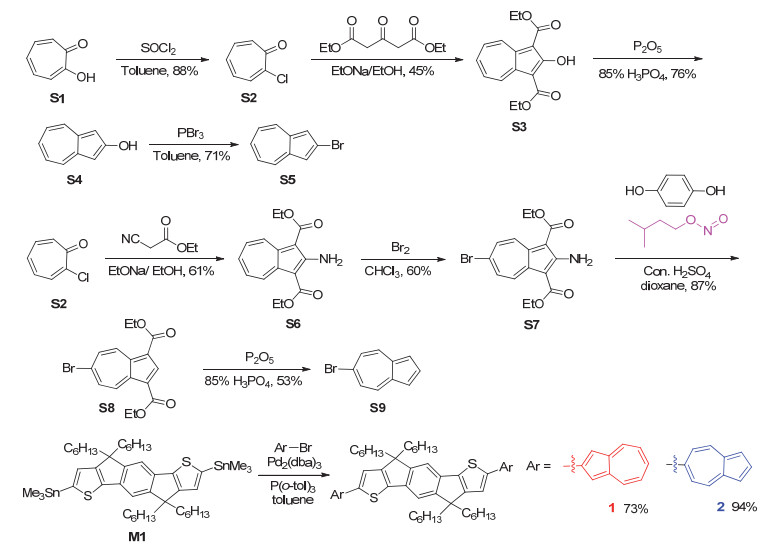
图 1.
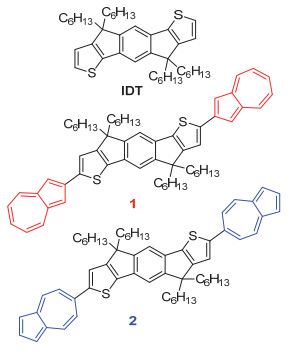
化合物IDT、1和2的化学结构式
Figure 1.
Chemical structures of compounds IDT, 1 and 2

薁, 与萘互为同分异构体, 是一种青蓝色的具有特殊分子结构的非苯芳香化合物.从分子结构来看, 薁是由缺电子的七元环和富电子的五元环骈合而成, 具有较大的分子偶极矩(约1.08 D)[1].与萘相比, 薁具有非镜面对称的分子前线轨道(HOMO/LUMO), 其第一激发单重态的电子排斥能较小, 具有较低的能隙.薁独特的分子结构使其具有不同于其他传统芳香化合物的物理化学性质[2].具有独特物理化学性质的薁类化合物已被应用于非线性光学材料[3]、有机场效应晶体管[4]、单分子器件[5]、有机太阳能电池(OPV)[6]、近红外材料[7]、电致发光材料[8]等领域. 2017年, 本课题组分别将薁的2-位和6-位通过炔键与缺电子的萘二酰亚胺(NDI)相连合成了两个同分异构体, 并将其应用于OFET的研究.两个化合物均表现出n-型半导体的传输性能, 其中通过薁缺电子的七元环与NDI相连的化合物具有更优的器件性能[9].最近, Hu等[10]分别通过薁的2-位和6-位与萘二噻吩酰二亚胺(NDTI)相连, 合成了两个同分异构体化合物NDTI-B2Az和NDTI-B6Az, 其中通过薁的2-位与NDTI相连的化合物NDTI-B2Az, 表现出p-型半导体的传输性能, 而通过薁的6-位与NDTI相连的化合物NDTI- B6Az, 表现出n-型半导体的传输性能.这些研究结果表明, 结构异构可以对相应材料的器件性能和载流子传输类型产生显著影响.
引达省并二噻吩(Indacenodithiophene, IDT)具有刚性的梯形平面结构和较低的HOMO能级, 有利于形成较强的分子间π-π相互作用, 从而获得较高的载流子迁移率[11].根据IDT的结构特点, 可以通过改变取代基、桥连碳原子以及两侧杂环等方式对其进行化学修饰和衍生, 进而调节其物理化学性质以及分子堆积方式等, 从而提高其器件性能[12].鉴于薁五元环的富电子性和七元环的缺电子性, 拟分别通过薁的五元环和七元环与富电子的IDT键连获得具有不同物理化学性质和分子堆积方式的有机半导体分子, 并研究其构效关系.基于此, 我们将薁的五元环(2-位)和七元环(6-位)溴代物分别与2, 7-二(三甲基锡)-4, 4, 9, 9-四己基-引达省并二噻吩进行Stille偶联反应, 得到两个同分异构体化合物1和2 (图 1), 并对其物理化学性质和OFET性能进行研究.
化合物1和2的合成路线如Scheme 1所示.参考文献[9]和[13]的合成方法, 从起始原料S1出发, 通过几步合成得到两个中间体S5和S9. S5和S9分别与2, 7-二(三甲基锡)-4, 4, 9, 9-四己基-引达省并二噻吩(M1)发生Stille偶联反应得到目标化合物1和2, 产率分别为73%和94%.
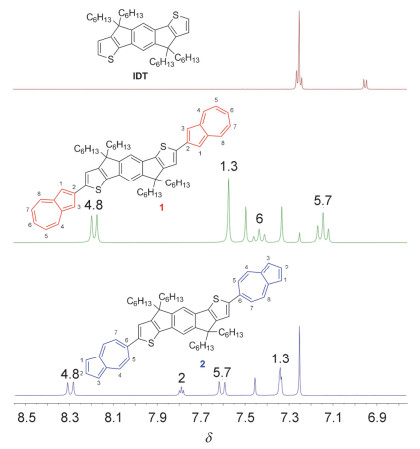
用1H NMR、13C NMR、质谱及元素分析等手段对目标化合物1和2的分子结构进行了表征. 图 2为化合物IDT、1和2的1H NMR谱图.与IDT相比, 化合物1和2化学位移由于薁单元的引入均向低场移动.化合物1中薁单元的化学位移依次为δ 8.29 (4, 8-位), 7.57 (1, 3-位), 7.43 (6-位), 7.16 (5, 7-位); 化合物2中薁单元的化学位移依次为δ 8.30 (4, 8-位), 7.79 (2-位), 7.60 (5, 7-位), 7.34 (1, 3-位).结果表明, 通过薁七元环与IDT键连的化合物2具有更低场的化学位移.室温条件下, 化合物1和2在二氯甲烷、氯仿、甲苯及四氢呋喃等有机溶剂中均具有良好的溶解性, 利于用溶液加工法制备其半导体器件.热重分析(TGA)曲线显示化合物1和2失重5%时的温度分别为416和412 ℃, 表明两个化合物均具有良好的热稳定性.差示扫描热量(DSC)曲线显示化合物2在室温到350 ℃之间没有明显的吸放热峰, 表明化合物2没有发生明显相变.而化合物1在53~90 ℃之间有微小的吸放热峰, 这种微小的固-固相变可能来自烷基链的扰动.
采用Gaussian 09在B3LYP/6-31G(d, p)计算平台对化合物1和2的分子构型进行优化, 并用密度泛函理论(DFT)计算了两个化合物的HOMO/LUMO轨道能级.如图 3所示, 两个化合物中IDT与薁环的二面角分别为0.9°和28.7°, 即化合物1具有更好的平面性.化合物1和2的HOMO轨道电子云主要集中在IDT和薁的五元环上, 而LUMO轨道电子云较均匀地离域在整个骨架上.与IDT母核的前线分子轨道能级(HOMO: -4.98 eV, LUMO: -1.23 eV)相比, 化合物1的HOMO能级升高, 为-4.70 eV; 而2的HOMO能级降低, 为-5.01 eV.与IDT母核相比, 化合物1和2的LUMO能级均有一定程度的降低, 分别为-2.25和-2.35 eV.因此, 通过薁的五元环与IDT相连的化合物1, 具有较好的平面性和较高的HOMO能级, 有利于分子间的电荷传输.

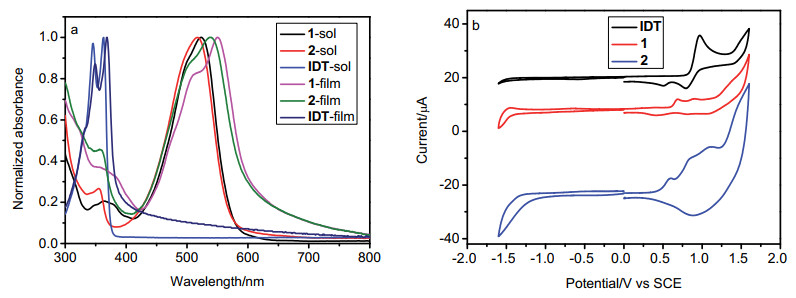
图 4a给出了化合物1、2和IDT在二氯甲烷溶液中和薄膜状态下的紫外-可见吸收光谱. 表 1列出了化合物1、2和IDT的光谱、电化学以及理论计算数据.与溶液中的吸收光谱相比, IDT在薄膜中的吸收光谱未发生明显变化, 其吸收峰在300~400 nm之间; 化合物1和2在二氯甲烷溶液中的末端吸收在400到650 nm之间, 其最大吸收峰分别为525和518 nm; 化合物1和2在薄膜中的吸收峰变宽, 最大吸收峰分别红移至551和538 nm, 分别红移了26和20 nm, 且伴随一个小的肩峰出现, 表明化合物1和2在薄膜状态下呈现J-型分子堆积.与IDT (6.43×10-4 mol•L-1•cm-1)相比, 化合物1和2由于薁单元的引入增加了其共轭程度, 从而增大了其摩尔消光系数ε(分别为1.36×10-5和1.12×10-5 mol• L-1•cm-1), 其中1的ε更高, 这可能是由于其分子中薁单元与IDT骨架间的二面角更小, 导致其共轭效应更强, 从而具有更高的摩尔消光系数.由薄膜的末端吸收计算出化合物1和2的光学带隙Eg分别为2.03和2.06 eV.化合物1相对于2具有更红移的吸收光谱和更窄的光学带隙.
 下载:
导出CSV
下载:
导出CSV
| Compd. | λmax/nm | EHOMOa/eV | ELUMOb/eV | Egc/eV | EHOMOd/eV | EHOMOd/eV | |
| Sol. | Film | ||||||
| IDT | 346, 365 | 349, 369 | -4.71 | -1.52 | 3.19 | -4.98 | -1.23 |
| 1 | 525 | 551 | -4.66 | -2.63 | 2.03 | -4.70 | -2.25 |
| 2 | 518 | 538 | -4.77 | -2.71 | 2.06 | -5.01 | -2.35 |
| a Calculated from EHOMO (eV)=-4.44-Eox1onset (calibration by ferrocene). b Estimated from ELUMO (eV)=EHOMO+Eg. c Estimated from the onset absorption of the thin film. d Estimated from DFT calculation. | |||||||
图 4b为化合物1、2和IDT在二氯甲烷溶液中测得的循环伏安(CV)曲线.三者均在正向呈现不可逆的氧化还原峰, 其起始氧化电位分别为0.22、0.33和0.27 V, 由公式EHOMO (eV)=-4.44-Eoxlonset(经二茂铁校正)计算出化合物1、2和IDT的HOMO能级分别为-4.66, -4.77及-4.71 eV.由HOMO能级结合光学带隙, 由公式ELUMO (eV)=EHOMO+Eg计算出三个化合物的LUMO能级分别为-2.63、-2.71和-1.52 eV.化合物1相对于化合物2具有更高的HOMO能级, 与理论计算结果相一致.
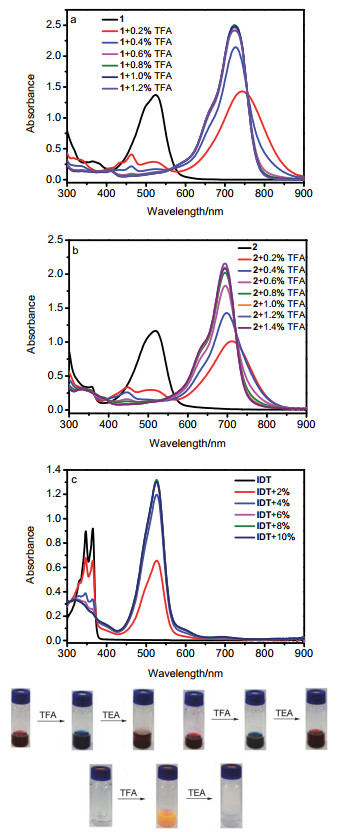
图 5为化合物1、2和IDT在TFA质子化过程中的紫外-可见吸收光谱及其质子响应照片.我们采用紫外-可见吸收光谱对化合物1、2和IDT质子化前后的光谱性质进行了研究.并对其在不同体积比的TFA下质子化的过程进行监测.化合物1和2的吸收光谱表现出相似的行为, 质子化之前的末端吸收峰在400到600 nm之间, 随着TFA的加入, 吸收峰红移至550到850 nm之间, 充分质子化时(TFA体积比约为1%)分别红移了约200 (525→725 nm)和177 nm (518→695 nm).化合物1和2质子化之前在二氯甲烷溶液中的颜色均为红色, 充分质子化后变为蓝色, 加入三乙胺恢复至原来的颜色, 表明两个化合物均具有可逆的质子响应特性.为了排除IDT母核对质子化的影响, 在相同条件下进行了IDT的质子响应研究, 发现IDT也具有可逆的质子响应性质.IDT在质子化之前的末端吸收峰在300到390 nm之间, 最大吸收峰为365 nm, 随着TFA的加入吸收峰红移至425到600 nm之间, 充分质子化时(TFA体积比约为8%)红移了约161 nm (365→526 nm). IDT质子化之前在二氯甲烷溶液中的颜色为无色, 充分质子化后变为橙黄色, 加入三乙胺恢复至无色.因此, 推测化合物1和2的质子响应特性可能是由薁五元环和IDT母核共同作用引起的, 相关机理还在进一步研究中.
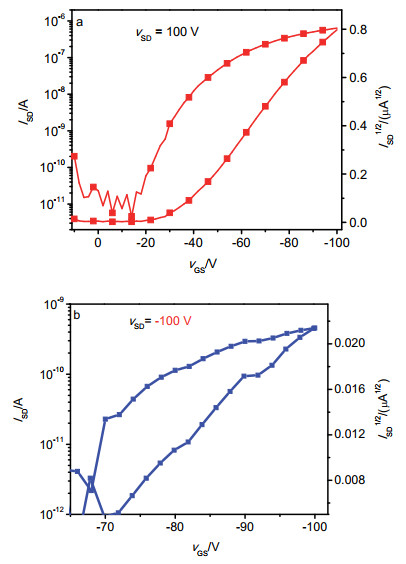
紫外-可见吸收光谱、电化学及理论计算的研究表明, 化合物1和2在物理化学性质上存在一定差异.为了进一步研究结构异构对其电荷传输的影响, 制备了两个化合物的OFET器件, 研究了其OFET性能.将化合物1和2配成溶液(5 mg•mL-1), 旋涂在十八烷基三甲氧基硅烷(OTMS)修饰的SiO2/Si基底上, 然后用掩模板法在薄膜上沉积金源漏电极, 制得底栅顶接触结构的OFET器件, 在N2氛围下用半导体测试仪测得OFET性能. 图 6为化合物1和2的OFET器件的转移曲线, 两个化合物均表现出p-型半导体传输特性. 表 2列出了化合物1和2的OFET器件性能的相关数据.从表中可以看出, 两个化合物均表现出p-型半导体传输性质, 其新制备的未经薄膜退火处理的OFET器件表现出最优性能.基于化合物1的OFET器件的空穴迁移率为4.14×10-3 cm2•V-1•s-1, 开关比为104~105; 基于化合物2的OFET器件的空穴迁移率为1.05×10-5 cm2•V-1•s-1, 开关比为105~106.从测试结果可以看出, 化合物1的空穴迁移率均比化合物2高出约两个数量级.其可能原因是:理论计算和电化学研究表明, 由薁富电子的五元环与IDT相连的化合物1具有更高的HOMO能级, 更有利于空穴的传输.而在我们之前的工作中[8], 分别将薁的2/6-位与缺电子的NDI相连, 两个化合物均表现出n-型半导体性能, 因通过薁的6-位与NDI相连的化合物的LUMO电子云在薁七元环具有较好的离域, 表现出较高的电子迁移率.本文中将薁2/6-位分别与富电子的IDT键连得到化合物1和2, 二者均表现出p-型半导体特性, 其中通过薁2-位连接的化合物1的HOMO电子云具有较好的离域, 而且化合物1相比于化合物2具有更好的分子骨架平面性, 这使得化合物1具有更高的空穴迁移率.因此, 可以通过改变末端薁单元所连接的共轭单元调控相应材料的载流子传输类型.
 下载:
导出CSV
下载:
导出CSV
| Compd. | Ta/℃ | μh(cm2•V-1•s-1) (Max) | VT/V | Ion/Ioff | |
| 1 | As-spun | 4.14×10-3 | -34.66 | 104~105 | |
| 60 | 1.29×10-3 | -52.84 | 104~105 | ||
| 2 | As-spun | 1.05×10-5 | -57.38 | 105~106 | |
| 60 | 9.59×10-6 | -63.88 | 105~106 |
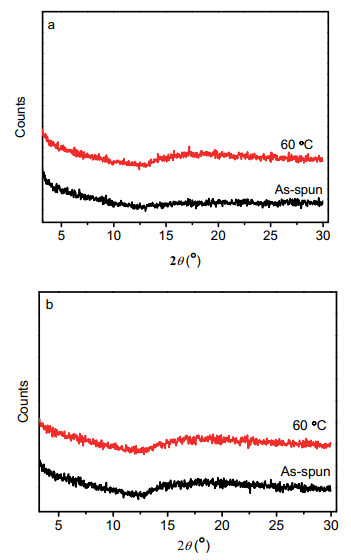
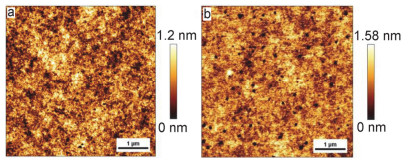
为了研究化合物薄膜的结构形貌和器件性能之间的关系, 采用X射线衍射(XRD)和原子力显微镜(AFM)对两个化合物的薄膜形貌进行了表征. 图 7为化合物1和2的XRD图, 化合物1和2在未经退火处理和60 ℃退火条件下均没有出现明显的衍射峰, 表明其薄膜结晶性较差. 图 8是化合物1和2新制备薄膜的AFM图, 从图中可以看出, 两个化合物的薄膜均具有良好的连续性, 但与化合物1相比, 化合物2的薄膜存在较多的缺陷, 导致大量载流子被捕获, 这可能是造成化合物2的OFET性能比化合物1差的原因之一.
根据薁五元环的富电子性和七元环的缺电子性, 分别用两个2-位薁和6-位薁对引达省并二噻吩(IDT)进行封端, 设计合成了两个基于薁和IDT的同分异构体化合物1和2, 并用TGA、DSC、CV、紫外-可见吸收光谱等手段研究了两个化合物的物理化学性质.化合物1和2在热力学、光谱和电化学等方面存在一定的差异.研究发现两个化合物均具有可逆的质子响应特性.化合物1和2质子化之后的末端吸收峰相对于质子化前分别红移了200和177 nm.两个化合物质子化之前的颜色均为红色, 充分质子化后变为蓝色, 加入三乙胺恢复至原来的红色.对其OFET性能进行了研究, 化合物1和2的OFET器件性能表现出数量级的差异, 其空穴迁移率分别为4.14×10-3和1.05×10-5 cm2• V-1•s-1.研究结果表明, 薁单元与IDT不同的键连方式较大地影响了其构筑的有机功能材料的器件性能和物理化学性质, 这为设计合成基于薁的有机功能材料提供了一种可行的思路.
核磁共振(1H NMR, 13C NMR)由Bruker DRX 400, Varian Mercury 300或JEOL 400核磁共振波谱仪测定, 采用CDCl3或acetone-d6作为溶剂, TMS作为内标.用Gaussian 09在B3LYP/6-311G+(d, p)计算平台上对化合物的分子优化构型和前线轨道能级进行DFT计算.质谱(MS) MALDI-TOF测定在Bruker Biflex Ⅲ MALDI- TOF质谱仪上进行.元素分析由Elementar Vario EL Ⅲ型元素分析仪测得.热重分析(TGA)由TGA Q500型热重分析仪测定, 在N2氛围下, 升温速度10 ℃/min.差热分析(DSC)由DSC Q2000仪器测定, 在N2氛围下升/降温速度为10 ℃/min.紫外-可见光谱(UV-vis)由U-2910或UH4150紫外可见光谱仪测得.电化学测试(CV)在计算机控制的CHI810D电化学分析仪上进行, 采用传统的三电极测试体系, 使用铂电极为工作电极, 饱和甘汞电极作为参比电极, 铂丝作为对电极, 样品溶于新蒸的二氯甲烷(浓度为1×10-3 mol/L), Bu4+NPF6- (0.1 mol/L)作为支持电解质, 扫描速度为100 mV/s.熔点由上海申光SGW X-4显微熔点仪测定.
参考文献[12a].将卓酚酮S1 (1 g, 8.19 mmol)溶于30 mL甲苯中, 然后加入二氯亚砜(1.07 g, 9 mmol), 有白色固体析出, 将反应温度升至110 ℃回流2 h, 白色固体消失, 体系变成深红色, 减压旋除过量的二氯亚砜和甲苯得到棕色固体, 柱层析分离[V(石油醚):V(乙酸乙酯)=5:1]得白色固体1.01 g, 产率88%. m.p. 63~64 ℃ (lit.[12a] m.p. 64~65 ℃); 1H NMR(400 MHz, acetone-d6) δ: 7.90 (d, J=9.3 Hz, 1H), 7.34 (dd, J=12.1, 8.3 Hz, 1H), 7.19 (dd, J=10.6, 8.3 Hz, 1H), 7.14~7.01 (m, 2H); 13C NMR (100 MHz, acetone-d6) δ: 179.23, 148.12, 138.19, 135.97, 135.65, 134.38, 131.51.
参考文献[12b].将金属钠(4 g, 174 mmol)溶解于50 mL无水乙醇, 将1, 3-丙酮二羧酸二乙酯(4.34 g, 21.5 mmol)加入上述溶液中, 溶液变为乳白色.将S2 (2.0 g, 14.0 mmol)溶于6 mL无水乙醇, 快速加入到上述溶液, 体系变成砖红色, 反应体系在室温下搅拌12 h.向体系中加入150 mL水, 有橙色固体析出, 减压过滤, 将所得橙色固体溶于25 mL冰乙酸中, 加入80 mL水稀释, 用50 mL二氯甲烷萃取3次, 合并有机相, 浓缩后柱层析[V(石油醚):V(乙酸乙酯)=5:1]分离得橙黄色固体1.82 g, 产率45%. m.p. 93~94 ℃ (lit.[12b] m.p. 95~96 ℃); 1H NMR (400 MHz, CDCl3) δ: 11.76 (s, 1H), 9.36 (d, J=6.1 Hz, 2H), 7.84~7.52 (m, 3H), 4.51 (q, J=7.0 Hz, 4H), 1.48 (t, J=7.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 172.19, 136.12, 134.65, 132.40, 60.50, 14.55.
参考文献[8]. 24 g五氧化二磷溶解于38 mL 85%磷酸中制得100%磷酸, 将化合物S3 (2.3 g, 8.0 mmol)加入到上述新制的100%磷酸中, 玻璃棒搅拌下升温至130 ℃, 反应20 min.反应液冷却至室温后倒入冰水中, 用30 mL甲苯萃取3次, 合并有机相, 浓缩后柱层析[V(石油醚):V(乙酸乙酯)=10:1]分离得红色固体880 mg, 产率76%. m.p. 110~111 ℃ (lit.[17c] m.p. 115.5~116.5 ℃); 1H NMR (400 MHz, acetone-d6) δ: 9.69 (s, 1H), 8.03 (d, J=10.0 Hz, 2H), 7.36 (t, J=9.8 Hz, 1H), 7.15 (dd, J=10.0 Hz, 9.8 Hz, 2H), 6.76 (s, 2H); 13C NMR (100 MHz, acetone-d6) δ: 167.64, 140.61, 131.49, 130.67, 123.98, 103.19.
参考文献[8].将化合物S4 (144 mg, 1.0 mmol)溶于30 mL甲苯中, 加入三溴化磷(836 mg, 3.0 mmol), 溶液变成棕色, 有绿色固体析出, 搅拌升温至90 ℃反应2 h, 待反应液冷却至室温, 向体系中加入少量水, 用30 mL甲苯萃取3次, 合并有机相, 浓缩后柱层析[V(石油醚):V(乙酸乙酯)=20:1]分离得紫色固体147 mg, 产率71%. m.p. 100~101 ℃ (lit.[17d] m.p. 104.2~104.8 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.25 (d, J=9.6 Hz, 2H), 7.63 (t, J=9.9 Hz, 1H), 7.35 (s, 2H), 7.23 (dd, J=9.6, 9.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 139.84, 137.27, 135.24, 127.48, 124.55, 118.75.
参考文献[12c].将乙醇钠(0.972 g, 14.3 mmol)溶于20 mL无水乙醇, 加入氰基乙酸乙酯(1.78 g, 1.57 mmol)于上述溶液中.将S2 (1.0 g, 7.14 mmol)溶于8 mL无水乙醇中, 冰水浴下缓慢滴加至反应体系中, 并保持体系温度低于5 ℃, 滴加完后在室温下搅拌反应4 h.反应结束后加入60 mL水, 用40 mL氯仿萃取3次, 合并有机相, 浓缩后柱层析[V(石油醚):V(乙酸乙酯)=5:1]分离得到1.23 g橙黄色固体, 产率61%. m.p. 91~92 ℃ (lit.[12b] m.p. 93~94 ℃); 1H NMR (300 MHz, CDCl3) δ: 9.15 (d, J=10.3 Hz, 2H), 7.80 (s, 2H), 7.56 (dd, J=10.3 Hz, 9.4 Hz, 2H), 7.44 (t, J=9.4 Hz, 1H), 4.47 (q, J=7.1 Hz, 4H), 1.48 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.52, 162.41, 146.05, 132.83, 132.58, 131.34, 99.68, 59.81, 14.65.
参考文献[12d].化合物S6 (2.87 g, 10.0 mmol)溶于50 mL氯仿, 冰盐浴下缓慢滴加稀释于10 mL氯仿中的Br2 (1.61 g, 10.1 mmol)到上述溶液中, 保持体系温度低于0 ℃.滴加结束后在冰盐浴下反应20 min, 撤去冰浴, 自然升温至室温, 搅拌反应30 min.反应结束后加入200 mL水, 用150 mL氯仿萃取3次, 合并有机相, 浓缩后柱层析[V(石油醚):V(乙酸乙酯)=5:1]分离得橙黄色固体3.6 g, 产率98%. m.p. 158~159 ℃ (lit.[12c] m.p. 160~161 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.79 (d, J=11.7 Hz, 2H), 7.78 (s, 2H), 7.77 (d, J=11.7 Hz, 2H), 4.44 (q, J=7.1 Hz, 4H), 1.45 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.14, 162.23, 144.14, 135.17, 129.28, 128.27, 100.83, 60.04, 14.59.
参考文献[12c].化合物S7 (2.0 g, 5.46 mmol)溶于300 mL二氧六环中, 加入浓硫酸(535 mg, 5.46 mmol)和对苯二酚(600 mg, 5.45 mmol).另取两个滴液漏斗, 其中一个漏斗中加入溶于200 mL二氧六环的对苯二酚(11.20 g, 102 mmol).另一个漏斗中加入溶于200 mL二氧六环的亚硝酸异戊酯(12.5 g, 107 mmol).然后在室温下同时等速率滴加至反应体系中, 溶液从橙色变成红色.反应4 h后, 加入500 mL 1 mol/L亚硫酸钠水溶液, 用300 mL正己烷萃取3次, 合并有机相, 浓缩后柱层析[V(石油醚):V(二氯甲烷)=1:1]分离得红色固体1.8 g, 产率93%. m.p. 199~200 ℃ (lit.[12c] m.p. 206~207 ℃); 1H NMR (400 MHz, CDCl3) δ: 9.49 (d, J=11.2 Hz, 2H), 8.81 (s, 1H), 8.03 (d, J=11.2 Hz, 2H), 4.42 (q, J=7.1 Hz, 4H), 1.44 (t, J=7.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 164.64, 143.58, 142.20, 138.51, 137.17, 133.73, 117.70, 60.28, 14.51.
参考文献[9].将24 g五氧化二磷溶于38 mL 85%磷酸中制得100%磷酸.将化合物S8 (295 mg, 1.0 mmol)加入至上述新制的100%磷酸中, 搅拌升温至130 ℃反应30 min.待反应液冷却至室温后倒入冰水中, 用30 mL甲苯萃取3次, 合并有机相, 浓缩后柱层析[V(石油醚):V(乙酸乙酯)=20:1]分离得蓝色固体110 mg, 产率53%. m.p. 105~106 ℃ (lit.[12c] m.p. 106.5~108 ℃); 1H NMR (300 MHz, acetone-d6) δ: 8.22 (d, J=10.6 Hz, 2H), 7.95 (t, J=3.8 Hz, 1H), 7.59 (d, J=10.6 Hz, 2H), 7.49 (d, J=3.8 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ: 138.69, 137.61, 134.58, 134.55, 126.18, 120.05.
将2, 7-二(三甲基锡)-4, 4, 9, 9-四己基-引达省并二噻吩(M1) (464 mg, 0.5 mmol), S5 (311 mg, 1.5 mmol), Pd2(dba)3 (10% mmol), P(o-tol)3 (20% mmol)溶于10 mL重蒸甲苯中, 抽换3次N2, 反应液在80 ℃下搅拌12 h, 待反应液冷却至室温, 向反应体系中加入少量水, 用5 mL二氯甲烷萃取3次, 合并有机相, 浓缩后柱层析[V(石油醚):V(二氯甲烷)=20:1]分离得绿色固体325 mg, 产率73%. m.p. 140~143 ℃; 1H NMR (400 MHz, CDCl3) δ: 1H NMR (400 MHz, CDCl3) δ: 8.18 (d, J=9.6 Hz, 2H), 7.57 (s, 2H), 7.49 (s, 1H), 7.43 (t, J=9.8 Hz, 1H), 7.33 (s, 1H), 7.14 (t, J=9.7 Hz, 2H), 2.06 (td, J=12.8, 4.4 Hz, 2H), 1.93 (td, J=12.8, 4.4 Hz, 2H), 1.12 (s, 12H), 1.03~0.83 (m, 4H), 0.77 (t, J=6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 156.81, 153.59, 144.54, 143.65, 142.8, 141.61, 136.18, 135.49, 134.78, 124.41, 120.56, 113.57, 113.36, 54.18, 39.41, 31.72, 29.85, 24.31, 22.70, 14.12; MS (MALDI-TOF) m/z: 855.4 [M+H]+; HRMS (MALDI-TOF) calcd for C60H70S2 854.4919, found 854.49135. Anal. calcd for C60H70S2: C 84.25, H 8.25; found C 84.03, H 8.42.
将2, 7-二(三甲基锡)-4, 4, 9, 9-四己基-引达省并二噻吩M1 (464 mg, 0.5 mmol), S9 (311 mg, 1.5 mmol), Pd2(dba)3 (10% mmol), P(o-tol)3 (20% mmol)溶于10 mL重蒸甲苯中, 抽换N2三次, 反应液在80 ℃下搅拌12 h, 反应液冷却至室温, 向反应体系中加入少量水, 5 mL二氯甲烷萃取3次, 合并有机相, 浓缩后, 柱层析[V(石油醚):V(二氯甲烷)=20:1]分离得绿色固体402 mg, 产率94%. m.p. 140~142 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.30 (d, J=10.4 Hz, 2H), 7.79 (t, J=3.6 Hz, 1H), 7.60 (d, J=10.4 Hz, 2H), 7.45 (s, 1H), 7.33 (d, J=2.3 Hz, 3H), 2.05~1.99 (m, 2H), 1.92~1.86 (m, 2H), 1.17~1.12 (m, 12H), 1.03~0.83 (m, 4H), 0.78 (t, J=6.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 156.92, 153.56, 149.76, 144.20, 143.92, 138.66, 136.38, 136.20, 135.69), 120.85, 120.64, 118.92, 113.61, 77.43, 77.11, 76.79, 54.46, 39.29, 31.70, 29.82, 24.34, 22.70. 14.12; MS (MALDI-TOF) m/z: 855.3 [M+H]+; HRMS (MALDI-TOF) calcd for C60H70S2 854.4919, found 854.49135. Anal. calcd for C60H70 S2: C 84.25, H 8.25; found C 83.98, H 8.32.
辅助材料(Supporting Information) 热重分析曲线、差示扫描热量曲线、核磁谱图和低分辨及高分辨质谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Lemal, D. M.; Goldman, G. D. J. Chem. Educ. 1988, 65, 923. doi: 10.1021/ed065p923
(a) Michl, J.; Thulstrup, E. W. Tetrahedron 1976, 32, 205.
(b) Dong, J.; Zhang, H. Chin. Chem. Lett. 2016, 27, 1097.
(c) Ou, L.; Zhou, Y.; Wu, B.; Zhu, L. Chin. Chem. Lett. 2019, 30, 1903.
(a) Cristian, L.; Sasaki, I.; Lacroix, P. G.; Donnadieu, B.; Asselberghs, I.; Clays, K.; Razus, A. C. Chem. Mater. 2004, 16, 3543.
(b) Migalska-Zalas, A.; El kouari, Y.; Touhtouh, S. Opt. Mater. 2012, 34, 1639.
(e) Wang, X.; Ng, J. K.-P.; Jia, P.; Lin, T.; Cho, C. M.; Xu, J.; Lu, X.; He, C. Macromolecules 2009, 42, 5534.
(a) Yamaguchi, Y.; Maruya, Y.; Katagiri, H.; Nakayama, K.-I.; Ohba, Y. Org. Lett. 2012, 14, 2316.
(b) Yamaguchi, Y.; Ogawa, K.; Nakayama, K.-I.; Ohba, Y.; Katagiri, H. J. Am. Chem. Soc. 2013, 135, 19095.
(c) Yamaguchi, Y.; Takubo, M.; Ogawa, K.; Nakayama, K.-I.; Koganezawa, T.; Katagiri, H. J. Am. Chem. Soc. 2016, 138, 1133.
(d) Xin, H.; Li, J.; Ge, C.; Yang, X.; Xue, T.; Gao, X. Mater. Chem. Front. 2018, 2, 975.
(e) Xin, H.; Ge, C.; Jiao, X.; Yang, X.; Rundel, K.; McNeill, C. R.; Gao, X. Angew. Chem. Int. Ed. 2018, 57, 1322.
(f) Xin, H.; Ge, C.; Gao, H.; Yang, X.; Gao, X. Chem. Sci. 2016, 7, 6701.
(a) Xia, J.; Capozzi, B.; Wei, S.; Strange, M.; Batra, A.; Moreno, J. R.; Amir, R. J.; Amir, E.; Solomon, G. C.; Venkataraman, L. Nano Lett. 2014, 14, 2941.
(b) Schwarz, F.; Koch, M.; Kastlunger, G.; Berke, H.; Stadler, R.; Venkatesan, K.; Lortscher, E. Angew. Chem. Int. Ed. 2016, 55, 11781.
(c) Yang, G.; Sara, S.; Liu, Z.; Li, X.; Hatef, S.; Tan, Z.; Li, R.; Zheng, J.; Dong, X.; Liu, J.; Yang, Y.; Shi, J.; Xiao, Z; Zhang, G.; Colin, L.; Hong, W.; Zhang, D. Chem. Sci. 2017, 8, 7505.
(d) Cai, S.; Deng, W.; Huang, F.; Chen, L.; Tang, C.; He, W.; Long, S.; Li, R.; Tan, Z.; Liu, J.; Xiao, Z.; Zhang, D.; Hong, W. Angew. Chem. Int. Ed. 2019, 58, 1.
(e) Huang, C.; Jevric, M.; Borges, A.; Olsen, S. T.; Hamill, J. M.; Zheng, J.-T.; Yang, Y.; Rudnev, A.; Baghernejad, M.; Broekmann, P.; Petersen, A. U.; Wandlowski, T.; Mikkelsen, K. V.; Solomon, G. C.; Brøndsted Nielsen, M.; Hong, W. Nat. Commun. 2017, 8, 15436.
Puodziukynaite, E.; Wang, H. W.; Lawrence, J.; Wise, A. J.; Rus-sell, T. P.; Barnes, M. D.; Emrick, T. J. Am. Chem. Soc. 2014, 136, 11043. doi: 10.1021/ja504670k
Zhou, Y.; Zhuang, Y.; Li, X.; Hans, A.; Yu, L.; Ding, J.; Zhu, L. Chem. Eur. J. 2017, 23, 7642. doi: 10.1002/chem.201700947
(a) Wang, X.; Ng, J. K.-P.; Jia, P.; Lin, T.; Cho, C. M.; Xu, J.; Lu, X.; He, C. Macromolecules 2009, 42, 5534.
(b) Tsurui, K.; Murai, M.; Ku, S.-Y.; Hawker, C. J.; Robb, M. J. Adv. Funct. Mater. 2014, 24, 7338.
辛涵申, 葛从武, 傅丽娜, 杨笑迪, 高希珂, 有机化学, 2017, 37, 711. doi: 10.6023/cjoc201609029Xin, H.; Ge, C.; Fu, L.; Yang, X.; Gao, X. Chin. J. Org. Chem. 2017, 37, 711(in Chinese). doi: 10.6023/cjoc201609029
Ran, H.; Duan, X.; Zheng, R.; Xie, F.; Chen, L.; Zhao, Z.; Han, R.; Lei, Z.; Hu, J. ACS Appl. Mater. Interfaces 2020, 12, 23225. doi: 10.1021/acsami.0c04552
翟文超, 周二军, 有机化学, 2016, 36, 276. doi: 10.6023/cjoc201604052Zhai, W..; Zhou, E. Chin. J. Org. Chem. 2016, 36, 2786(in Chinese). doi: 10.6023/cjoc201604052
(a) Lin, Y.; He, Q.; Zhao, F.; Huo, L.; Mai, J.; Lu, X.; Su, C.; Li, T.; Wang, Y.; Zhu, J.; Sun, Y.; Wang, C.; Zhan, X. J. Am. Chem. Soc. 2016, 138, 2973.
(b) Cai, L. P.; Moehl, T.; Moon, S. J.; Decoppet, J. D.; Robin, H. B.; Xue, Z. S.; Liu, B.; Zakeeruddin, S. M.; Grätzel, M. Org. Lett. 2014, 16, 106.
(a) Nozoe, T.; Takase, K.; Shimazaki, N. Bull. Chem. Soc. Jpn. 1964, 37, 1644.
(b) Nozoe, T.; Seto, S.; Matsumura, S.; Murase, Y. Bull. Chem. Soc. Jpn. 1962, 35, 1179.
(c) McDonald, R. N.; Richmond, J. M.; Curtis, J. R.; Petty, H. E.; Hoskins, T. L. J. Org. Chem. 1976, 41, 1811.
(d) Gao, H.; Yang, X.; Xin, H.; Gao, T.; Gong, H.; Gao, X. Chin. J. Org. Chem. 2018, 38, 2680(in Chinese).
(高洪磊, 杨笑迪, 辛涵申, 高铁阵, 龚和贵, 高希珂, 有机化学, 2018, 38, 2680.)
(e) Zhang, J.; Petoud, S. Chem. Eur. J. 2008, 14, 1264.
(f) Huang, H.; Du, Z.; Huang, C.; Zhang, J.; Liu, S.; Fu. N.; Zhao, B.; Yang, R.; Huang, W. Polymer 2019, 162, 11.
(g) Bai, H.; Wang, Y.; Cheng, P.; Li, Y.; Zhu, D.; Zhan, X. ACS Appl. Mater. Interfaces 2014, 6, 8426.

图 3 化合物IDT、1和2的前线分子轨道能级及电子云分布情况
Figure 3 Frontier molecular orbital energy levels and electron cloud distribution of compounds IDT, 1 and 2

图 4 (a) 化合物1、2和IDT在二氯甲烷溶液及薄膜中的紫外-可见吸收光谱; (b)化合物1、2和IDT在二氯甲烷(支持电解质: 0.1 mol/L Bu4+NPF6-, 参比电极:饱和甘汞电极, 扫描速率: 100 mV/s)中测得的循环伏安曲线
Figure 4 (a) UV-vis absorption spectra of compounds 1, 2 and IDT in dichloromethane and in thin film; (b) cyclic voltammograms of compounds 1, 2 and IDT in dichloromethane (supporting electrolyte: 0.1 mol/L Bu4+NPF6-, reference electrode: SCE, scan rate: 100 mV/s)

图 5 化合物1和2及IDT在TFA质子化过程中的紫外-可见吸收光谱变化及其可逆的质子响应照片
Figure 5 UV-Vis absorption spectrum and reversible proton- response photos of compounds 1, 2 and IDT during TFA protonation

图 6 基于化合物1和2的OFET器件转移曲线
Figure 6 Transfer curves of OFET device based on compounds 1 and 2

图 7 化合物1 (a)和2 (b)的薄膜新制备和60 ℃退火条件下的XRD图
Figure 7 XRD patterns of thin films of compounds 1 (a) and 2 (b) as-spun and annealed at 60 ℃

图 8 化合物1 (a)和2 (b)薄膜未经退火处理的AFM图
Figure 8 AFM images of thin films of compounds 1 (a) and 2 (b) without annealing treatment
表 1 化合物1、2和IDT的光谱、电化学和理论计算数据
Table 1. Optical, electrochemical and DFT calculation data for compounds 1, 2 and IDT
| Compd. | λmax/nm | EHOMOa/eV | ELUMOb/eV | Egc/eV | EHOMOd/eV | EHOMOd/eV | |
| Sol. | Film | ||||||
| IDT | 346, 365 | 349, 369 | -4.71 | -1.52 | 3.19 | -4.98 | -1.23 |
| 1 | 525 | 551 | -4.66 | -2.63 | 2.03 | -4.70 | -2.25 |
| 2 | 518 | 538 | -4.77 | -2.71 | 2.06 | -5.01 | -2.35 |
| a Calculated from EHOMO (eV)=-4.44-Eox1onset (calibration by ferrocene). b Estimated from ELUMO (eV)=EHOMO+Eg. c Estimated from the onset absorption of the thin film. d Estimated from DFT calculation. | |||||||
 下载: 导出CSV
下载: 导出CSV
表 2 化合物1和2的OFET器件性能
Table 2. Performance of OFET devices based on compounds 1 and 2
| Compd. | Ta/℃ | μh(cm2•V-1•s-1) (Max) | VT/V | Ion/Ioff | |
| 1 | As-spun | 4.14×10-3 | -34.66 | 104~105 | |
| 60 | 1.29×10-3 | -52.84 | 104~105 | ||
| 2 | As-spun | 1.05×10-5 | -57.38 | 105~106 | |
| 60 | 9.59×10-6 | -63.88 | 105~106 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们