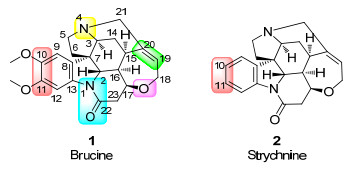
图 1.
马钱子碱与番木鳖碱结构式
Figure 1.
Structures of brucine and strychnine

马钱子是一种传统中药, 始载于《本草纲目》, 多种现代中药的配方中均含其成分[1].普遍认为, 中国古代毒药“牵机药”即未经炮制的马钱子.马钱子的主要活性成分是马钱子碱(1, Brucine)和番木鳖碱(2, 又名士的宁, Strychnine)[2]. 1819年, 法国化学家Pelletier和Caventou在马钱子树皮中发现了马钱子碱[3].直到1884年, 化学家Hanssen才通过实验, 将马钱子碱和士的宁转化为相同的分子, 确定了马钱子碱与番木鳖碱拥有非常相似的结构[4].将马钱子碱苯环上10和11两个位置的甲氧基换为氢原子, 就是番木鳖碱(图 1)[5].虽然马钱子碱的毒性比番木鳖碱弱, 但成人摄入2 mg纯马钱子碱, 也会表现出类似于番木鳖碱中毒的症状[6].有研究表明, 甘草等中药有缓解马钱子碱中毒症状的作用[7].此外, 马钱子碱还是目前最苦的生物碱.当饮水中马钱子碱的含量为7 mg/L时, 便可明显感觉到苦味[8].
马钱子碱具有抗炎镇痛、抑制肿瘤血管生成等作用[9], 但其高肾毒性、神经中枢毒性、低水溶性以及较窄的治疗窗口限制了该生物碱在医药领域的应用[10].使用传统炮制的方法可以将少部分马钱子碱(1)转化成相应的N-氧化物3 (Scheme 1), 后者在保留生物活性的同时毒性大幅度削弱, 拓宽了马钱子的治疗窗口并增强了药用安全性[11].但不同产地以及不同方法炮制的马钱子中生物碱的含量差异较大, 仍需使用高效液相色谱等现代分析仪器准确检测其含量[12].因为马钱子碱具有刺激中枢神经的效果, 也曾作为兴奋剂被不正当地运用在体育运动领域[13].

尽管马钱子碱在医药领域受到诸多限制, 但其在有机化学领域却流光异彩.对其丰富的官能团研究促进了其化学合成方法学的发展; 对其结构进行修饰得到诸多具有潜在药用价值的马钱子碱衍生物, 并对其他药物后期修饰提供了可借鉴的方法; 作为天然的手性碱可以运用到手性拆分和不对称合成中.本文将从马钱子碱各个反应位点的结构修饰, 以及其本身和其衍生物作为手性拆分剂、手性催化剂、手性配体和手性助剂的实际应用两个角度进行分类综述.
马钱子碱具有6个手性中心, 一个芳香环, 一个酰胺官能团, 一个单独的叔胺位点, 一个C=C双键和两个烯丙型位点(图 1).在不引入额外保护基的情况下实现对其单一位点的后期结构修饰是合成化学的一大挑战.
因此, 对其丰富的官能团转化研究能够极大地促进化学合成方法学的发展.尽管新开发出的有机合成方法在短时间内不能促进社会生产, 但这些方法的潜在应用价值仍值得我们注意.很多早期开发的合成方法已经广泛应用于制药行业[14], 对社会的进步起到了积极的作用.此外, 对马钱子碱的后期结构修饰往往只需一两步反应就能得到天然产物的类似物, 相比于从头合成更高效、快速.这些类似物大多能够改变天然分子的部分生物活性, 如改变与受体的结合能力、在生物体内的代谢时间、水相或油相中的溶解度等.生物活性的改变有可能会大幅提升天然分子的药用价值[15].如上文所述, 将马钱子碱转化为相应的N-氧化物后, 药用价值会有极大的提升[11].
本节将综述马钱子碱的结构修饰及部分衍生物的重要应用价值.
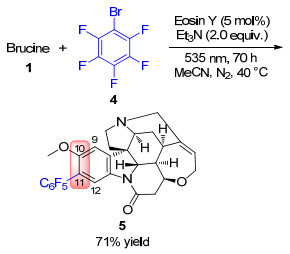
由于马钱子碱反应活性位点丰富, 选择性对苯环结构单元修饰较为困难.虽然有较多关于番木鳖碱的苯环衍生化的报道[16], 但直接对马钱子碱苯环的修饰却非常少. 2016年, König课题组[17]以曙红Y (Eosin Y)为光催化剂, 三乙胺作为电子给体, 在波长为535 nm的绿光照射下, 实现了对马钱子碱芳环上11号位点上的脱甲氧基多氟芳基化(Scheme 2).在光催化体系的介导下, 五氟溴苯(4)的CAr—Br键均裂生成五氟苯基自由基(C6F5•), 之后与马钱子碱反应, 以71%的收率获得了对11号位甲氧基选择性取代的产物5.
该反应实现了温和条件下对马钱子碱的区域选择性反应, 并且前期不需要官能团保护.因此, 该实验方法在天然分子后期功能化方面具有较大的发展潜力.由于包括五氟苯基在内的多氟芳基也是重要的药物活性结构基元[18], 故经过五氟苯基修饰的马钱子碱衍生物也很可能具有潜在的药用价值.尽管该官能团的引入会大幅度改变马钱子碱芳环的电性, 但对叔胺、酰胺、C=C双键等官能团影响较小.此外, 五氟苯基也是具有一定化学活性的官能团, 可以在多种条件下实现脱氟官能团化[19], 为未来进一步研究马钱子碱衍生物的生物活性和手性应用做出了铺垫.
马钱子碱酰胺部分能发生缩合偶联和还原反应.由于其空间结构较为复杂, 酰胺官能团的羰基部分裸露在外.羰基氧的亲核性及其碳的亲电性是该位点能够发生化学反应的内因.此外受酰胺官能团的影响, 其α-H具有一定的酸性, 也可以发生一系列反应.
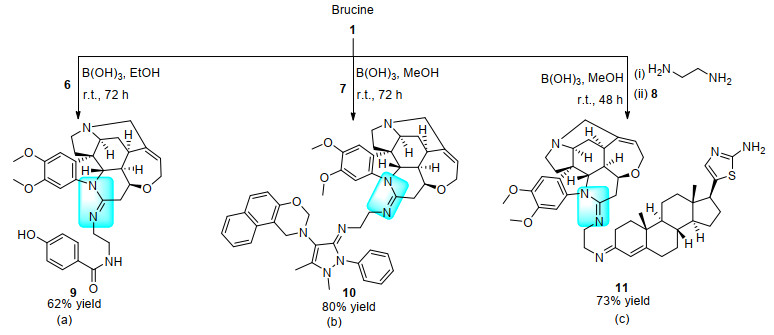
2013年, Figueroa-Valverde课题组[20a]以硼酸为催化剂, 用马钱子碱与含伯胺官能团化合物6(图 2)缩合偶联, 以62%的收率得到了相应的脒类化合物9 (Scheme 3a).次年, 该课题组利用相同的方法, 实现了马钱子碱与含有伯胺的噁嗪类化合物7[20b]、含有羰基的甾类化合物8[20c]的组装反应(Schemes 3b, 3c).
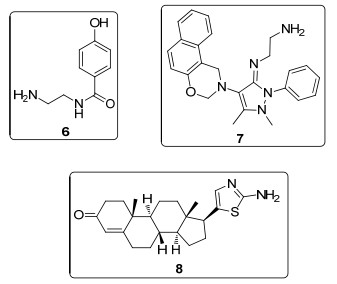
该实验方法条件温和简单, 无需昂贵的催化剂, 反应收率普遍中等偏上, 实用价值较高.通过该方法可以高效地将两种复杂分子偶联在一起, 官能团容忍性强.该方案获得的马钱子碱衍生物的药用价值还有待进一步探究.
马钱子碱双键和酰胺部分均可发生还原反应, 若使用氢化铝锂、硼氢化钠等常见的强还原剂难以实现酰胺部分的选择性还原.使用适当的亲电试剂将羰基活化, 可促进选择性还原反应.具有亲电性和亲氧性的硅烷类化合物是理想的活化试剂之一.
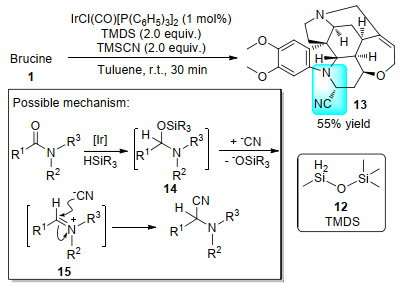
2017年, Dixon课题组[21]以四甲基二硅氧烷(12, TMDS)为还原剂, 实现了铱催化的酰胺部分选择性硅氢化; 硅氢化产物14通过亚胺正离子中间体15, 进一步发生Strecker型反应, 以55%的中等产率得到了氰基化产物13 (Scheme 4).由于氰基具有线性、大极性、可与含活泼氢物质形成氢键等性质, 故被氰基修饰的有机物也往往具有重要的药物活性[22].除了潜在的药用价值外, 由于氰基也是重要的活性官能团[23], 也可以在13的氰基位点上进一步结构修饰.
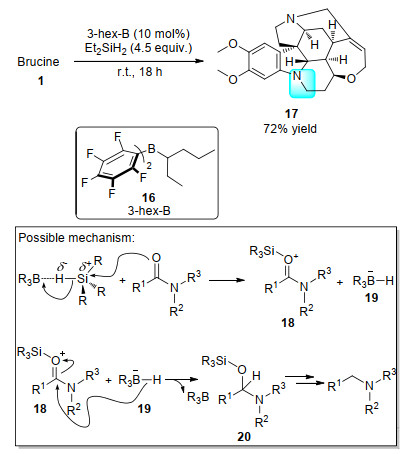
同年, Gagné课题组[24]用特定的有机硼试剂16作为Lewis酸活化硅烷上的Si—H键, 得到了超亲电性(super-electrophilic)的硅正离子, 很好地和酰胺氧原子络合(活化酰胺), 最终, 将酰胺部分还原成了胺17 (Scheme 5).该方法反应条件温和, 区域选择性强, 对其它天然产物后期结构修饰有高价值的借鉴意义.
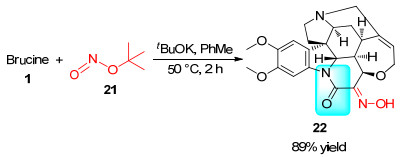
2003年Zlotos课题组[25]用叔丁醇钾对马钱子碱酰胺α-位去质子化, 再对叔丁基亚硝酸酯(21)亲核取代, 得到了相应的亚硝基化合物, 经互变异构, 最终以89%的产率得到了带有肟官能团的马钱子碱衍生物22 (Scheme 6).将22进一步衍生化可得到潜在的药用分子(详见第1.3.5节).
马钱子碱的叔胺部分富有电子, 具有一定的还原性、碱性和亲核性.因此, 这部分可以发生氧化、亲核取代等多种化学反应.此外, 氮原子有三个α-C, 选择性α-C—H键官能团化也受到许多化学方法学研究者的青睐.
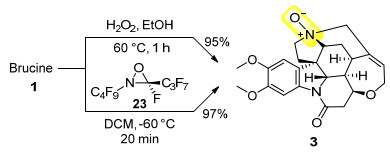
1994年, 蔡宝昌课题组[26]发现马钱子碱(1)可以被过氧化氢氧化成相应的N-氧化物3, 该反应产率可达95% (Scheme 7). 1998年, Resnati课题组[27]实现了用全氟代的氧杂吖丙啶试剂23对马钱子叔胺部分的氧化, 以97%的收率获得了马钱子碱N-氧化物(3) (Scheme 7). N-氧化物3的重要药用价值在前文中已有描述[11].
马钱子碱的N-氧化物可以被多种还原剂还原. 1987年, Jousseaume课题组[28]用六正丁基二锡将马钱子碱N-氧化物(3)还原成了马钱子碱, 该反应产率为84% (Scheme 8a).同年, 周洵钧课题组[29]以碲氢化钠作为温和的还原剂, 在弱酸性条件下以84%的收率实现了对3的选择性还原(Scheme 8b). 1994年, 蔡宝昌课题组[26]研究马钱子碱被过氧化氢氧化的过程中, 发现了其N-氧化物(3)可以被亚硫酸氢钠还原(Scheme 8c). 2009年, Oh课题组[30]使用蔡宝昌课题组发展的氧化条件, 获得了马钱子碱N-氧化物(3).随后, 以五氯化铌为催化剂, 锌为还原剂, 以95%的产率实现了对3的还原(Scheme 8d).以上还原中, 亚硫酸氢钠还原法廉价、安全、便捷、绿色, 不失为一种优秀的还原反应.
2012年, Davies课题组[31]在无过渡金属催化情况下实现了卡宾对马钱子碱的形式C—N键插入反应.在该反应中, 首先富电子的叔胺对游离卡宾24进行亲核进攻得到氮叶立德中间体25.随后发生Stevens重排, 在活性更高的烯丙型位点生成形式上C—N键插入产物26 (Scheme 9).
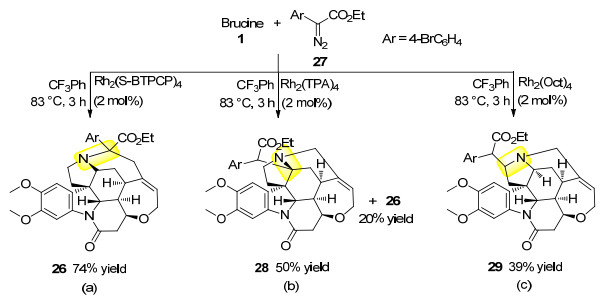
2015年, Beckwith课题组[32]以不同配体的铑(Ⅱ)为催化剂, 用卡宾前体重氮化合物27和马钱子碱反应, 选择性实现了氮原子α-C—H键官能团化, 高效地制备了三种马钱子碱衍生物:一种和Davies等的研究相似, 是形成氮叶立德中间体之后再发生Stevens重排的产物26 (Schemes 10a, 10b), 另外两种是直接对氮原子的α-C—H键直接插入的产物28和29 (Schemes 10b, 10c).反应选择性受Rh催化剂配体的控制.
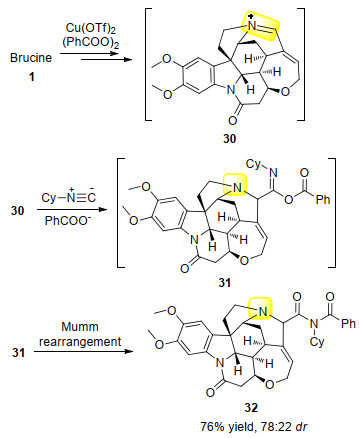
2019年, Mancheño课题组[33]以Cu(Ⅱ)为催化剂, 用有机过氧化物逐步将叔胺选择性地氧化成了亚胺正离子中间体30.随后, 亚胺正离子、异腈、羧酸负离子发生三组分反应得到对应的O-酰基亚胺酸酯中间体31; 进一步发生Mumm重排, 得到了选择性官能团化的马钱子碱衍生物32, 反应位点为被氮原子活化的烯丙位(Scheme 11).
以上C—N键或C—H键插入反应, 均实现了在不改变原有重要官能团的基础上引入新的官能团.除了得到更多生物活性可能改变的马钱子碱衍生物外, 更重要的意义在于扩展了对天然产物非活泼位点衍生化的方法, 有助于提高基于天然产物结构修饰的药物开发效率.未来也可以将引入官能团进一步设计, 以作为生物探针深入研究药物作用机制[32].
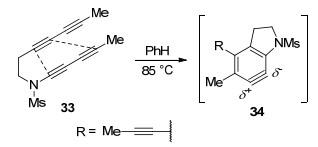
2017年, Hoye课题组[34]使用含有三个(或更多)C≡C叁键的底物33, 在加热条件下实现了分子内去六氢Diels-Alder反应, 得到了相应的苯炔中间体34 (Scheme 12).
随后利用叔胺氮原子的亲核性与苯炔中间体34反应, 得到了中间体35.经过分子内的氢转移得到氮叶立德中间体36.该中间体的20号位点带有负电荷, 夺取酚羟基上的氢.随后, 酚氧负离子与中间体36的5号位发生SN2反应, 得到双键保持产物37.相应地, 酚氧负离子与22号位通过SN2'反应得到端烯型产物38.氮叶立德中间体36的20号位点与C=C形成了三中心四电子的离域体系, 使负电荷得以从20号位过渡到22号位. 22号位夺取酚羟基上的氢之后, 酚氧负离子与5号位发生SN2反应生成了双键迁移产物39 (Scheme 13, 图 3).
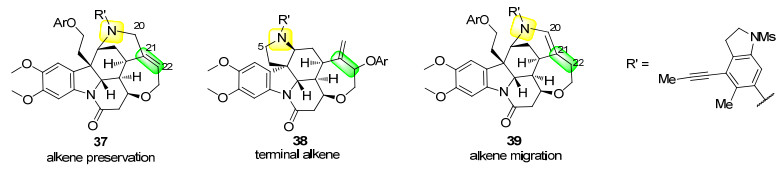
该反应能生成的产物类型和产率与酚的结构有关(图 4).氮叶立德中间体36的20号位电子云密度高, 但空间位阻较大; 与之相反, 22号位电子云密度低, 但空间位阻较小.如果使用雌二醇(40)这类酚羟基位点附近位阻小的试剂, 将会对36的20号位质子化, 随后以55%的中等收率得到单一的双键保持型产物37.使用含有大位阻基团的2, 6-二叔丁基对甲基苯酚(41), 将会对36的22号位质子化, 并伴随双键迁移; 随后酚氧负离子与5号位发生SN2反应, 得到单一的双键迁移型产物39; 受位阻影响, 该反应收率较低, 仅为39%.与40相比, 4-甲氧基苯酚(42)分子结构更简单、空间位阻更小, 故更容易和36发生反应.该反应中双键保持型产物37的产率提升至61%, 同时还得到了收率为5%的双键迁移型产物39.使用空间位阻适中的均三甲基苯酚(43)和维生素E (44), 除了生成37和39外, 还可以从22号位点发生SN2'反应得到了端烯型产物38.用43和44参与的反应相比较, 不难发现, 随着分子复杂程度的增加, 产物的总收率有所下降.该反应实现了三种物质的组装反应, 为天然产物的后期修饰提供了高价值的借鉴方法.
2013年, Romo课题组[35]以双(新戊酰氧)碘苯(45, (tBuCO2)2IPh)为氧化剂, Rh(Ⅱ)为催化剂, 用马钱子碱和磺酰胺46反应, 得到了氮-氮叶立德产物47.在该反应中(tBuCO2)2IPh将磺酰胺可逆地氧化成了PhI=NR型亚胺基碘烷49[36]. 49与Rh(Ⅱ)催化剂作用得到了[Rh]=NR型铑乃春中间体50, 再分别与马钱子碱的叔胺氮原子和其邻位叔碳C—H键反应, 最终得到了收率为28%的氮-氮叶立德47和收率为21%的开环产物内酰胺48 (Scheme 14).
值得一提的是, 含有C≡C叁键的天然分子衍生物52可与带标记的叠氮化物51反应, 生成带有标记的三唑化合物53.带有标记的三唑类化合物53可以作为细胞探针, 用于深入研究天然分子在生物体内的作用机制(Scheme 15)[35].上述37、38、39、47等含有C≡C叁键的马钱子衍生物都具有这种应用的潜质.
叶立德47在10% Cd/Pb[37]催化下, 可脱除烷氧基磺酰基保护基R得到相应的叶立德54, 产率可达76% (Scheme 16) [35].由于54正负电荷处于分离状态, 故与马钱子碱1相比更易溶于水, 可大幅度改善马钱子碱1由于水溶性差导致的药用受限[10d].该化合物的药用价值还有待进一步探究.

2017年, Pérez课题组[38]改进了Romo等的方法, 以催化性能较Rh(Ⅱ)低的Ag(Ⅰ)为催化剂, 在不添加氧化剂的情况下, 直接用亚胺基碘烷PhI=NTs与马钱子碱反应, 以83%的收率得到了唯一的叶立德产物55 (Scheme 17).该反应过程中也产生了与[Rh]=NR中间体相似的[Ag]=NTs中间体.与Romo课题组的方法相比, 该方法反应效率更高, 无副产物生成, 实用价值更高.
叔胺氮原子亲核性较高, 可与卤代烃发生亲核取代反应, 生成多种具有实际应用价值的马钱子碱衍生物.本文列举几处典型的应用.
1999年, Birdsall课题组[39]用马钱子碱和几种简单的卤代烃反应, 得到了相应的季铵盐衍生物56~59 (Scheme 18).除和卤代物反应外, 还和磺酰羟胺反应得到了叔胺位点胺化的产物60.该研究证实马钱子碱季铵盐可以作为神经递质乙酰胆碱(61)受体蛋白变构剂, 对五种毒蕈碱型受体(Muscarine受体, 简称M受体, 有M1~M5五种亚型)都有一定的亲和力.衍生物56~60对M受体产生变构作用后, 都能选择性地增强乙酰胆碱和M3受体的结合能力(正协同作用)(图 5), 并抑制乙酰胆碱和其他四种亚型M受体相结合(负协同作用).未经衍生化的马钱子碱(1)仅在M1亚型受体上与乙酰胆碱有正协同作用, 而马钱子碱氮氧化物(3)在M3和M4受体上与乙酰胆碱均有正协同作用.这些研究证明, 马钱子碱季铵盐衍生物具有治疗阿尔茨海默病的潜在效果.
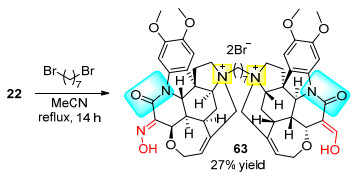
2003年Zlotos课题组[25]用端位二卤代烷将两分子的马钱子碱相连, 得到了多个双马钱子碱季铵盐衍生物62 (Scheme 19).这些化合物可以作为毒蕈碱M2受体的变构剂, 能够增加相应拮抗剂与M2受体的亲和力.该方法也适用于之前所得肟类化合物22的偶联, 得到相应的马钱子碱衍生物63 (Scheme 20).通过实验对比分析, 带有肟官能团的63与猪心M2受体的结合能力比马钱子碱1高56倍左右, 62这类双马钱子碱衍生物与猪心M2受体的结合能力比马钱子碱1高115倍左右.

2005年, Král课题组[40]用马钱子碱和苄基卤代烷修饰的卟啉衍生物反应, 得到了两种四马钱子碱-卟啉衍生物66和67, 产率分别为77%和48% (Scheme 21).四马钱子碱-卟啉衍生物含有多个芳香环, 能与其他含有芳香环的化合物产生π-π堆积作用; 化合物上含有多个氧、氮杂原子, 可以和其他含有活泼氢的物质形成氢键; 卟啉环氮原子上带有氢, 也可以和其他含有氮、氧的化合物形成氢键; 马钱子碱季铵部分带有正电, 可与带有负电物质结合.四马钱子碱-卟啉衍生物由于具有上述多种性质, 可以作为甲醇、乙腈等溶剂的高效凝结剂[40a].除此之外, 卟啉-马钱子碱衍生物在溶剂中还可以通过非共价键作用选择性识别腺苷三磷酸(ATP)等单核苷酸[40b]. ATP等单核苷酸具有芳环且含杂原子的嘌呤基团、带负电的磷酸基团, 且可以提供氢原子形成氢键.单核苷酸这些特性和含有特定空间结构的四马钱子碱-卟啉衍生物形成了互补, 通过非共价键作用实现了在ADP和AMP存在下对ATP的选择性识别.由于卟啉衍生物本身具有荧光性质, 它们与单核苷酸形成复合物后, 可以用于核苷酸的化学传感研究[40b-40d].
C=C双键具有还原和氧化双重性质.此外, 双键富有电子, 具有一定的亲核性.马钱子碱C=C双键能基于以上三种性质发生化学反应.
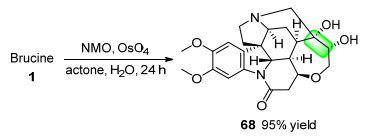
2009年, Oh课题组[41a]以四氧化锇为催化剂, 用N-甲基吗啉-N-氧化物(NMO)[42]对马钱子碱进行了双羟化, 以95%的收率获得了衍生物68 (Scheme 22).
双羟化衍生物68上的氮原子依然有较强的亲核性, 可以和苄溴发生亲核取代反应, 以82%的收率得到相应的季铵盐衍生物69 (Scheme 23).
68中的羟基去质子化可以和乙酰氯反应, 得到两种乙酰化的衍生物70和71, 二者产率均为55% (Scheme 24).这些马钱子碱衍生物可以作为手性配体, 用于过渡金属催化的不对称合成(见第3.2节)[41].
2016年, Hartwig课题组[43]以Togni试剂72[44]为三氟甲基化试剂, 四甲基硅基叠氮(TMSN3)为叠氮化试剂, 实现了马钱子碱C=C双键的双官能团化. Togni试剂在Fe(Ⅱ)催化下产生亲电的三氟甲基自由基, 进而与C=C双键的较富电子一端连接, 得到的自由基中间体被TMSN3捕获, 实现叠氮化.在手性配体73的作用下, 该反应实现了区域选择性和立体选择性的双重控制, 以70%的收率得到了单一构型的马钱子碱衍生物74 (Scheme 25).该方法同样适用于其他含有C=C双键的天然分子, 所得结构修饰产物可供药物筛选, 为药物发现提供更多的机会.
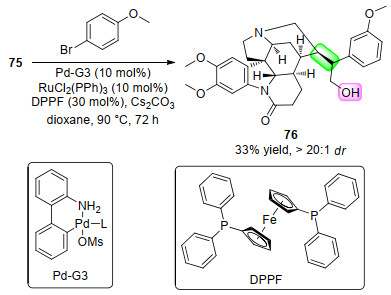
2019年, Dydio课题组[45]用铂催化剂在高温高压下实现了对马钱子碱C=C双键的氢化反应.在该反应中, 双键和醚键被同时氢化, 得到了22%产率的马子碱的醇类衍生物75 (Scheme 26).
醇衍生物75可以在Pd(Ⅱ)和Ru(Ⅱ)催化剂的共同作用下与卤代芳烃反应, 得到了羟基β-C—H键选择性芳基化的产物76, 产率为33% (Scheme 27).对复杂产物75的成功修饰, 突出了该双催化体系的合成应用价值.
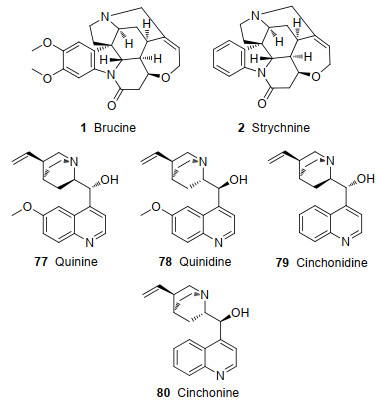
从马钱子属植物中提取的马钱子碱(1)、番木鳖碱(2), 以及从金鸡纳亚科植物中提取的奎宁(77, Quinine)、奎尼丁(78, Quinidine)、辛可尼丁(79, Cinchonidine)、辛可宁(80, Cinchonine)(图 6), 都是常见的具有光学活性的天然生物碱.这些手性生物碱可用于对羧酸、膦(磷)酸、酚等酸性外消旋体进行拆分, 主要原因是:首先, 碱性的氮杂环结构可以和羧酸、膦(磷)酸、酚等酸性物质反应, 易生成相应的固体盐.其次, 含有的大极性官能团和芳香环体系易在分子间产生范德华力、氢键、π-π堆积效应等非共价作用.由于不同马钱子碱盐之间非共价作用程度不同, 故在溶剂中的溶解度也不同, 得以通过结晶实现不同马钱子碱盐的拆分.通过对马钱子碱盐进行碱化、酸化处理, 可拆分出酸性有机物.最后, 它们都含有多个手性中心和复杂并环、螺环或桥环的立体结构.天然的光学活性和独特的空间结构是拆分外消旋化合物的重要诱因.一些情况下, 仅仅用马钱子碱(1)并不能完全实现两个对映异构体的拆分, 还需同时借助手性碱2、77~80.
早在1916年, Wren和Williams[46]就使用马钱子碱对外消旋的苯基琥珀酸进行了拆分.随后的数十年里, 马钱子碱常被用于拆分手性氨基酸[47]和含有羧基的手性药物分子[48].此外, 早期还有报道使用马钱子碱直接拆分手性醇[49]和手性卤代烃[50]的案例, 随着不对称合成及手型拆分方法的多样化, 这些方法已不再是研究的热点.新世纪以来, 除了传统的拆分应用外, 马钱子碱更多地被用于拆分手性配体和手性配体前体.
历经一百多年的研究和发展, 马钱子碱的手性应用已数不胜数. Jaen[51]在2001年对马钱子碱以往的手性应用进行了很好的总结, 本文主要对更新和更具有实际应用价值的研究进展进行综述.
马钱子碱可用于拆分螺旋手性和轴手性羧酸或酚类化合物. 2008年, Wild课题组对前人的方法[52a]进行了改进, 更高效地实现了用马钱子碱对手性螺环化合物81的拆分[52b].首先, 将81和马钱子碱在乙醇溶液中回流加热2 h, 随后经过热过滤除去不溶杂质.将滤液在热油浴中缓慢冷却, (S)-羧酸与马钱子碱形成的盐82结晶析出.对盐82多次重结晶, 随后酸化处理可得(S)-羧酸83.对母液多步处理可得(R)-羧酸84 (Scheme 28).将每个83羧基都连接上多个氮原子的官能团, 可以得到一个六齿的金属离子螯合剂85 (Scheme 29).
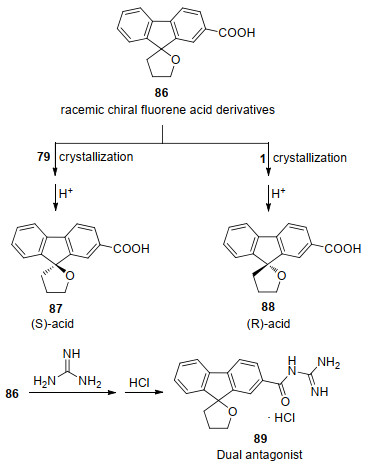
2012年, Yoshida课题组[53a]使用辛可尼丁(79)与马钱子碱(1)对外消旋芴酸衍生物86进行了拆分, 而手性芴的酰胍类(carbonyl guanidine)衍生物89可作为5-HT2B和5-HT7受体双重拮抗剂(Dual antagonist) (Scheme 30). 2014年, Moritomo课题组[53b]使用了Yoshida课题组的手性拆分方法, 合成了一系列的酰胍类5-HT2B和5-HT7受体双重拮抗剂.
2004年到2005年, Solladié课题组[54a-54b]用马钱子碱拆分了轴手性联苯二甲酸90, 接着和长连烷基联二苯酚反应生成相应的轴手性酯92 (Scheme 31).这种酯类化合物可以作为向列相液晶(nematic liquid crystals)手性掺杂剂(chiral dopants). 2014年, Haino课题组[54c]用同样的方法拆分了外消旋体90, 用以研究非对映超分子复合物.

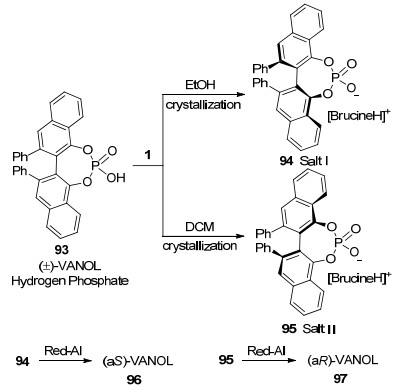
2009年, Polavarapu课题组[55a]发现外消旋膦酸93能与马钱子碱生成两种光学活性的盐, 并测定了两种盐的晶体结构.由于两种盐结构不同, 在不同溶剂中析出晶体的先后顺序也有差异.在乙醇中盐I (94)首先析出, 在二氯甲烷中盐Ⅱ (95)先结晶.使用铝试剂Red-Al[55b]还原, 得到了两种单一光学活性的手性联萘酚VANOL 96, 97(Scheme 32).
2002年, Král课题组[56]开发了马钱子碱-硅胶复合的手性识别支架101 (Scheme 33).该课题组用马钱子碱和6-溴己酸(98)反应得到了相应的季铵盐衍生物99, 再和3-氨基丙基硅胶100缩合得到了带有正电荷的马钱子碱-硅胶复合支架101.该支架可以作为色谱固定相, 对含有羧基结构的轴手性二萘酚102和103进行拆分.需要注意的是, 该拆分方法的作用机制为阴离子交换作用, 与直接用马钱子碱进行拆分的机制不同, 并不涉及季胺部分与酸性基团的作用.
除了用手性色谱拆分轴手性联萘酚外, 也可以直接用马钱子碱拆分.
2009年, Tanaka和Urbanczyk-Lipkowska课题组[57a]实现了马钱子碱对含有两个羧基的联萘酚104的手性拆分(Scheme 34).两个羧基作为活性基团, 在拆分过程中起到与马钱子碱成盐的作用.
2016年, Bedekar课题组[57b]实现了对不含羧基结构的手性联萘酚107的拆分(Scheme 35).该拆分属于真正意义上对酚类化合物的拆分, 萘-2, 7-二酚部分的两个羟基与马钱子碱叔胺部分通过氢键作用缔合.该拆分方法操作简单, 只需将107与马钱子碱在甲醇中回流, 析出的晶体为108与马钱子碱的缔合体, 母液中含有未反应的109.
使用马钱子碱还可以对其它手性羧酸进行拆分. 2004年, Otto课题组[58]使用马钱子碱从外消旋的S-苄基-N-甲酰基-β-高半胱氨酸(110)中拆分出了S-构型的对映异构体111.去甲酰基之后可得(S)-S-苄基-β-高半胱氨酸, 该化合物经过多步反应之后可制得手性β-磺酰内胺112 (Scheme 36).
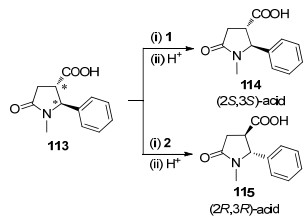
2008年, Czarnocki课题组[59]将含有羧基的γ-内酰胺113分别和马钱子碱、番木鳖碱(2)作用, 得到了2S, 3S和2R, 3R构型的对映异构体114和115 (Scheme 37). 114可以协助对外消旋醇进行拆分(详见第2.2节).
2005年, Doyle课题组[60]实现了对trans-2-苯基环丙基甲酸(116)的拆分(Scheme 38).之后将2-苯基环丙基甲酸逐步转化为咪唑酮类衍生物119和120 (Scheme 39), 可以作为Rh(Ⅱ)催化剂配体用于铑卡宾参与的不对称C—H键官能团化反应和醛与双烯的不对称杂-Diels- Alder环加成反应.

2011年, White课题组[61]用马钱子碱拆分出了桥环结构的手性二甲酸122, 之后逐步合成了C2-对称的四齿手性配体123 (Scheme 39), 可用于Cr(Ⅲ)催化的醛与双烯的不对称杂-Diels-Alder环加成反应和醛与烯丙基溴的不对称Nozaki-Hiyama-Kishi反应.
2007年, Carlier课题组[62]用马钱子碱拆分手性酸124后, 用相应的羧酸125合成了手性格氏试剂127 (Scheme 40).
2014年, 董广彬课题组[63]使用马钱子碱拆分了手性酸128 (Scheme 41), 用以研究酮类化合物α-C—H键活化反应中的立体效应.
Thiele’s酸(130)是一种具有独特刚性夹角的分子, 又因为其两端的羧基易于衍生化, 故可以利用130制作应用于超分子或生物化学领域的分子支架[64]. 2017年, Wulff课题组[65]筛选了辛可尼丁(79)、L-色氨酸乙酯、L-酪氨酸酰肼等手性胺类化合物, 均无法有效拆分外消旋的Thiele’s酸.最终, 使用马钱子碱作为拆分试剂时, (-)-Thiele’s酸(131)与两分子马钱子碱以盐的形式结晶析出, (+)-Thiele’s酸(132)保留在了母液中.最后进行酸化处理, 实现了对130的手性拆分(Scheme 42).
2017年, 姚祝军课题组[66]用马钱子碱和奎宁(77)配合拆分了外消旋的2, 3-二甲基酒石酸(133).在乙醇中, 以奎宁(77)为拆分试剂可以得到(2S, 3S)-2, 3-二甲基酒石酸(134); 在水溶液中, 以马钱子碱为拆分试剂可以得到(2R, 3R)-2, 3-二甲基酒石酸(135).被拆分的2, 3-二甲基酒石酸在手性拆分、手性催化等领域具有一定的应用潜力.
由于醇类不能与手性生物碱成盐, 故需要一定的策略对醇修饰.目前对醇的拆分主要有三种策略.第一, 将外消旋的醇和手性酸反应, 制备成非对映异构的酯, 经分离、水解, 也可以间接地拆分对映异构的醇.例如, 利用Scheme 43中的(2S, 3S)-羧酸114和外消旋醇反应可以得到一对非对映异构的酯, 通过柱层析的方式可使二者分离; 再经过皂化反应分别得到一对互为对映异构体的醇[59].第二, 部分特定结构的醇可以通过与手性生物碱的氢键作用, 实现直接拆分.第三, 将醇和邻苯二甲酸酐反应, 得到邻苯二甲酸半酯.通过用手性生物碱拆分邻苯二甲酸半酯, 再水解得醇, 间接地实现了对醇的对映异构体的拆分.这是最常用的方法.由于第一种方法不涉及马钱子碱的应用, 第二种方法限制性强且鲜有报道, 故本节仅介绍最后一种策略.
1926年, Kenyon[67]将利用手性生物碱分离邻苯二甲酸半酯, 水解半酯间接得到手性醇的方法公布, 并对该方法做了详尽的描述(Scheme 44).首先用外消旋的1-甲基庚醇(137)和邻苯二甲酸酐(136)反应得到相应的半酯138, 接着用马钱子碱与半酯成盐139, 实现拆分.使用盐酸可以使马钱子碱解离, 经过皂化水解得到了两种互为对映异构体的脂肪醇140和141.
自Kenyon公布他的方法之后的80年里, 该方法被广泛地应用于多种外消旋醇的拆分, 在Jaen的综述中已有所列举[51].更值得关注的是, 也可以使用该方法拆分邻苯二甲酸半酰胺,随后经过水解,得到手性伯胺化合物[68].
马钱子碱可以对外消旋药物进行拆分, 该方法同样要求药物分子含有酸性的羧基或酚羟基.虽然该部分实质上还是对羧酸和酚的拆分(第2.1节), 但鉴于目前药物合成在有机化学研究中的重要地位, 本文将外消旋药物的拆分单独列出来.
5, 5-二苯乙内酰脲(145, PHT)是一种抗惊厥药物, 在生物体内可代谢成一个苯基对位被羟基化的衍生物(142, 简称HPPH). 1975年, Claesen课题组[69a]用马钱子碱对HPPH进行了手性拆分, 研究了相关的生物代谢作用(Scheme 45). 2004年, Vogel课题组[69b]实现了对HPPH的人工合成, 并用Claesen课题组的方法进行了手性拆分.
2003年, Nielsen课题组[70]实现了对抑菌药山芬霉素(146, Saphenamycin)的全合成, 使用马钱子碱拆分出了(-)-山芬霉素(147) (Scheme 46).通过对其马钱子碱盐的X光衍射分析, 确定了所得(-)-山芬霉素为R构型.
2005年, Challenger课题组[71]合成了内皮素拮抗剂UK-350, 926 (148).在热丙酮中可结晶出R型异构体的马钱子碱盐, 浓缩母液可以提取出S型异构体150 (Scheme 47).

2009年, Kim课题组[72]使用两种不同的生物碱, 实现了对抗炎镇痛药依托度酸(151, Etodolac)外消旋体的拆分.依托度酸与辛可尼丁(79)生成盐, 结晶酸化得到药物活性较低的(—)-依托度酸(152).与之相反, 与马钱子碱作用得到了比外消旋体药物活性更高的(+)-依托度酸(153)(Scheme 48).
2012年, Kuo课题组[73]合成了可作为神经毒素的手性硫代膦酸酯.该课题组将硫代膦酸154和马钱子碱溶于热的丙酮溶剂中, 冷却后S构型的硫代膦酸马钱子碱盐155以固体析出, R构型的盐156被保留在溶液中, 将溶液蒸发得到了两种构型的粗盐.将两种粗盐分别溶于氢氧化钠的醇溶液中, 加水会使马钱子碱固化析出.分别用四氯化碳洗涤后, 水相用盐酸酸化使硫代膦酸游离.用二氯甲烷分别萃取水相后, 减压浓缩可得一对硫代膦酸对映异构体157和158.使用二环己基胺(160)稳定硫代磷酸157后, 用碘乙烷乙基化得到手性的硫代膦酸酯159 (Scheme 49).

马钱子碱是天然生物碱, 与质子酸结合生成盐是其基本性质, 此处不再赘述.马钱子碱盐大多具有固定的晶型, 非固体的酸可以通过与马钱子碱成盐来确定构型[74].此外马钱子碱还可以作为催化剂, 参与碱催化的有机化学反应[75].由于马钱子碱作为有机碱的非手性应用并不是重点, 本小节仅介绍一例.
2019年, Gates课题组[74b]用马钱子碱对五配位的有机膦羧酸161去质子化, 得到了与马钱子碱成盐的六配位膦化合物162.通过X射线衍射确定了所合成化合物的立体结构(Scheme 50).这种六配位的膦化合物负离子是潜在的烯烃阳离子聚合引发剂[74b].
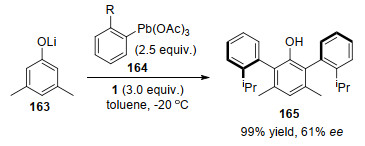
2002年, Yamamoto课题组[76]以马钱子碱为手性配体, 在实现了芳基铅(Ⅳ)化合物164对3, 5-二甲基苯酚的高效不对称双官能团化反应, 几乎以定量的产率得到了酚165 (Scheme 51).马钱子碱通过与铅(Ⅳ)配位起到手性诱导效果, 对应异构体过剩(enantiomeric excess, ee)值为61%.
2009年到2010年, Oh课题组[41]用马钱子碱双羟基化的衍生物68作为手性配体, 用不同金属催化剂和相同的底物反应, 得到了对映选择性不同的产物.由此推测不同金属和马钱子碱双羟基化衍生物的配位方式不同.用Ag(Ⅰ)为催化剂时, 有两分子马钱子碱双羟基化衍生物作为配体, 且只有氮原子和银原子配位(166); 以Cu(Ⅰ)和Cu(Ⅱ)为催化剂时, 氮原子和氮β位氧原子参与络合(167, 169); 而每分子的马钱子碱双羟基化衍生物可以和两个Zn(Ⅱ)原子配位(168) (Scheme 52).

2009年, Oh课题组[41a]用Ag(Ⅰ)或Cu(Ⅰ)为催化剂, 马钱子碱双羟基化衍生物作为配体, 实现了氮杂1, 3-偶极化合物对C=C双键的不对称环加成反应(Scheme 53).当使用Ag(Ⅰ)催化剂166时, 以较好的立体选择性得到构型为2S, 4S, 5R的产物172, 反应收率为79%, ee值为74%.当使用Cu(Ⅰ)催化剂167时, 能够以极高的对应选择性和极高的产率获得构型为2R, 4R, 5S的产物173, 产率和ee值分别为98%和95%.

同年, Oh课题组[41b]使用Cu(Ⅰ)或Zn(Ⅱ)为催化剂, 实现了硝基甲烷(175)与醛174的不对称加成反应(Scheme 54).以2-呋喃甲醛为底物参与反应, 使用Cu(Ⅰ)催化剂167可得到S构型产物176, 产率为82%, ee值高达97%.使用Zn(Ⅱ)为催化剂时, 可得到R构型的产物177, 产率和ee值分别为88%和90%.

2010年, Oh课题组[41c]以Cu(Ⅱ)为催化剂, 实现了吲哚类化合物178与硝基烯烃179的高对映选择性加成(Scheme 55).当R1为氢且R2为苯基时, 能够以50%的反应收率和86%的ee值获得S构型产物180.

1904年, Marckwald[77a]用马钱子碱作催化剂, 实现了2-甲基-2-乙基丙二酸(181)在高温下的不对称去羰基化反应(Scheme 56).在该反应中, 质子化的马钱子碱季铵盐中间体182是手性质子源. 1987年, Toussaint, Capdevielle和Maumy[77b]用Cu(Ⅰ)和马钱子碱为催化剂, 实现了苯基丙二酸单酯(184)在温和条件下的不对称去羰基化反应(Scheme 56).上述两种产物的光学纯度(optical purity, op)值均为在10%左右.

1985年, Mikołajczyk课题组[78]以马钱子碱为手性助剂实现了亚磺酸酯187的不对称合成.在该反应中, 具有手性和亲核性的马钱子碱与格氏试剂形成了手性络合物[79], 接着和亚硫酸二酯186发生加成-消除反应, 以84%的收率得到R构型的亚磺酸酯187, ee值为62% (Scheme 57).
2009年, Kolesińska课题组[80]用马钱子碱和2-氯-4, 6-二甲氧基-1, 3, 5-三嗪(189)反应, 得到了能够活化单一光学构型氨基酸的手性助剂190.使用助剂190实现了在反应过程中将外消旋的氨基酸191进行拆分, 进而与另一分子氨基酸194缩合成二肽195 (Scheme 58).该反应产物对应异构体浓度比(enantiomeric ratio, er)值最高可达99.6:0.4 (D构型:L构型).
2010年, Kimachi课题组[81]用对三氟甲基苄氯(196)和马钱子碱反应得到了铵盐中间体197, 该中间体与苯甲醛发生Darzens反应生成S, S构型的环氧化合物198.该反应依托马钱子碱铵盐中间体的光学活性和空间位阻, 实现了对映和非对映选择性的双重控制(Scheme 59).该反应产物的非对映异构体浓度比(diastereomeric ratio, dr)值为17:83 (cis:trans), ee值为65%.
2017年, Nicolaou课题组[82]使用Oh课题组的方法得到了马钱子碱双羟基化的衍生物68.随后, 利用叔胺氮的亲核性, 与对三氟甲基苄溴(199)反应, 以92%的收率得到了相应的铵盐200 (Scheme 60).
铵盐200可以作为手性相转移催化剂, 催化蒽酮201的不对称烯丙基化反应(Scheme 61).在该反应中, 蒽酮201的10号位被去质子化得到相应的碳负离子.由于手性铵盐200的氮原子带有正电荷, 可以与碳负离子作用, 依靠手性铵盐200的手性实现了在10号位置的不对称烯丙基化.通过高效液相色谱测得该反应的选择性较低, dr (10S:10R)值仅为56:44.然而, 通过奎宁类的相转移催化剂可以实现很好的手性诱导效果[82].
本文综述了关于马钱子碱结构修饰和手性应用的最新进展.马钱子碱具有芳基、醚、酰胺、叔胺、C=C双键等结构单元和复杂的并环、螺环、桥环立体结构, 对其进行选择性结构修饰具有很高的难度, 目前已有多种方法实现了在其特定位点的选择性反应.结构修饰后的产物具有独特的生物活性, 可作为手性配体或助剂应用到不对称合成中.手性应用方面, 马钱子碱作为手性拆分试剂, 广泛应用于外消旋羧酸、磷(膦)酸、酚、醇和药物的拆分.此外, 马钱子碱本身也可作为不对称合成中的手性配体、手性催化剂或手性助剂.
目前针对马钱子碱的研究仍有不完善之处.第一, 大部分工作者只对马钱子碱进行了结构修饰, 并没有进一步测试马钱子碱衍生物的生物活性.第二, 大部分马钱子碱衍生物还有待进一步发掘其合成应用价值, 如肟衍生物22本身具有羟基, 有作为新型手性配体的潜质.第三, 作为手性拆分试剂拆分药物时, 由于马钱子碱毒性较高, 一般不会优先采用.从马钱子碱衍生物中筛选易制备且低毒性的拆分试剂是值得研究的方向.
对马钱子碱的研究有很大的实际应用意义, 已证实马钱子碱及其衍生物具有一定的潜在医用价值.得益于专业研究人员的贡献, 马钱子碱结构修饰的产物数目得到了一定的扩展.然而, 碍于不同专业人员之间的交流限制, 鲜有对其药理、毒理测试的报道.因此将马钱子碱衍生物应用于实际药物, 依然任重而道远, 未来还需要来自不同学科的研究人员的通力合作.
(a) Hai, P.; Wang, C.; Luo, G. Chin. J. Pharm. 2016, 47, 1394(in Chinese).
(海平, 王慧春, 骆桂法, 中国医药工业杂志, 2016, 47, 1394.)
(b) Zhang, J.-J.; Yang, Y.-J.; Qi, L.-P.; Dong, Y.-Y.; Zhang, J.; Li, R.-K.; Liu, Y.-M. Rheum. Arthritis 2017, 6, 38(in Chinese).
(张景姣, 杨艳娇, 祁利平, 董艳艳, 张健, 李瑞柯, 刘雅敏, 风湿病与关节炎, 2017, 6, 38.)
(c) Duan, C.-X.; Guo, C.-J. Chin. J. Pharm. Anal. 2018, 38, 331(in Chinese).
(段存贤, 郭承军, 药物分析杂志, 2018, 38, 331.)
(d) Zhou, S.; Zhou, H.; Chen, Y.; Meng, F. Acta Chin. Med. 2018, 33, 1302(in Chinese).
(周淑娟, 周红敏, 陈岩岩, 孟菲, 中医学报, 2018, 33, 1302.)
(e) Liu, M.; Cui, X.; Shi, H.; Wang, X.; Chen, Z.; Niu, A.; Gao, R.; Cao, X. China Pharm. 2019, 30, 2980(in Chinese).
(刘满军, 崔小敏, 石会丽, 王晓萍, 陈志永, 牛安琦, 高蓉, 曹小平, 中国药房, 2019, 30, 2980.)
(a) Wu, X.-J.; Ma, F.-S.; Yu, Y. Lishizhen Med. Mater. Med. Res. 2016, 27, 2145(in Chinese).
(吴小娟, 马凤森, 喻炎, 时珍国医国药, 2016, 27, 2145.)
(b) Yan, J.; Lin, J.-T.; Liu, Z.-Q.; Zhen, X.-Y. Chin. J. Pharm. Anal. 2020, 40, 132(in Chinese).
(闫静, 林佳娣, 刘志强, 甄晓宇, 药物分析杂志, 2020, 40, 132.)
(c) Dong, Z.; Yang, Z.; Xu, J. Univ. Chem. 2020, DOI:10.3866/PKU.DXHX202005056(in Chinese).
(董子阳, 杨占会, 许家喜, 大学化学, 2020, DOI:10.3866/PKU.DXHX202005056.)
Wormley, T. Micro-chemistry of Poisons Including Their Physiological, Pathological, and Legal Relations: Adapted to the Use of the Medical Jurist, Physician, and General Chemist, Wood, W., New York, 1869.
Buckingham, J. Bitter Nemesis:The Intimate History of Strychnine, CRC Press, Boca Raton, USA, 2007, p. 225.
Frédérich, M.; Choi, Y. H.; Verpoorte, R. Planta Med. 2003, 69, 1169. doi: 10.1055/s-2003-818014
Malone, M. H.; St. John-Allan, K. M.; Bejar, E. J. Ethnopharmacol. 1992, 35, 295. doi: 10.1016/0378-8741(92)90028-P
(a) Ren, J.; Zhang, X.; Chu, Z. J. Liaoning Univ. Tradit. Chin. Med. 2016, 18, 221(in Chinese).
(任佳佳, 张学顺, 褚志杰, 辽宁中医药大学学报, 2016, 18, 221.)
(b) Zhang, M.; Wang, C.; Wen, Q.; Fang, P.-F. Chin. J. Clin. Pharmacol. Ther. 2017, 33, 2282(in Chinese).
(张敏, 王超, 温菁, 方平飞, 中国临床药理学杂志, 2017, 33, 2282.)
Shiba, T. Kagaku Sosetsu 1976, 14, 129.
(a) Yin, W.; Wang, T.-S.; Yin, F.-Z.; Cai, B.-C. J. Ethnopharmacol. 2003, 88, 205.
(b) Li, G.; Zhang, C.; Li, Z. Famous Doctor 2018, 147(in Chinese).
(李国璋, 张超, 李泽, 名医, 2018, 147.)
(c) Li, Y.-F.; Ren, W. Chin. J. Pain Med. 2019, 25, 94(in Chinese).
(李永丰, 任维, 中国疼痛医学杂志, 2019, 25, 94.)
(d) Li, Y.; Zhou, J. Chin. J. Mod. Appl. Pharm. 2019, 36, 2805(in Chinese).
(李阳杰, 周敬, 中国现代应用药学, 2019, 36, 2805.)
(e) Shi, X.; Zhu, M.; Kang, Y.; Yang, T.; Chen, X.; Zhang, Y. Phytomedicine 2018, 241.
(f) Suo, M.; Li, P.; Zhang, M.; Zhu, Y.; Xu, M.; Li, W. China Oncol. 2018, 28, 241(in Chinese).
(索明珠, 李平, 张梅, 朱耀东, 徐梦冉, 李为雨, 中国癌症杂志, 2018, 28, 241.)
(g) Xu, M.; Li, P. Acta Univ. Med. Anhui 2020, 55, 195(in Chinese).
(徐萌, 李平, 安徽医科大学学报, 2020, 55, 195.)
(a) Zhang, M.; Wang, C.; Cai, H.-L.; Wen, J.; Fang, P.-F. Curr. Med. Sci. 2019, 39, 890.
(b) Wu, X.; Ma, F.; Zheng, G. Pharmacol. Clin. Chin. Mater. Med. 2016, 32, 231(in Chinese).
(吴小娟, 马凤森, 郑高利, 中药药理与临床, 2016, 32, 231.)
(c) Li, S.; Wang, X.-P. Int. J. Nanomed. 2017, 12, 5797.
(d) Qin, J.; Yang, L.; Sheng, X.; Sa, Z.; Huang, T.; Li, Q.; Gao, K.; Chen, Q.; Ma, J.; Shen, H. Oncol. Lett. 2018, 15, 6137.
(e) Ma, J.-B.; Qiu, H.-W.; Rui, Q.-H.; Liao, Y.-F.; Chen, Y.-M.; Xu, J.; Zhang, Y.; Zhu, Y.; Zhao, Y.-G. Anal. Chim. Acta 2018, 1020.
(a) Cai, B.-C.; Hattori, M.; Namba, T. Chem. Pharm. Bull. 1990, 38, 1295.
(b) Wang, D.-D.; Li, J.-S.; Cai, B.-C. Chin. Arch. Tradit. Chin. Med. 2009, 27, 435(in Chinese).
(王丹丹, 李俊松, 蔡宝昌, 中华中医药学刊, 2009, 27, 435.)
(c) Chen, X.; Liu, L.; Zhu, W.; Chen, H.; Guan, Y. Jiangxi J. Tradit. Chin. Med. 2018, 49, 63(in Chinese).
(陈谢谢, 刘丽丽, 朱卫丰, 陈丽华, 管咏梅, 江西中医药, 2018, 49, 63.)
(a) Wu, F.; Li, A.; Guo, J. Pharm. Today 2017, 27, 355(in Chinese).
(吴菲, 李阿荣, 郭洁文, 今日药学, 2017, 27, 355.)
(b) Wang, M.; Qi, W. Acta Chin. Med. 2017, 32, 1236(in Chinese).
(王明昭, 齐武强.中医学报, 2017, 32, 1236.)
(c) Nie, Y.; Cao, L.; Xu, Z.; Xin, B. World Latest Medicine Information 2018, 18, 155(in Chinese).
(聂彦彦, 曹璐璐, 徐志龙, 辛波, 世界最新医学信息文摘, 2018, 18, 155.)
(d) Zhang, Y.-J.; Zhang, Z.-P.; Zhai, J.-L.; Tian, L. Guangzhou Chem. Ind. 2019, 47, 121(in Chinese).
(张云静, 张自品, 张雪燕, 翟佳丽, 田蕾, 广州化工, 2019, 47, 121.)
刘守清, 李慧玲, 蒋勉, 李培标, 分析测试学报, 1998, 17, 5. https://www.cnki.com.cn/Article/CJFDTOTAL-KJYJ201507006.htmLiu, S.; Li, H.; Jiang, M.; Li, P. J. Instrum. Anal. 1998, 17, 5(in Chinese). https://www.cnki.com.cn/Article/CJFDTOTAL-KJYJ201507006.htm
(a) Blakemore, D. C.; Castro, L.; Churcher, I.; Rees, D. C.; Thomas, A. W.; Wilson, D. M.; Wood, A. Nat. Chem. 2018, 10, 383.
(b) Nicolaou, K. C.; Rigol, S. Angew. Chem. Int. Ed. 2019, 58, 11206.
(a) Cernak, T.; Dykstra, K. D.; Tyagarajan, S.; Vachal, P.; Krska, S. W. Chem. Soc. Rev. 2017, 46, 1760.
(b) White, M. C.; Zhao, J. J. Am. Chem. Soc. 2018, 140, 13988.
(c) Richardson, J.; Sharman, G.; Martínez-Olid, F.; Cañellas, S.; Gomez, J. E. React. Chem. Eng. 2020, 5, 779.
(d) Feng, K.; Quevedo, R. E.; Kohrt, J. T.; Oderinde, M. S.; Reilly, U.; White, M. C. Nature (London, U. K.) 2020, 580, 621.
(a) Mohsen, A. M. Y.; Mandour, Y. M.; Sarukhanyan, E.; Breitinger, U.; Villmann, C.; Banoub, M. M.; Breitinger, H.-G.; Dandekar, T.; Holzgrabe, U.; Sotriffer, C.; Jensen, A. A. Zlotos, D. P. J. Nat. Prod. 2016, 79, 2997.
(b) Svejstrup, T. D.; Ruffoni, A.; Julia, F.; Aubert, V. M.; Leonori, D. Angew. Chem. Int. Ed. 2017, 56, 14948.
(c) Wang, J.; Li, R.; Dong, Z.; Liu, P.; Dong, G. Nat. Chem. 2018, 10, 866.
(d) Ruffoni, A.; Juliá, F.; Svejstrup, T. D.; McMillan, A. J.; Douglas, J. J.; Leonori, D. Nat. Chem. 2019, 11, 426.
(e) Berger, F.; Plutschack, M. B.; Riegger, J.; Yu, W.; Speicher, S.; Ho, M.; Frank, N.; Ritter, T. Nature (London, U. K.) 2019, 567, 223.
(f) Nishii, Y.; Ikeda, M.; Hayashi, Y.; Kawauchi, S.; Miura, M. J. Am. Chem. Soc. 2020, 142, 1621.
Meyer, A. U.; Slanina, T.; Yao, C.-J.; König, B. ACS Catal. 2016, 6, 369. doi: 10.1021/acscatal.5b02410
(a) Borie, C.; Mondal, S.; Arif, T.; Briand, M.; Lingua, H.; Dumur, F.; Gigmes, D.; Stocker, P.; Barbarat, B.; Robert, V.; Nicoletti, C.; Olive, D.; Maresca, M.; Nechab, M. Eur. J. Med. Chem. 2018, 148, 306.
(b) Cierpiał, T.; Kiełbasiński, P.; Kwiatkowska, M.; Łyzwa, P.; Lubelska, K.; Kuran, D.; Dąbrowska, A.; Kruszewska, H.; Mielczarek, L.; Chilmonczyk, Z.; Wiktorska, K. Bioorg. Chem. 2020, 94, 103454.
(a) Otsuka, S.; Yorimitsu, H.; Osuka, A. Chem.-Eur. J 2015, 21, 14703.
(b) Chen, W.; Hooper, T. N.; Ng, J.; White, A. J. P.; Crimmin, M. R. Angew. Chem. Int. Ed. 2017, 56, 12687.
(c) Shigeno, M.; Okawa, T.; Imamatsu, M.; Nozawa-kumada, K.; Kondo, Y. Chem.-Eur. J. 2019, 25, 10294.
(a) Figueroa-Valverde, L.; Diaz-Cedillo, F.; Garcia-Cervera, E.; Pool-Gomez, E.; Camacho-Luis, A.; Rosas-Nexticapan, M.; Lopez-Ramos, M.; May-Gil, I.; Sarao-Alvarez, A.; Naal-Dzib, C. Asian J. Chem. 2013, 25, 6783.
(b) Figueroa-Valverde, L.; Diaz-Cedillo, F.; Rosas-Nexticapa, M.; Garcia-Cervera, E.; Pool-Gomez, E.; Lopez-Ramos, M.; Hau-Heredia, L.; Sarabia-Alcocer, B. J. Chem. 2014, 757953/1-757953/10.
(c) Figueroa-Valverde, L.; Diaz-Cedillo, F.; Garcia-Cervera, E.; Gomez, E. P.; Rosas-Nexticapa, M.; Lopez-Ramos, M. Asian J. Chem. 2014, 26, 4959.
Fuentes de Arriba, A. L.; Lenci, E.; Sonawane, M.; Formery, O.; Dixon, D. J. Angew. Chem. Int. Ed. 2017, 56, 3655. doi: 10.1002/anie.201612367
(a) Fleming, F. F.; Yao, L.; Ravikumar, P. C.; Funk, L.; Shook, B. C. J. Med. Chem. 2010, 53, 7902.
(b) Klein, B. A.; Robertson, I. M.; Reiz, B.; Kampourakis, T.; Li, L.; Sykes, B. D. ACS Med. Chem. Lett. 2019, 10, 1007.
(c) Lameira, J.; Bonatto, V.; Cianni, L.; dos Reis Rocho, F.; Leitão, A.; Montanari, C. A. Phys. Chem. Chem. Phys. 2019, 21, 24723.
(a) Qi, L.; Hu, K.; Yu, S.; Zhu, J.; Cheng, T.; Wang, X.; Chen, J.; Wu, H. Org. Lett. 2017, 19, 218.
(b) Seo, B.; Kim, Y. G.; Lee, P. H. Org. Lett. 2016, 18, 5050.
(c) Kouznetsov, V. V.; Galvis, C. E. P. Tetrahedron 2018, 74, 773.
Bender, T. A.; Payne, P. R.; Gagné, M. R. Nat. Chem. 2018, 10, 85. doi: 10.1038/nchem.2863
Zlotos, D. P. Buller, S. Holzgrabea, U. Mohr, K. Bioorg. Med. Chem. 2003, 11, 2627. doi: 10.1016/S0968-0896(03)00146-9
蔡宝昌, 何亚维, 张永清, 吴皓, 中国药学杂志, 1994, 29, 169. https://www.cnki.com.cn/Article/CJFDTOTAL-SHXJ201402006.htmCai, B.-C.; He, Y.-W.; Zhang, Y.-Q.; Wu, H. Chin. Pharm. J. 1994, 29, 169(in Chinese). https://www.cnki.com.cn/Article/CJFDTOTAL-SHXJ201402006.htm
Arnone, A.; Metrangolo, P.; Novo, B.; Resnati, G. Tetrahedron 1998, 54, 7831. doi: 10.1016/S0040-4020(98)00417-7
Jousseaume, B.; Chanson, E. Synthesis 1987, 155.
陈坚, 吕平, 周洵钧, 有机化学, 1987, 7, 459. http://sioc-journal.cn/Jwk_yjhx/CN/Y1987/V7/I6/459Chen, J.; Lü, P.; Zhou, X.-J. Chin. J. Org. Chem. 1987, 7, 459(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y1987/V7/I6/459
Oh, K.; Knabe, W. E. Tetrahedron 2009, 65, 2966. doi: 10.1016/j.tet.2009.02.013
Hansen, S. R.; Spangler, J. E.; Hansen, J. H.; Davies, H. M. L. Org. Lett. 2012, 14, 4626. doi: 10.1021/ol3020754
He, J.; Hamann, L. G.; Davies, H. M. L.; Beckwith, R. E. J; Nat. Commun. 2015, 6, 5943. doi: 10.1038/ncomms6943
Brandhofer, T.; Gini, A.; Stockerl, S.; Piekarski, D. G.; Mancheño, O. G. J. Org. Chem. 2019, 84, 12992. doi: 10.1021/acs.joc.9b01765
Ross, S. P.; Hoye, T. R. Nat. Chem. 2017, 9, 523. doi: 10.1038/nchem.2732
Li, J.; Cisar, J. S.; Zhou, C.-Y.; Vera, B.; Williams, H.; Rodríguez, A. D.; Cravatt, B. F.; Romo, D. Nat. Chem. 2013, 5, 510. doi: 10.1038/nchem.1653
Fiori, K. W.; Du Bois, J. J. Am. Chem. Soc. 2007, 129, 562. doi: 10.1021/ja0650450
Dong, Q.; Anderson, C. E.; Ciufolini, M. A. Tetrahedron Lett. 1995, 36, 5681. doi: 10.1016/00404-0399(50)1122X-
Maestre, L.; Dorel, R.; Pablo, O.; Escofet, I.; Sameera, W. M. C.; Álvarez, E.; Maseras, F.; Diaz-Requejo, M. M.; Echavarren, A. M.; Pérez, P. J. J. Am. Chem. Soc. 2017, 139, 2216. doi: 10.1021/jacs.6b08219
(a) Gharagozloo, P.; Lazareno, S.; Popham, A.; Birdsall, N. J. M. J. Med. Chem. 1999, 42, 438.
(b) Birdsall, N. J. M.; Farries, T.; Gharagozloo, P.; Kobayashi, S.; Lazareno, S.; Sugimoto, M. Mol. Pharmacol. 1999, 55, 778.
(a) Král, V.; Pataridis, S.; Setnička, V.; Záruba, K.; Urbanová, M.; Volka, K. Tetrahedron 2005, 61, 5499.
(b) Kejík, Z.; Záruba, K.; Michalík, D.; Šebek, J.; Dian, J.; Pataridis, S.; Volka, K.; Král, V. Chem. Commun. (Cambridge, U. K.) 2006, 1533.
(c) Rezanka, P.; Záruba, K.; Král, V. Tetrahedron Lett. 2008, 49, 6448.
(d) Záruba, K.; Králová, J.; Řezanka, P.; Poučková, P.; Veverková, L.; Král, V. Org. Biomol. Chem. 2010, 8, 3202.
(a) Kim, H. Y.; Shi, H.-J.; Knabe, W. E.; Oh, K. Angew. Chem. Int. Ed. 2009, 48, 7420.
(b) Kim, H. Y.; Oh, K. Org. Lett. 2009, 11, 5682.
(c) Kim, H. Y.; Kim, S.; Oh, K. Angew. Chem. Int. Ed. 2010, 49, 4476.
Van Rheenen, V.; Kelly, R. C.; Cha, D. Y. Tetrahedron Lett. 1976, 1973.
Karimov, R. R.; Sharma, A.; Hartwig, J. F. ACS Cent. Sci. 2016, 2, 715. doi: 10.1021/acscentsci.6b00214
(a) Eisenberger, P.; Gischig, S.; Togni, A. Chem.-Eur. J. 2006, 12, 2579.
(b) Parsons, A. T.; Buchwald, S. L. Angew. Chem. Int. Ed. 2011, 50, 9120.
(c) Wang, X.; Ye, Y.; Zhang, S.; Feng, J.; Xu, Y.; Zhang, Y.; Wang, J. J. Am. Chem. Soc. 2011, 133, 16410.
(d) Mizuta, S.; Galicia-López, O.; Engle, K. M.; Verhoog, S.; Wheelhouse, K.; Rassias, G.; Gouverneur, V. Chem.-Eur. J. 2012, 18, 8583.
(e) Shimizu, R.; Egami, H.; Hamashima, Y.; Sodeoka, M. Angew. Chem. Int. Ed. 2012, 51, 4577.
(f) Wang, F.; Qi, X.; Liang, Z.; Chen, P.; Liu, G. Angew. Chem. Int. Ed. 2014, 53, 1881.
Lichosyt, D.; Zhang, Y.; Hurej, K.; Dydio, P. Nat. Catal. 2019, 2, 114. doi: 10.1038/s41929-018-0207-1
Wren, H.; Williams, H. J. Chem. Soc., Trans. 1916, 109, 572. doi: 10.1039/CT9160900572
Abderhalden, E.; Faust, W.; Haase, E. Z. Physiol. Chem. 1934, 228, 187. doi: 10.1515/bchm2.1934.228.3-6.187
Toki, K. Bull. Chem. Soc. Jpn. 1958, 31, 333. doi: 10.1246/bcsj.31.333
(a) Toda, F.; Tanaka, K.; Ueda, H. Tetrahedron Lett. 1981, 22, 4669.
(b) Toda, F.; Tanaka, K.; Mori, K. Chem. Lett. 1983, 827.
Tanner, D. D.; Ruo, T. C. S.; Meintzer, C. P. J. Org. Chem. 1985, 50, 2573. doi: 10.1021/jo00214a032
Jaen, J. C. e-EROS Encycl. Reagents Org. Synth. 2001, doi. org/10. 1002/047084289X. rb334. doi: 10.1002/047084289X.rb334
(a) Hagishita, S.; Kuriyama, K.; Hayashi, M.; Nakano, Y.; Shingu, K.; Nakagawa, M. Bull. Chem. Soc. Jpn. 1971, 44, 496.
(b) Warr, R. J.; Willis, A. C.; Wild, S. B. Inorg. Chem. 2008, 47, 9351.
(a) Yoshida, S.; Kasai, M.; Kimura, T.; Akiba, T.; Takahashi, T.; Sakamoto, S. Org. Process Res. Dev. 2012, 16, 654.
(b) Moritomo, A.; Yamada, H.; Matsuzawa-Nomura, T.; Watanabe, T.; Itahana, H.; Oku, M.; Akuzawa, S.; Okada, M. Bioorg. Med. Chem. 2014, 22, 6026.
(a) Holzwarth, R.; Bartsch, R.; Cherkaoui, Z.; Solladié, G. Chem.-Eur. J. 2004, 10, 3931.
(b) Holzwarth, R.; Bartsch, R.; Cherkaoui, Z.; Solladié, G. Eur. J. Org. Chem. 2005, 3536.
(c) Tsunoda, Y.; Fukuta, K.; Imamura, T.; Sekiya, R.; Furuyama, T.; Kobayashi, N.; Haino, T. Angew. Chem. Int. Ed. 2014, 53, 7243.
(a) Polavarapu, P. L.; Petrovic, A. G.; Vick, S. E.; Wulff, W. D.; Ren, H.; Ding, Z.; Staples, R. J. Org. Chem. 2009, 74, 5451.
(b) Yang, Z. Univ. Chem. 2020, 35, 185(in Chinese).
(杨占会, 大学化学, 2020, 35, 185)
Záruba, K.; Král, V. Tetrahedron:Asymmetry 2002, 13, 2567. doi: 10.1016/S0957-4166(02)00715-2
(a) Tanaka, K.; Oda, S.; Nishihote, S.; Hirayama, D.; Urbanczyk-Lipkowska, Z. Tetrahedron: Asymmetry 2009, 20, 2612.
(b) Sundar, M. S.; Bedekar, A. V. RSC Adv. 2016, 6, 46258.
Röehrich, T.; Abu Thaher, B.; Manicone, N.; Otto, H.-H. Monatsh. Chem. 2004, 135, 979.
Piwowarczyk, K.; Zawadzka, A.; Roszkowski, P.; Szawkalo, J.; Leniewski, A.; Maurin, J. K.; Kranz, D.; Czarnocki, Z. Tetrahedron:Asymmetry 2008, 19, 309. doi: 10.1016/j.tetasy.2008.01.014
Doyle, M. P.; Morgan, J. P.; Fettinger, J. C.; Zavalij, P. Y.; Colyer, J. T.; Timmons, D. J.; Carducci, M. D. J. Org. Chem. 2005, 70, 5291. doi: 10.1021/jo050609o
White, J. D.; Shaw, S. Org. Lett. 2011, 13, 2488. doi: 10.1021/ol2007378
Carlier, P. R.; Zhang, Y. Org. Lett. 2007, 9, 1319. doi: 10.1021/ol070149g
Mo, F.; Dong, G. Science (Washington, DC, U. S.) 2014, 345, 68. doi: 10.1126/science.1254465
Chen, J.; Kilpatrick, B.; Oliver, A. G.; Wulff, J. E. J. Org. Chem. 2015, 80, 8979. doi: 10.1021/acs.joc.5b01332
Chen, J.; Sun, X.; Oliver, A. G.; Wulff, J. E. Can. J. Chem. 2017, 95, 234. doi: 10.1139/cjc-2016-0125
Zhu, J.; Yuan, Y.; Wang, S.; Yao, Z.-J. ACS Omega 2017, 2, 4665. doi: 10.1021/acsomega.7b00749
Kenyon, J. Org. Synth. 1926, 6, 68. doi: 10.15227/orgsyn.006.0068
Hartmann, R. W.; Batzl, C.; Pongratz, T. M.; Mannschreck, A. J. Am. Chem. Soc. 1992, 35, 2210.
(a) Poupaert, J. H.; Cavalier, R.; Claesen, M. H.; Dumont, P. A. J. Med. Chem. 1975, 18, 1268.
(b) Riedner, J.; Vogel, P. Tetrahedron: Asymmetry 2004, 15, 2657.
Laursen, J. B.; Jorgensen, C. G.; Nielsen, J. Bioorg. Med. Chem. 2003, 11, 723. doi: 10.1016/S0968-0896(02)00472-8
Ashcroft, C. P.; Challenger, S.; Clifford, D.; Derrick, A. M.; Hajikarimian, Y.; Slucock, K.; Silk, T. V.; Thomson, N. M.; Williams, J. R. Org. Process Res. Dev. 2005, 9, 663. doi: 10.1021/op050102f
Dung, P. T.; Trung, T. Q.; Kim, K. H. Arch. Pharmacal Res. 2009, 32, 1425. doi: 10.1007/s12272-009-2012-5
Kuo, L. Y.; Glazier, S. K. Inorg. Chem. 2012, 51, 328. doi: 10.1021/ic2016897
(a) Movassaghi, M.; Piizzi, G.; Siegel, D. S.; Piersanti, G. Angew. Chem. Int. Ed. 2006, 45, 5859.
(b) Hazin, K.; Patrick, B. O.; Gates, D. P. Inorg. Chem. 2019, 58, 188.
(a) Matsumoto, K.; Uchida, T. Chem. Lett. 1981, 1673.
(b) Ikemoto, T.; Nagata, T.; Yamano, M.; Ito, T.; Mizuno, Y.; Tomimatsu, K. Tetrahedron Lett. 2004, 45, 7757.
Kano, T.; Ohyabu, Y.; Saito, S.; Yamamoto, H. J. Am. Chem. Soc. 2002, 124, 5365. doi: 10.1021/ja012287l
(a) Marckwald, W. Ber. 1904, 37, 349.
(b) Toussaint, O.; Capdevielle, P.; Maumy, M. Tetrahedron Lett. 1987, 28, 539.
(a) Drabowicz, J.; Legędź, S.; Mikołajczyk, M. J. Chem. Soc., Chem. Commun. 1985, 23, 1670.
(b) Drabowicz, J.; Legędź, S.; Mikołajczyk, M. Tetrahedron 1988, 44, 5243.
Spek, A. L.; Voorbergen, P.; Schat, G.; Blomberg. C.; Bickelhaupt, F. J. Organomet. Chem. 1974, 77, 147. doi: 10.1016/S0022-328X(00)81313-3
(a) Kolesińska, B.; Kamiński, Z. J. Org. Lett. 2009, 11, 765.
(b) Kolesińska, B.; Kasperowicz, K.; Sochacki, M.; Mazur, A.; Jankowski, S.; Kamiński, Z. J. Tetrahedron Lett. 2010, 51, 20.
Kinoshita, H.; Ihoriya, A.; Ju-Ichi, M.; Kimachi, T. Synlett 2010, 2330.
Nicolaou, K. C.; Liu, G.; Beabout, K.; McCurry, M. D.; Shamoo, Y. J. Am. Chem. Soc. 2017, 139, 3736. doi: 10.1021/jacs.6b12654

图式 1 马钱子中药炮制过程中的反应
Scheme 1 Reaction involved in the traditional processing of Strychnos nux-vomica L.

图 2 Figueroa-Valverde课题组使用的含伯胺官能团的化合物
Figure 2 Primary amine-containing compounds used by Figueroa-Valverde's group

图式 10 铑(Ⅱ)催化下马钱子碱和重氮化合物的反应
Scheme 10 Rhodium(Ⅱ)-catalyzed reactions of brucine with diazo compounds

图式 11 铜(Ⅱ)催化下马钱子碱和异腈的反应
Scheme 11 Copper(Ⅱ)-catalyzed reaction of brucine with isonitrile

图 3 马钱子碱与苯炔中间体反应的产物结构
Figure 3 Structures of the reaction product of brucine with benzyne intermediate

图 4 与氮叶立德中间体36发生反应的酚类及产物
Figure 4 Phenols applied in the reactions with aminimides intermediates 36

图式 16 10% Cd/Pb催化脱除烷氧基磺酰基保护基
Scheme 16 10% Cd/Pb-catalyzed removal of alkoxysulfonyl protecting groups

图 5 马钱子碱季铵衍生物的变构作用
Figure 5 Allosteric interactions of brucine quaternary ammonium derivatives

图式 19 双马钱子碱季铵盐62的合成
Scheme 19 Synthesis of bisquaternary ammonium dimers of brucine 62

图式 23 马钱子碱双羟基化衍生物和苄溴的反应
Scheme 23 Reaction of dihydroxylated derivatives of brucine with benzyl bromide

图式 27 马钱子碱衍生物75羟基的β-C—H芳基化
Scheme 27 Arylation of the β-C—H bond of the hydroxyl group in brucine derivative 75

图 6 常用于手性拆分的马钱子属和金鸡纳树属生物碱
Figure 6 Application of strychnos and cinchona alkaloids in chiral resolution

图式 29 由螺旋手性羧酸合成六齿配体
Scheme 29 Synthesis of hexadentate ligand from spiro chiral carboxylic acid

图式 31 向列相液晶手性掺杂剂92的合成
Scheme 31 Synthesis of chiral dopants 92 for nematic liquid crystals

图式 32 手性膦酸93的拆分及衍生化
Scheme 32 Resolution of chiral phosphoric acid 93 and its derivatization

图式 38 2-苯基环丙基甲酸的拆分及衍生化
Scheme 38 Chiral resolution and derivatization of trans-2- phenylcyclopropane-1-carboxylic acid

图式 49 神经毒素手性硫代膦酸酯159的合成
Scheme 49 Synthesis of chiral phosphonothioate 159 as neurotoxin

图式 51 3, 5-二甲基苯酚的不对称双官能团化
Scheme 51 Asymmetric bifunctionalization of 3, 5-dimethyl- phenol

图式 52 不同金属离子和马钱子碱衍生物的络合形式
Scheme 52 Complexing forms of different metal ions and brucine derivatives

图式 53 化合物170和烯丙酸叔丁酯(171)的不对称环加成反应
Scheme 53 Asymmetric cycloaddition of compound 170 with tert-butyl acrylate (171)

图式 54 硝基甲烷与醛174的不对称加成反应
Scheme 54 Asymmetric addition reaction of nitromethane with aldehyde 174

图式 55 吲哚类化合物与硝基烯烃的对映选择性加成
Scheme 55 Enantioselective addition of indoles and nitroalkenes

图式 56 马钱子碱作用下的不对称去羰基化反应
Scheme 56 Asymmetric decarbonylation under the action of brucine

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
































 下载:
下载:










































