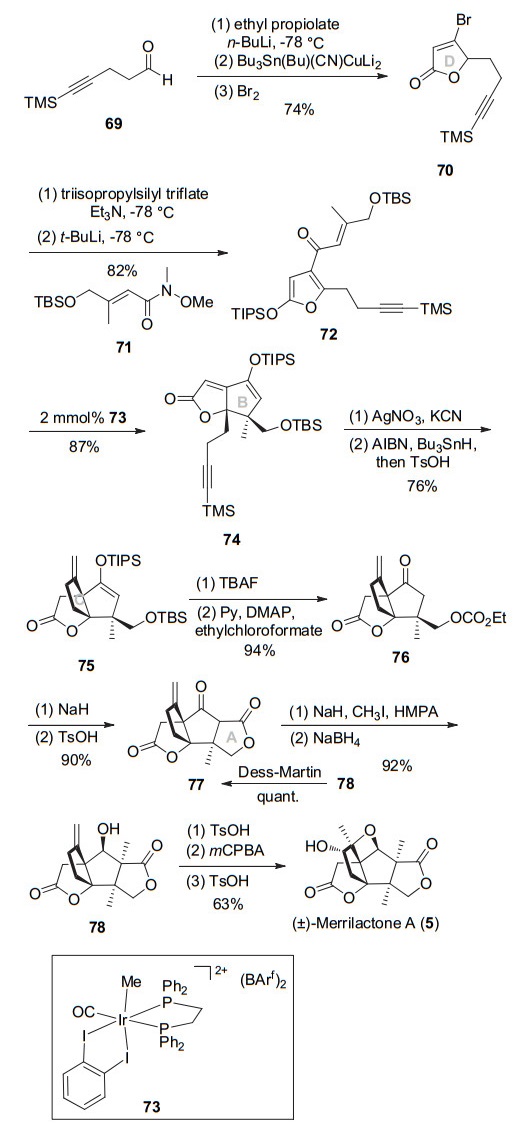
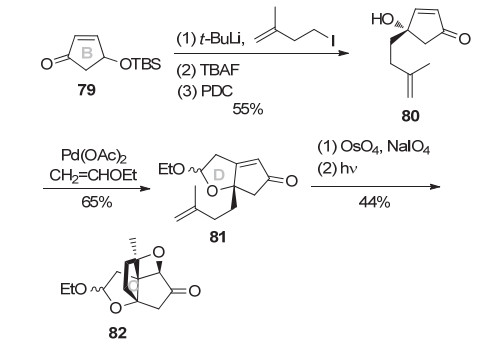
2005年, Greaney小组[14]发展出分子内Paternò- Büchi光促加成构建氧杂环丁烷E环策略, 完成了merrilactone的BCDE四环构建.该路线如Scheme 10所示, 1-碘-3-甲基-3-丁烯制备的锂试剂与烯酮79发生1, 2-加成, TBAF脱除TBS保护, 然后PDC氧化得到烯酮80.醋酸钯催化80的醇和乙烯基乙醚的串联氧钯化/分子内碳钯环化反应构建D环, 得到烯酮81.然后经氧化切断末端烯烃得到甲基酮, 光促进Paternò-Büchi环化可得到四环产物82, 从而完成merrilactone的BCDE四环构建.这是目前为止, 报道的第二种构建氧杂环丁烷E环的新策略.
2010年Greaney小组[15]利用光促[2+2]环化构建连续手性季碳中心及扩环反应巧妙的构筑出含有边链的A环.接着烷基化得到自由基环化前体, 经Ti(Ⅲ)诱导的自由基环化反应实现C环的构筑以及选择性氧化构建A环, 最终完成了(±)-merrilactone A的形式合成及(±)-anislactone A的首次全合成.具体如Scheme 11所示, 二甲基顺丁烯二酸酐9与乙烯酮缩二甲醇83进行光促[2+2]环化构建出连续手性季碳中心, 接着锂铝氢还原酸酐, 苄基保护伯醇, 酸性条件下脱缩酮保护得到环酮84.应用Tiffeneau-Demjanov扩环反应构建B环, 乙酯与烯丙醇交换为烯丙酯, 然后β-酮酯α位烯丙基化, 最后用Tsuji-Trost脱羧-脱氢反应得到不饱和酮85.该化合物将末端烯烃氧化切断、还原醛得到醇86.化合物86用次氯酸钠氧化不饱和酮得到环氧87 (α/β=2.2:1), 其中β型产物可经还原脱氧重新转为不饱和酮86.环氧87用TBS保护羟基得到产物, 经过立体选择性烷基化、脱除末端炔基TMS保护得到自由基环化前体89.在过量[Cp2TiCl2]/Zn促进下, 环氧开环产生三级自由基, 经过5-exo-dig环化立体专一性构建出C环, 得到双环中间体90.用Ley氧化仲羟为酮、TBAF脱除TBS保护及进一步Ley氧化构建内酯得到三环产物91.该产物利用Inoue小组报道的策略, 即酸性条件将末端双键异构化, 钠/液氨条件脱除双苄基, Fetizon氧化区域选择性构建内酯, 可得已知四环中间体40, 从而完成了(±)-merri- lactone A的形式合成.
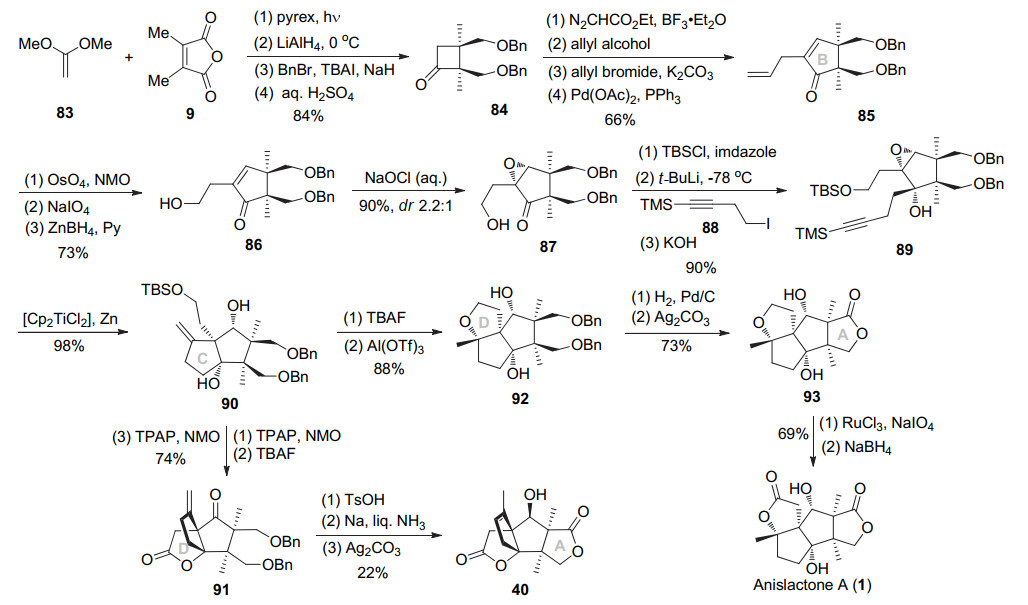
另一方面, 二醇90用TBAF脱除TBS保护基, Al(OTf)3催化分子内环醚化得到三环产物92.进而氢化脱除苄基, Fetizon氧化二醇得到γ-丁内酯93.用RuO4氧化将呋喃环转化为γ-丁内酯, 同时伯醇被氧化为酮, 最后用NaBH4还原酮(dr 5:1), 以22步, 5.7%收率, 完成了(±)-anislactone A的首次合成.
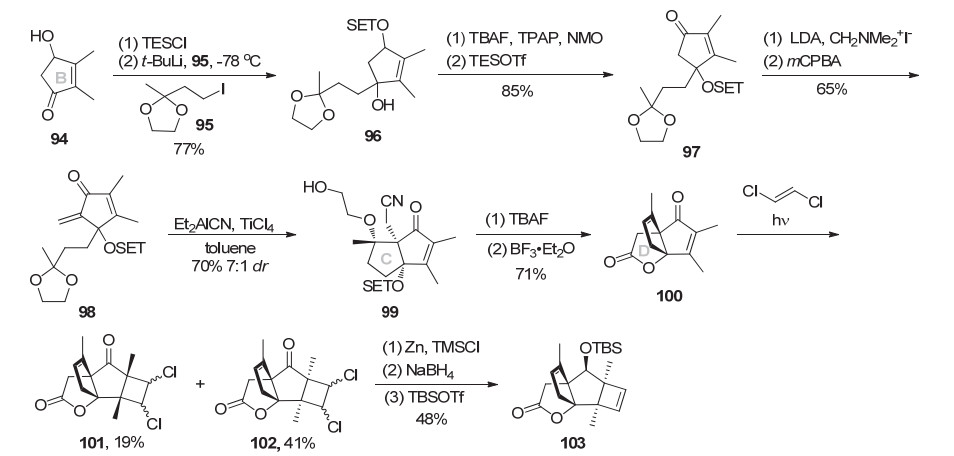
2012年, Greaney小组[15b]报道了一条新的(±)-mer- rilactone A形式合成路线.具体如Scheme 12所示, 烯酮94用TES保护伯醇, 烷基碘95与叔丁基锂交换得到烷基锂试剂与烯酮1, 2-亲核加成生成醇96.用TBAF脱除TES保护, Ley氧化烯丙醇, TES保护叔醇得到烯酮97.二异丙基氨基锂(LDA)为碱拔去烯酮97的α位质子与Eschenmoser’s试剂发生Mannich反应, 然后用mCPBA氧化消除叔胺得到二烯酮98.在TiCl4作用下, 烯酮98发生串联的氰基Michael加成/分子内aldol缩合, 立体选择性构建出C环(dr 7:1).脱除TES保护以及三氟化硼促进的叔醇与腈基的内酯化, 同时消除乙二醇单醚得到烯酮100, 完成D环的构建.三环化合物100与反式二氯乙烯进行光促[2+2]环加成反应制备出四环中间体102.锌粉消除脱氯、NaBH4还原羰基、TBS保护伯醇得到Mehta小组[12]的合成路线已知中间体103, 从而完成了(±)-merri- lactone A的形式合成.
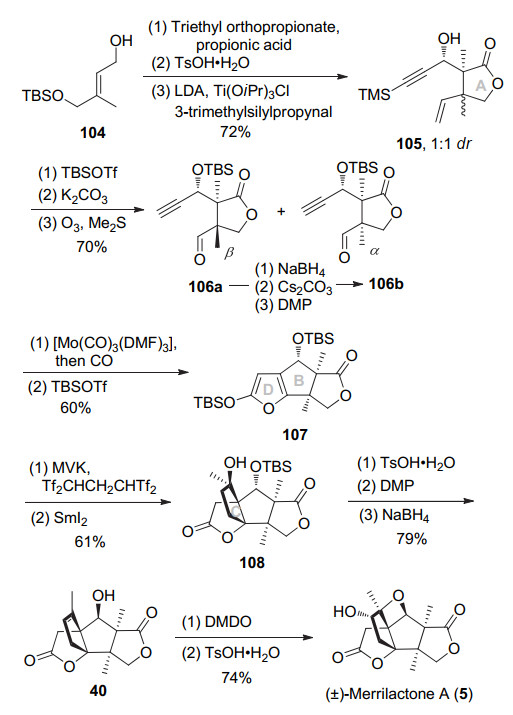
2012年, 兰州大学翟宏斌教授课题组[16]以Johnson- Claisen重排构建出连续季碳中心, 杂Pauson-Khand反应构筑A环以及内酯D环, 并且用酮羰基作为自由反应前体进行自由基加成反应构建C环, 最终15步, 以11%的收率完成了(±)-merrilactone A的合成, 为目前所报道的最短的合成路线之一.具体如Scheme 13所示, 烯丙醇104与原丙酸三乙酯发生Johnson-Claisen重排, 酸性条件下脱TBS同时内酯化构建A环.然后LDA做碱, Ti(OiPr)3Cl为促进剂与TMS保护丙炔醛发生aldol反应得到内酯105, 产物中与烯基相连碳的dr值为1:1.接着TBS保护羟基, 碱性条件下脱除炔基TMS保护基, 臭氧切断末端双键得到可用柱层析分离醛106a与106b.其中β异构体106a经过还原、Cs2CO3促进的酯交换(dr 1.6:1)、Dess-Martin氧化可转化为醛106b.炔醛106b在Mo(CO)3(DMF)3促进下发生杂Pauson-Khand反应同时构建BD环得到不饱和内酯, 然后TBSOTf将不饱和内酯转化为硅氧基呋喃107.使用Tf2CHCH2CHTf2促进硅氧基呋喃107和甲基乙烯基酮的Mukaiyama-Michael加成反应, 然后SmI2介导的酮和不饱和内酯的还原偶联反应即可立体选择性构建C环, 得到四环中间体108.对甲苯磺酸促进的叔醇消除反应同时脱除TBS保护, 然后经过Dess-Martin氧化、NaBH4还原可将7位羟基经构型翻转, 得到已知中间体40.最后, 应用Inoue教授的方法, 经过DMDO环氧化、酸性条件关氧杂环丁烷可完成(±)-merrilactone A的合成.
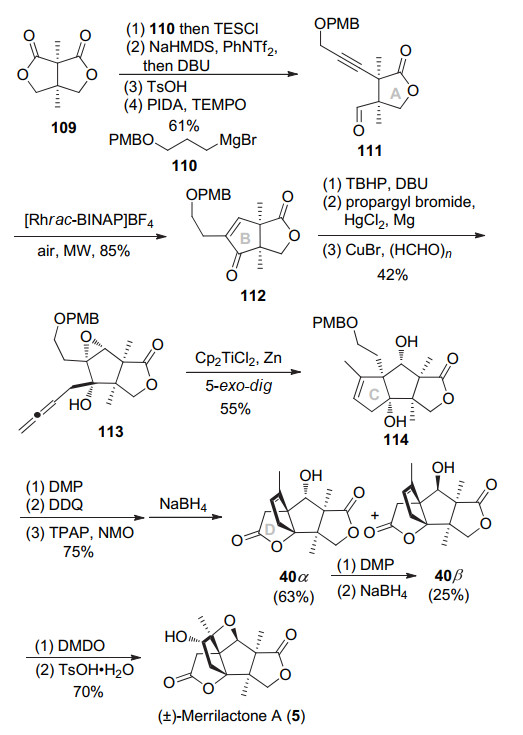
2018年, 王博课题组[17]采取去对称化策略构建连续季碳中心, Rh(I)催化分子内氢酰化构筑B环, Ti(Ⅲ)诱导环氧联烯的分子内自由基环化构建C环, 最后以15步, 5.5%总收率完成(±)-merrilactone的合成.具体如Scheme 14所示, 双内酯109与格氏试剂110选择性和一个酯基亲核加成, 去对称化构建连续季碳中心.接着TES保护羟基, 然后羰基转化为烯基OTf, 用DBU消除得到内炔.脱除TES, TEMPO/PIDA条件氧化伯醇得到醛111.醛111用[Rhrac-BINAP]BF4催化分子内醛和炔的氢酰化反应即可构建B环, 得到烯酮112.烯酮环氧化, 炔丙基格氏试剂和酮加成, 然后在CuBr催化下, 端炔转化为联烯, 得到关环前体113.现场生成的Ti(Ⅲ)促进环氧和联烯的5-exo-dig自由基关环构建C环, 以55%收率得到三环中间体114. Dess-Martin试剂氧化仲醇, 2, 3-二氯-5, 6-二氰基-1, 4-苯醌(DDQ)脱除对甲氧基苄基(PMB)保护, 然后用Ley氧化构筑D环. NaBH4还原羰基以63%和25%收率得到两个可柱层析分离的异构体40α和40β, 其中40α可通过氧化还原转化为40β.最后参照Inoue的DMDO氧化、开环氧, 即可将40转化为(±)-merrilactone A.
本文总结归纳了anislactone型倍半萜类天然产物的全合成工作, 其中merrilactone A的全合成研究最受追捧, 国内外共有7个小组完成了其合成工作, 其中有两个小组实现了该天然产物的不对称全合成, 而只一个研究小组完成了anislactone A的全合成.该家族的其他成员如anislactone B、henrylactone G、merrilactone B和merrilactone C的全合成工作尚未见报道.在该类天然产物的合成工作组, 连续季碳中心的构建基本上都是采用[4+2]或[2+2]环加成策略构建.另外, 在已完成的merrilactone A合成工作中, 氧杂环丁烷的构建都是采用Danishefsky的合成策略, 即双键环氧化及酸促进开环反应, 其他策略还未见用于这类天然产物合成.最后, 虽然目前最短合成路线为十五步(翟宏斌小组以及王博小组), 但这几条路线均为消旋体合成路线, 由于关键反应的限制, 很难通过这两条路线实现该类天然产物的不对称合成.随着有机合成化学的飞速反展, 特别是最近几年C—H键氧化反应的研究的深入, 可以预见在不久将来, 通过C—H键的直接氧化完成该类天然产物的路线将会被报道.另外, 由于天然来源的有限, anislactone型倍半萜天然产物的多种药理活性仍然是一个未得到细致研究.因此, 通过发展高效合成路线完成此类天然产物的合成, 并以此为基础对其进行药物化学研究, 将推动该类天然产物的药物应用研究.
熊燕子, 龚正礼, 中国调味品, 2008, 9, 28.Xiong, Y.-Z.; Gong, Z. L. China Condiment. 2008, 9, 28(in Chinese).
Kouno, I.; Mori, K.; Kawano, N.; Sato, S. Tetrahedron Lett. 1989, 30, 7451. doi: 10.1016/S0040-4039(00)70722-2
Huang, J.; Yokoyama, R.; Yang, C.; Fukuyama, Y. Tetrahedron Lett. 2000, 41, 6111. doi: 10.1016/S0040-4039(00)01023-6
Huang, J.; Yang, C.; Masami T.; Fukuyama, Y. Tetrahedron Lett. 2001, 57, 4691. doi: 10.1016/S0040-4020(01)00418-5
Liu, J.-F.; Wang, Y.-F.; Bi, Y.-P.; Li, H.-J.; Jia, L.; Bi, Y.-F.; Zhang, Y.-B. Tetrahedron Lett. 2013, 54, 4834. doi: 10.1016/j.tetlet.2013.06.081
Birman, V. B.; Danishefsky, S. J. J. Am. Chem. Soc. 2002, 124, 2080. doi: 10.1021/ja012495d
Meng, Z.; Danishefsky, S. J. Angew. Chem. Int. Ed. 2005, 44, 1511. doi: 10.1002/anie.200462509
Inoue, M.; Sato, T.; Hirama, M. J. Am. Chem. Soc. 2003, 125, 10772. doi: 10.1021/ja036587+
Inoue, M.; Sato, T.; Hirama, M. Angew. Chem. Int. Ed. 2006, 45, 4843. doi: 10.1002/anie.200601358
Inoue, M.; Lee, N.; Kasuya, S.; Sato, T.; Hirama, M.; Moriyama, M.; Fukuyama, Y. J. Org. Chem. 2007, 72, 3065. doi: 10.1021/jo0700474
Mehta, G.; Singh, S. R. Tetrahedron Lett. 2005, 46, 2079. doi: 10.1016/j.tetlet.2005.01.133
Mehta, G.; Singh, S. R. Angew. Chem. Int. Ed. 2006, 45, 953. doi: 10.1002/anie.200503618
(a) He, W.; Huang, J.; Sun, X.; Frontier, A. J. J. Am. Chem. Soc. 2007, 129, 498.
(b) He, W.; Huang, J.; Sun, X.; Frontier, A. J. J. Am. Chem. Soc. 2008, 130, 300.
Iriondo-Alberdi, J.; Perea-Buceta, J. E.; Greaney, M. F. Org. Lett. 2005, 7, 3969. doi: 10.1021/ol0514496
(a) Shi, L.; Meyer, K.; Greaney, M. F. Angew. Chem. Int. Ed. 2010, 49, 9250.
(b) Nazef, N.; Davies, R. D. M.; Greaney, M. F. Org. Lett. 2012, 14, 3720.
Chen, J.; Gao, P.; Yu, F.; Yang, Y.; Zhu, S.; Zhai, H. Angew. Chem. Int. Ed. 2012, 51, 5897. doi: 10.1002/anie.201200378
Liu, W.; Wang, B. Chem. Eur. J. 2018, 24, 16511. doi: 10.1002/chem.201804195

图式 2 Anislactone B的转化关系及其机理
Scheme 2 Transformations of anislactone B and its mechanism

图式 3 Danishefsky小组的(±)-merrilactone A路线
Scheme 3 Danishefsky's synthesis of (±)-merrilactone A

图式 4 Danishefsky小组的(-)-merrilactone A不对称合成路线
Scheme 4 Danishefsky's asymmetric synthesis of (-)-merri- lactone A

图式 5 Inoue和Hirama的(±)-merrilactone A合成路线
Scheme 5 Inoue and Hirama's synthesis of (±)-merrilactone A

图式 6 Inoue小组的merrilactone A不对称合成路线
Scheme 6 Inoue's asymmetric synthesis of merrilactone A

图式 9 Frontier小组的(±)-merrilactone A合成路线
Scheme 9 Frontier's synthesis of (±)-merrilactone A

图式 11 Greaney小组的(±)-merrilactone A形式合成及(±)-anislactone A首次合成路线
Scheme 11 Greaney's formal synthesis of (±)-merrilactone A and first synthesis of (±)-anislactone A

图式 12 Greaney组(±)-merrilactone A的形式合成
Scheme 12 Greaney's formal synthesis of (±)-merrilactone A

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:





