图 1.
苯丙素苷的代表性结构
Figure 1.
Structures of typical phenylpropanoid glycosides

苯丙素苷化合物(Phenylpropanoid Glycosides, 简称PPGs)是一类在自然界中广泛存在的糖苷化合物, 尤其在玄参科和唇形科植物中最为常见[1]. PPGs的核心结构通常是2-苯乙基-β-D-吡喃葡萄糖苷, 其2, 4位或6位羟基通过酯键连接反式的取代肉桂酰基, 2, 3, 4或6位羟基再通过糖苷键连接L-鼠李糖(Rha)、D-葡萄糖(Glc)和D-木糖(Xyl)等(图 1)[2].因其苷元为取代的苯乙基, 亦被称为苯乙醇苷类化合物(Phenylethanoid glycosides, PhEGs).由于取代单糖的种类和连接位置的不同, 以及肉桂酰基和苯乙基上取代基团的变化, 该类化合物呈现丰富的结构多样性.
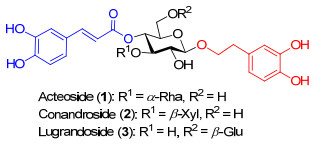
毛蕊花苷(Acteoside), 作为PPGs家族中的明星分子, 最早由Scarpati等[2a]在1963年从毛蕊花属植物Verbascum sinuatum中提取得到并命名为“Verbasco- side”.直到1968年, Birkofer等[3]才确定了该分子的结构, 并命名为“Acteoside”(图 1).迄今, Acteoside已经被证实是多种中药(例如肉苁蓉、地黄、大叶紫珠等)的重要有效成分之一, 在抗氧化[4]、抗炎杀菌[5]、抗肿瘤[6]、抗病毒[7]和保肝护肝[8]等方面具有广泛的生物活性.但是, 其在植物中的含量低微(0.02%~0.4%), 因此, 如何通过化学方法高效合成Acteoside受到科学家的关注[9].
苯丙素苷类分子中存在α, β-不饱和酯基, 限制了糖苷合成中常用保护基的使用, 导致目前对该类天然产物的化学合成路线存在保护基操作繁琐、步骤较多等缺陷[10-16]. 1999年, Kawada课题组[10]和van Boom课题组[11]先后报道了Acteoside的全合成.前者在合成中使用了苄基保护酚羟基, 其脱除条件易破坏咖啡酸结构, 因此, 总产率较低(最长线性11步, 总收率3.5%); 而后者虽然选取了较易脱除的保护基, 但是经历了较多保护基的转换操作, 合成路线较长(最长线性16步, 总收率7.1%).为了减少保护基操作, 2017年, Judeh小组[16b]使用2-氨基乙基二苯基硼酸酯活化多羟基受体, 与卤苷给体进行区域选择性糖苷化生成相应原酸酯, 乙酰基保护2, 4位羟基后, 在三氟甲磺酸三甲基硅酯(TMSOTf)促进下转化为1→3连接的二糖, 并完成了苯丙素苷Osmanthuside-B6的合成.我们拟利用葡萄糖上各个羟基的反应活性的差别[17], 应用我们实验室发展的一价金催化的糖苷化反应[18], 尝试区域选择性的糖苷化来合成关键的α-(1→3)连接二糖7-1, 以完成Acteoside和其它苯丙素苷类分子的高效合成.
按照文献方法制备得到鼠李糖邻炔基苯甲酸酯给体5[19]和6位羟基乙酰化的葡萄糖硫苷受体6[20].随后, 我们尝试在三苯基膦金(I)双(三氟甲磺酰基)亚胺盐(Ph3PAuNTf2)催化下, 实现糖基炔酯给体5对2, 3, 4-三羟基裸露的受体6的区域选择性糖苷化反应(表 1).考虑到糖环上不同位置羟基的反应活性差异一般在低温下较明显, 反应首先选择在-70 ℃下进行(Entry 1), 以32%的收率得到了期望的α-(1→3)糖苷化产物7-1, 其主要副产物为α-(1→4)糖苷化二糖产物7-2, 收率为5%, 说明该反应具有较好的区域选择性.产率较低的原因是反应不完全, 有较多的给体剩余.为了提高给体的转化率, 我们升高反应温度至-30 ℃, 薄层色谱(TLC)监测发现有新的副产物产生, 而在糖苷化反应基本结束后将反应体系升至室温搅拌, 该新的副产物大部分消失, 因此, 怀疑上述副产物是原酸酯化合物, 升至室温后转化为正常糖苷化产物或水解产物, 目标产物7-1的产率为46% (Entry 2).考虑到原酸酯一般可以在路易斯酸催化下转化为正常的糖苷化产物[21], 分别在糖苷化反应开始和结束后加入0.1 equiv.的TMSOTf, 考察催化量的路易斯酸在反应的不同时期促进上述副产物向二糖7-1的转化情况.我们发现在反应开始即加入TMSOTf并不能在反应过程中有效促进原酸酯的转化(Entry 4);而在给体5基本消耗完全后升至室温再加入TMSOTf可以有效促进原酸酯向7-1的转化(Entry 5).最后, 以61%的收率得到了所需的α-(1→3)糖苷化产物7-1.
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | Temp./℃ | TMSOTf/equiv. | Yieldd/% | |
| 7-1 | 7-2 | |||
| 1 | -70 | 32% | 5% | |
| 2 | -30~r.t. | 46% | Trace | |
| 3 | -30~r.t. | 0.1b | 48% | Trace |
| 4 | -30~r.t. | 0.1c | 61% | Trace |
| a Reaction conditions: 5 (0.1 mmol), 6 (0.1 mmol), 4 Å MS (250 mg), CH2Cl2 (5 mL), 20 h; b TMSOTf was added at the beginning of the reaction; c TMSOTf was added after consumption of the donor; d Isolated yields. | ||||
在顺利得到α-(1→3)糖苷化产物7-1后, 尝试在葡萄糖单元的2位选择性引入保护基(表 2).考虑到目标分子中含有咖啡酸这个α, β-不饱和酯基结构, 尝试使用可以正交脱除的硅基进行保护.但是, 在实验中发现7-1中葡萄糖2位羟基位阻较大, 即便使用叔丁基二甲硅基三氟甲磺酸酯(TBSOTf)或TMSOTf在碱性条件下也难以对该羟基进行硅基保护(Entries 1~4).而且还发现氯乙酰基也同样难以引入, 并且对2, 4位羟基的选择性较差.经过初步筛选反应条件, 仅以约30%的收率得到期望的产物8 (Entries 5~7).综合考虑后, 选择体积较小的乙酰基进行选择性保护, 在上述选择性酰化条件筛选的基础上, 使用乙酰氯在4-二甲氨基吡啶(DMAP)促进下以最高51%的收率得到葡萄糖2位乙酰基保护的二糖硫苷9.另外, 也尝试了直接使用咖啡酸与7-1上的4位羟基进行选择性酯化.但是, 发现使用1-乙基-3(3-二甲基丙胺)碳二亚胺(EDCI)/二环己基碳二亚胺(DCC)等缩合剂均无法很好地区分葡萄糖的2, 4位羟基.
下载:
导出CSV
 |
|||
| Entry | Reagent | Base | Product |
| 1 | TBSCl | Imidazole | NR |
| 2 | TBSOTf | 2, 6-Lutidine | Trace |
| 3 | TBSOTf | NEt3 | Trace |
| 4 | TMSOTf | 2, 6-Lutidine | Trace |
| 5 | ClCOCH2Cl | NEt3 | 8 (23%)b |
| 6 | ClCOCH2Cl | Pyridine | Trace |
| 7 | ClCOCH2Cl | DMAP | 8 (35%)b (26%~32%) |
| 8 | ClCOCH3 | DMAP | 9 (51%)b |
| a 7-1 (0.1 mmol), base (0.13~0.2 mmol), 0 ℃~r.t., 15 h; b Isolated yields. | |||
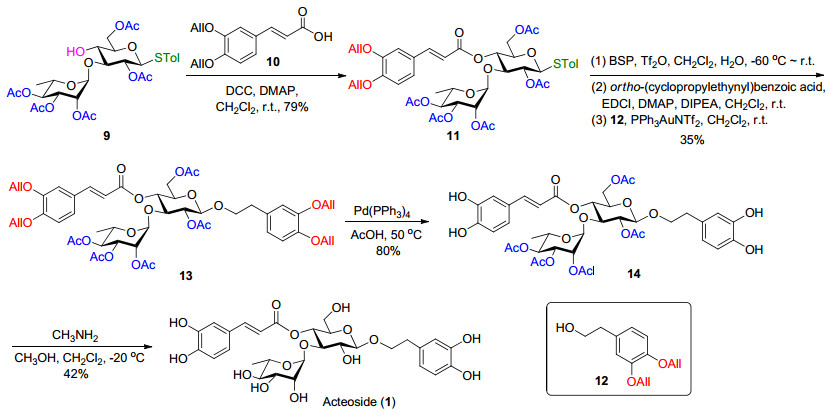
接下去, 按照文献方法制备了烯丙基保护的咖啡酸片段10[22], 并尝试其与二糖9中的葡萄糖4位羟基在缩合条件下酯化.首先使用常用的EDCI作为缩合剂, 发现该反应收率较低, 有较多的杂质生成, 可能是由于乙酰基的迁移所造成.改用DCC作缩合剂, 可有效抑制该类副产物的生成, 以79%的收率得到二糖11.随后, 制备了烯丙基保护的苯乙醇片段12[23], 并尝试其与硫苷11的糖苷化反应.使用常规的N-碘代丁二酰亚胺(NIS)/路易斯酸活化条件时, 在低温(-30~-20 ℃)下, 该二糖硫苷不易活化; 而升高反应温度, 则导致硫苷的降解.使用1-(苯基磺酰基)哌啶(BSP)/三氟甲磺酸酐(Tf2O)[24]作为活化剂时, 给体水解严重, 糖苷化的产率不理想.因此, 尝试将硫苷转变为相应的邻炔基苯甲酸酯给体.尝试使用N-溴代丁二酰亚胺(NBS)或NIS水解硫苷, 却发现化合物11基本不反应; 额外加入三氟乙酸则导致反应混乱.最终, 使用BSP/Tf2O反应体系成功水解了二糖硫苷11; 随后, 将其转化成邻炔基苯甲酸酯给体, 并直接与苯乙醇12进行金催化糖苷化反应, 顺利得到化合物13 (Scheme 1).
最后, 只需要进行烯丙基和乙酰基这两种保护基的脱除便能得到目标产物Acteoside.考虑到脱除烯丙基所使用的钯试剂和三苯基膦配体可能不易与终产物分离, 选择先烯丙基后乙酰基的脱除顺序.在实际操作中, 化合物13在四三苯基膦钯[(PPh3)4Pd]的作用下, 以80%的收率顺利脱除四个烯丙基得到化合物14.最后, 以甲醇和二氯甲烷的混合溶剂, 化合物14在甲胺条件下脱除所有乙酰基[25], 以42%的收率得到目标产物Acteoside (Scheme 1).乙酰基脱除这一步反应产率并不理想, 主要是因为该条件导致部分咖啡酸支链断裂; 除此之外, 文献报道类似的碱性条件可能导致4→6位酰基迁移, 收率也不理想[26].
发展了一条合成毛蕊花苷(Acteoside)的新路线, 以已知化合物5和6为起始原料, 最长线性步骤为8步, 总产率为3%.其中, 关键步骤是PPh3AuNTf2催化下的全乙酰化鼠李糖邻炔基苯甲酸酯给体5与2, 3, 4-三羟基葡萄糖硫苷6的区域选择性糖苷化反应, 以较好的收率得到α-(1→3)二糖7-1.该合成路线减少了保护基操作, 缩短了合成步数, 并可以用于其它苯丙素苷类化合物的合成.
核磁共振数据由BrukerAV-400和Agilent-500型核磁共振仪测定(以TMS作为内标); 高分辨质谱由Agilent 6224 TOF LC/MS和Agilent TOF LC/MS 1260- 6230型仪器测定; 比旋光度由Anton Paar MCP 5500旋光仪测定, 光源为钠光灯, 589 nm; 薄层色谱监测采用质量分数为10%的浓硫酸乙醇加热显色或是UV254 nm显色.未特别注明的商业化试剂并未进行进一步纯化.常用的溶剂使用微波炉活化的4 Å分子筛进行干燥.
将鼠李糖给体5 (46 mg, 0.1 mmol)和硫苷6 (33 mg, 0.1 mmol)溶于干燥的二氯甲烷(4 mL)中, 加入活化好的4 Å MS (250 mg), 氩气保护下室温搅拌0.5 h.反应体系降温至-30 ℃, 通过注射器逐滴加入PPh3AuNTf2的二氯甲烷溶液(0.01 mol/L, 1 mL).在-30 ℃下反应20 h, 加入TMSOTf (1.8 µL, 0.01 mmol).体系升温至室温反应3 h, 加入Et3N淬灭反应, 反应液经硅藻土过滤并浓缩, 柱层析[V(石油醚):V(乙酸乙酯)=4:1至2:1)得白色泡沫状固体7-1 (37 mg, 产率61%), 并分离得到少量的7-2 (<5%).
7-1:
7-2:
将化合物7-1 (300 mg, 0.50 mmol)和DMAP (73 mg, 0.60 mmol)溶于干燥的二氯甲烷(5 mL)中, 氩气保护, 0 ℃下逐滴加入氯乙酰氯(186 μL, 2.50 mmol), 保持0 ℃反应1 h后升至室温反应10 h, 乙酸乙酯稀释, 反应液用饱和食盐水洗, 有机相用无水Na2SO4干燥, 过滤, 浓缩, 柱层析[V(石油醚):V(乙酸乙酯)=6:1至4:1]得白色泡沫状固体8 (120 mg, 产率35%):
以化合物7-1 (330 mg, 0.55 mmol)为原料, 按照化合物8的合成方法制备得到白色化合物9 (160 mg, 产率48%):
将化合物9 (180 mg, 0.28 mmol), 咖啡酸片段10 (146 mg, 0.56 mmol), DMAP (51 mg, 0.42 mmol)和DCC (145 mg, 0.70 mmol)溶于干燥的二氯甲烷(5 mL)中, 室温反应至原料反应完全. 反应液垫硅藻土过滤, 有机相用饱和食盐水洗, 无水Na2SO4干燥, 过滤, 浓缩, 柱层析[V(石油醚):V(丙酮)=6:1至4:1]得白色固体11 (194 mg, 产率79%):
将化合物11 (210 mg, 0.24 mmol)和BSP (63 mg, 0.3 mmol)溶于二氯甲烷(10 mL)中.将反应体系冷却至-60 ℃, 通过注射器缓慢加入三氟甲磺酸酐(64 μL, 0.36 mmol). 10 min后, 滴加20 μL水.保持-60 ℃反应20 min后, Et3N淬灭反应.体系升至室温, 反应液用饱和食盐水溶液洗, 乙酸乙酯萃取两次, 合并有机相, 无水Na2SO4干燥, 浓缩, 快速柱层析得淡黄色糖浆.
将上述粗品, 邻-环丙乙炔基苯甲酸(56 mg, 0.28 mmol), EDCI (54 mg, 0.28 mmol)和DMAP (34 mg, 0.28 mmol)溶于干燥的二氯甲烷(4 mL)中, 氩气保护, 通过注射器滴加N, N-二异丙基乙胺(DIPEA) (50 μL, 0.28 mmol), 室温反应至原料基本消耗完全.乙酸乙酯稀释, 饱和食盐水洗, 乙酸乙酯萃取两次, 合并有机相, 无水Na2SO4干燥, 浓缩, 快速柱层析得浅黄色泡沫状固体.
将上述给体粗品和化合物12 (112 mg, 0.48 mmol)溶于干燥的二氯甲烷(4 mL)中, 加入活化好的4 Å MS (300 mg), 氩气保护下室温搅拌0.5 h.通过注射器加入PPh3AuNTf2的二氯甲烷溶液(0.05 mol/L, 1 mL), 室温下反应至给体消耗完全.加入Et3N淬灭反应, 反应液垫硅藻土过滤并浓缩, 柱层析[V(石油醚):V(乙酸乙酯)=4:1至3:1]得白色泡沫状固体13 (79 mg, 三步产率35%):
将化合物13 (104 mg, 0.10 mmol)溶于脱气乙酸(5 mL)中, 氩气保护下加入Pd(PPh3)4 (121 mg, 0.10 mmol), 体系升温至50 ℃, 反应至原料消耗完全(约2 h, TLC监测).反应液浓缩, 柱层析[V(二氯甲烷):V(甲醇)=60:1至30:1]得浅黄色固体14 (70 mg, 产率80%):
将化合物14 (58 mg, 0.07 mmol)溶于二氯甲烷(2 mL)中, -30 ℃下缓慢滴加质量分数为30%~33%的甲胺甲醇溶液(3 mL).体系维持在-25 ℃至-20 ℃反应10 h, 迅速浓缩除去甲胺. C18反相柱层析[V(水):V(甲醇)=4:1至2:1]得棕黄色固体Acteoside (1) (18 mg, 产率42%):
辅助材料(Supporting Information) 化合物7-1, 7-2, 8, 9, 11, 13, 14和Acteoside (1)的1H NMR, 13C NMR和HRMS谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)下载.
(a) Molgaard, P.; Ravn, H. Phytochemistry 1988, 27, 2411.
(b) Park, H. J.; Jung, W. T.; Basnet, P.; Kadota, S.; Namba, T. J. Nat. Prod. 1996, 59, 1128.
(a) Scarpati, M. L.; Monache, D. Ann. Chim. (Rome, Italy) 1963, 53, 356.
(b) Nonaka, G.; Nishioka, I. Phytochemistry 1977, 16, 1265.
(c) Baudouin, G.; Skaltsounis, A.L.; Tillequin, F.; Koch, M. Planta Med. 1988, 54, 321.
Birkofer, L.; Kaiser, C.; Thomas, U. Z. Naturforscher 1968, 23b, 1051.
Leporini, L.; Menghini, L.; Foddai, M.; Petretto, G. L.; Chessa, M.; Tirillini, B.; Pintore, G. Nat. Prod. Res. 2015, 29, 899. doi: 10.1080/14786419.2014.955490
Rao, K. Y.; Lien, H.-M.; Lin, Y.-H.; Hsu, Y.-M.; Yeh, C.-T.; Chen, C.-C.; Lai, C.-H.; Tzeng, Y.-M. Food Chem. 2012, 132, 780. doi: 10.1016/j.foodchem.2011.11.037
(a) Inoue, M.; Sakuma, Z.; Ogihara, Y.; Saracoglu, I. Biol. Pharm. Bull. 1998, 21, 81.
(b) Li, J.; Zheng, Y.; Zhou, H.; Su, B.; Zheng, R. Planta Med. 1997, 63, 499.
Kernan, M. R.; Amarquaye, A.; Chen, J. L.; Chan, J.; Sesin, D. F.; Parkinson, N.; Ye, Z.; Barrett, M.; Bales, C.; Stoddart, C. A.; Sloan, B.; Blanc, P.; Limbach, C.; Mrisho, S.; Rozhon, E. J. J. Nat. Prod. 1998, 61, 564. doi: 10.1021/np9703914
Morikawa, T.; Pan, Y.; Ninomiya, K.; Imura, K.; Matsuda, H.; Yoshikawa, M.; D. Yuan D.; Muraoka, O. Bioorg. Med. Chem. 2010, 18, 1882. doi: 10.1016/j.bmc.2010.01.047
He, J.; Hu, X.-P.; Zeng, Y.; Li, Y.; Wu, H.-Q.; Qiu, R.-Z.; Ma, W.-J.; Li, T.; Li, C.-Y.; He, Z.-D. J. Asian Nat. Prod. Res. 2011, 13, 449. doi: 10.1080/10286020.2011.568940
Kawada, T.; Asano, R.; Hayashida, S.; Sakuno, T. J. Org. Chem. 1999, 64, 9268. doi: 10.1021/jo9906983
Duynstee, H. I.; de Koning, M. C.; Ovaa, H.; van der Marel, G. A.; van Boom, J. H. Eur. J. Org. Chem. 1999, 10, 2623.
(a) Zhang, S.-Q.; Li, Z.-J.; Wang, A.-B.; Cai, M.-S.; Feng, R. Carbohydr. Res. 1997, 299, 281.
(b) Zhang, S.-Q.; Li, Z.-J.; Wang, A.-B.; Cai, M.-S.; Feng, R. Carbohydr. Res. 1998, 308, 281.
(c) Li, Q.; Li, S.-C.; Li, H.; Cai, M.-S.; Li, Z.-J. Carbohydr. Res. 2005, 340, 1601.
Das, S. K.; Reddy, K. A.; Mukkanti, K. Carbohydr. Res. 2007, 342, 2309. doi: 10.1016/j.carres.2007.06.022
Liu, Y.-G.; Li, X.; Xiong, D.-C.; Yu, B.; Pu, X.; Ye, X.-S. Eur. J. Med. Chem. 2015, 95, 313. doi: 10.1016/j.ejmech.2015.03.038
(a) Shu, P.; Xiao, X.; Zhao, Y.; Xu, Y.; Yao, W.; Tao, J.; Wang, H.; Yao, G.; Lu, Z.; Zeng, J.; Wan, Q. Angew. Chem., Int. Ed. 2015, 54, 14432.
(b) Zhao, Y.; Zeng, J.; Liu, Y.; Xiao, X.; Sun, G.; Sun, J.; Shu, P.; Fu, D.; Meng, L.; Wan, Q. J. Carbohydr. Chem. 2018, 37, 471.
(a) Khong, D. T.; Judeh, Z. M. A. Carbohydr. Res. 2016, 436, 50.
(b) Khong, D. T.; Judeh, Z. Org. Biomol. Chem. 2017, 15, 2638.
(c) Khong, D. T.; Judeh, Z. M. A. Tetrahedron Lett. 2017, 58, 109.
Yu, B.; Li, B.; Xing, G.; Hui, Y. J. Comb. Chem. 2001, 3, 404. doi: 10.1021/cc010014g
(a) Yu, B. Acc. Chem. Res. 2018, 51, 507.
(b) Zhu, D.; Yu, B. Chin. J. Chem. 2018, 36, 681.
(c) Ehianeta, T. S.; Shen, D.; Xu, P.; Yu, B. Chin. J. Chem. 2019, 37, 827.
(d) Shao, W. B.; An, Q. L.; Cao, X.; Yu, B. Acta Chim. Sinica 2019, 77, 999(in Chinese).
(邵文博, 安泉林, 曹鑫, 俞飚, 化学学报, 2019, 77, 999.)
(e) Shen, R. Z.; Cao, X.; Yu, B. Acta Chim. Sinica 2018, 76, 278(in Chinese).
(沈仁增, 曹鑫, 俞飚, 化学学报, 2018, 76, 278.)
(a) Li, Y.; Yang, Y.; Yu, B. Tetrahedron Lett. 2008, 49, 3604.
(b) Yoshimura, F.; Itoh, R.; Torizuka, M.; Mori, G.; Tanino, K. Angew. Chem., Int. Ed. 2018, 57, 17161.
Liang, P.-H.; Lu, Y.-J.; Tang, T.-H. Tetrahedron Lett. 2010, 51, 6928. doi: 10.1016/j.tetlet.2010.10.135
Kong, F. Carbohydr. Res. 2007, 342, 345. doi: 10.1016/j.carres.2006.09.025
Rakesh, J.; Dickman, M. H.; Kuhnert, N. Org. Biomol. Chem. 2012, 10, 5266. doi: 10.1039/c2ob25124h
Hu, Z. F.; Silipo, A.; Li, W.; Molinaro, A.; Yu, B. J. Org. Chem. 2019, 84, 13733. doi: 10.1021/acs.joc.9b01956
(a) Crich, D.; Smith, M. J. Am. Chem. Soc. 2002, 124, 8867.
(b) Codée, J. D. C.; Litjens, R. E. J. N.; den Heeten, R.; Overkleeft, H. S.; van Boom, J. H.; van der Marel, G. A. Org. Lett. 2003, 5, 1519.
Xu, J.; Liu, Y.; Dupouy, C.; Chattopadhyaya, J. J. Org. Chem. 2009, 74, 6534. doi: 10.1021/jo901009w
Kawada, T.; Asano, R.; Makino, K.; Sakuno, T. J. Wood. Sci. 2002, 48, 512. doi: 10.1007/BF00766648
表 1 受体6与给体5的区域选择性糖苷化a
Table 1. Optimization of the regioselective glycosylation of acceptor 6 with donor 5
|
||||
| Entry | Temp./℃ | TMSOTf/equiv. | Yieldd/% | |
| 7-1 | 7-2 | |||
| 1 | -70 | 32% | 5% | |
| 2 | -30~r.t. | 46% | Trace | |
| 3 | -30~r.t. | 0.1b | 48% | Trace |
| 4 | -30~r.t. | 0.1c | 61% | Trace |
| a Reaction conditions: 5 (0.1 mmol), 6 (0.1 mmol), 4 Å MS (250 mg), CH2Cl2 (5 mL), 20 h; b TMSOTf was added at the beginning of the reaction; c TMSOTf was added after consumption of the donor; d Isolated yields. | ||||
 下载: 导出CSV
下载: 导出CSV
表 2 二糖7-1中2位羟基的选择性保护a
Table 2. Selective protection of the 2-OH in 7-1
|
|||
| Entry | Reagent | Base | Product |
| 1 | TBSCl | Imidazole | NR |
| 2 | TBSOTf | 2, 6-Lutidine | Trace |
| 3 | TBSOTf | NEt3 | Trace |
| 4 | TMSOTf | 2, 6-Lutidine | Trace |
| 5 | ClCOCH2Cl | NEt3 | 8 (23%)b |
| 6 | ClCOCH2Cl | Pyridine | Trace |
| 7 | ClCOCH2Cl | DMAP | 8 (35%)b (26%~32%) |
| 8 | ClCOCH3 | DMAP | 9 (51%)b |
| a 7-1 (0.1 mmol), base (0.13~0.2 mmol), 0 ℃~r.t., 15 h; b Isolated yields. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们