Scheme 1.
Stereoselective addition reactions of alkynes with O-containing nucleophiles
Silver-Catalyzed Stereoselective Addition of Organic Hypervalent Iodine(III) Reagents to Alkynes
Xinyu Chen , Xueyan Liu , Fang Ma , Xianfang Hong , Hongji Li

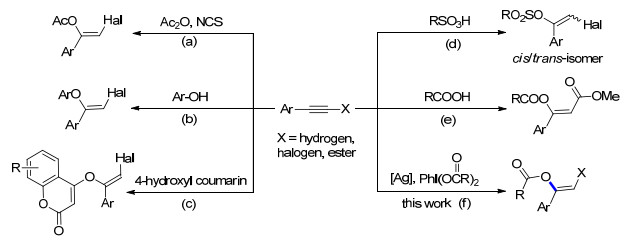
The functionalization of alkynes represents an extremely powerful tool for the construction of chemical bonds, which has widespread application in organic synthesis.[1] Alkynes usually are used as feedstock to afford a series of valuable commodity chemicals, such as acetaldehydes, ketones, vinyl esters and some unique compounds that are not easy to obtain by conventional synthetic methods.[2] Particularly, in the case of functionalized alkynes, a number of vinyl esters can be prepared from the addition reaction of carbon-carbon triple bond, in which the pursuit of good selectivity remains a significant challenge. In recent years, considerable effort has been devoted to explore the versatile additions of alkynes toward achieving high selectivity.[3] For example, Jiang's group[4] has realized the difunctionalization of terminal alkynes by silver catalysis, generating a variety of 1-halo-1-alkenes in good yields and selectivity (Scheme 1a). Hou and coworkers[5] have established the halogenation of internal alkynes using quaternary ammonium salt as mediators. Then it is found that both phenols and 4-hydroxyl coumarins are effective O-containing nucleophiles, which can enable the stereoselective addition of alkynes (Schemes 1b and 1c).[6] Very recently, Xu et al.[7] reported an efficient addition of sulfonic acids to haloalkynes, in which the reactivity of sulfonic acids can be tuned by varying the hydrogen bonding cluster (Scheme 1, d). Moreover, it should be noted that the direct addition of carboxyl acid to propiolates also proceeds well under palladium catalysis (Scheme 1, e).[8] Although the reported strategies have been well developed for the alkyne addition, most of which suffer from poor selectivity or critical reaction conditions. Hence, the development of an efficient and alternative methodology for the alkyne addition with good selectivity is highly desirable.
Organic hypervalent iodine reagents (HIRs) have gained increasing interest as coupling partners, oxidants and both of them in organic synthesis, mainly due to their selective, versatile and environment-benign properties.[9] Several typical organoiodine(III) reagents have been successfully employed for the construction of organic molecules.[9-10] Among them, [bis(acyloxy)iodo]arenes (DIB) and [hydroxy(tosyloxy)iodo]benzene (HTIB, Koser's reagent), belong to one such reagent that has been frequently explored in organic transformations.[10] For example, we recently have utilized the organoiodine(III) reagents to realize the selective C(sp3)—H acetoxylation of 2H-aziri-dines[11c] and radical cyclization of unsaturated oximes, [11d] respectively. During the course of the investigation, it was found that the cleavage of I—O bond counld be facilitated by iron(II/III) salts. We then postulated the possible cleavage model of I—O bond mediated by other transition metals, such as silver salts. In this contribution, we will report an example of silver-catalyzed I—O cleavage within HIRs derived from DIB and the corresponding addition to functionalized alkynes (Scheme 1f).
Based on previous work, [11] we commenced this study with the reaction of (bromoethynyl)benzene 1a with hypervalent iodine reagent 2a to evaluate the feasibility of the process, and the results are summarized in Table 1. Initially, Ag2O was selected as a catalyst for the catalytic system.[6b] The model reaction proceeded slightly in CH3CN at 90 ℃ for 6 h, and the desired product 3a was generated in 21% isolated yield (Entry 1). It shoud be noted that the NOSEY spectra of 3a further supported the observed stereoslectivity. Other silver salts, such as AgOAc, AgOTf and AgNO3 were then tried for the addition reaction of 1a, but failed to give the product 3a (Entries 2~4). Gratifyingly, it was found that Ag2CO3 exhibited high efficiency in this reaction system, affording 3a in 86% yield (Entry 5). Instead of Ag2CO3, the use of some other catalysts including CuO and Cu2O, Fe(acac)2, Fe(acac)3 and Pd(OAc)2 did not promote this addition process (Entries 6~10). No product 3a was isolated in absence of Ag2CO3 (Entry 11). Following, it was found that the reaction of 1a with 2a was considerably affected by the reaction medium. For instance, using ethanol or acetic acid as a solvent did not remarkably accelerate the addition process, and only 15% and 22% yields were achieved under the above conditions, respectively (Entries 12 and 13). Moreover, some other polar solvents were also examined in the model reaction, and resulted into inferior yields (Entries 13~17). The reaction did proceed in toluene (Entry 18). Finally, the evaluation of reaction time, temperature and catalyst loading gave the optimal reaction conditions for the addition reaction of 1a with 2a. The experiment results indicated that longer reaction time and higher temperature would lead to partial decomposition of 3a with lower isolated yield (Entries 19~24).
 下载:
导出CSV
下载:
导出CSV
 |
|||
| Entry | Catalyst | Solvent | Yieldb/% |
| 1 | Ag2O | CH3CN | 21 |
| 2 | AgOAc | CH3CN | 0 |
| 3 | AgOTf | CH3CN | 0 |
| 4 | AgNO3 | CH3CN | 0 |
| 5 | Ag2CO3 | CH3CN | 86 |
| 6 | CuO | CH3CN | 0 |
| 7 | Cu2O | CH3CN | Trace |
| 8 | Fe(acac)2 | CH3CN | 0 |
| 9 | Fe(acac)3 | CH3CN | 0 |
| 10 | Pd(OAc)2 | CH3CN | 0 |
| 11 | — | CH3CN | 0 |
| 12 | Ag2CO3 | EtOH | 15 |
| 13 | Ag2CO3 | AcOH | 22 |
| 14 | Ag2CO3 | THF | Trace |
| 15 | Ag2CO3 | DMF | n.r. |
| 16 | Ag2CO3 | DMSO | 17 |
| 17 | Ag2CO3 | 1, 4-Dioxane | Trace |
| 18 | Ag2CO3 | PhMe | 0 |
| 19 | Ag2CO3 | CH3CN | 65c |
| 20 | Ag2CO3 | CH3CN | 83d |
| 21 | Ag2CO3 | CH3CN | 72e |
| 22 | Ag2CO3 | CH3CN | 58f |
| 23 | Ag2CO3 | CH3CN | 78g |
| 24 | Ag2CO3 | CH3CN | 64h |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol) and catalyst (2.5 mol%) in CH3CN (1 mL) at 90 ℃ under ambient air for 6 h.b Isolated yield.cAg2CO3 (1 mol%). dAg2CO3 (5 mol%). e 80 ℃. f100 ℃. g 8 h. h5 h. | |||
With the optimal conditions in hand, we next investigated the applicability of this method across a series of bromoalkynes, and the result is listed in Table 2. The bromoalkynes bearing aryl group could undergo the reaction with 2a to deliver the related products in good yields. Notably, the para-substitued substrates with an electron-donating group such as Me, nPr, nBu, nPent, MeO and EtO, were well tolerated under the standard conditions (3b~3g). The incorporation of F and Br into the para position did not affect the silver-catalyzed system, generating the desired product in 89% (3h) and 86% (3i) yields, respectively. We then found that the bromoalkynes with electron-deficient groups including CN, NO2 and CF3 afforded the products in excellent yields (3j~3l). It was found that 4-(bromoethynyl)-1, 1'-biphenyl could react with 2a to give 76% yield of 3m. The use of meta-substituted bromoalkynes gave the according products in comparable yields, indicating weak steric hindrance exists in this process (3n and 3o). Introducing thienyl group into bromoalkynes still generated the desired products, albeit with moderate yields (3p~3q). Finally, it was found that 1-bromopent-1-yne still reacted with 2a to afford the desired product 3r in 75% isolated yield.
下载:
导出CSV
 |
 |
 |
 |
 |
 |
| a Reaction conditions: 1a (0.2 mmol), 2 (0.2 mmol) and Ag2CO3 (2.5 mol%) in CH3CN (1 mL) at 90 ℃ under ambient air for 6 h. b Isolated yield. |
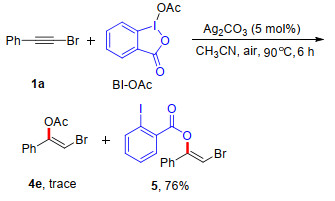
To explore the scope of this method, we next turned our attention to the HIRs, and the result is shown in Table 3. Firstly, a number of PhI(O2CR)2 were readily synthesized through a ligand exchange of DIB with carboxylate acids in boiling toluene. Notably, most of the HIRs derived from aryl carboxylic acid reacted with (bromoethynyl)benzene 1a under optimized conditions to afford the addition products in satisfactory yields (4a~4d). The scope was also expanded to HIRs derived from alkyl carboxylic acid, and which produced the addition products in lower yields (4e~4f). This result can be explained by an intramolecular competition experiment of BI-OAc with bromoalkyne 1a under standard reaction conditions, generating 5 as a major product in 76% yield (Scheme 2). It was found that replacing the bromide with Cl or I within alkynes still gave the corresponding products in acceptable yields (4g and 4h).
下载:
导出CSV
 |
 |
 |
 |
| a Reaction conditions: 1 (0.2 mmol), 2 (0.2 mmol) and Ag2CO3 (2.5 mol%) in CH3CN (1 mL) at 90 ℃ under ambient air for 6 h. bIsolated yield. c24 h. d10 h. |
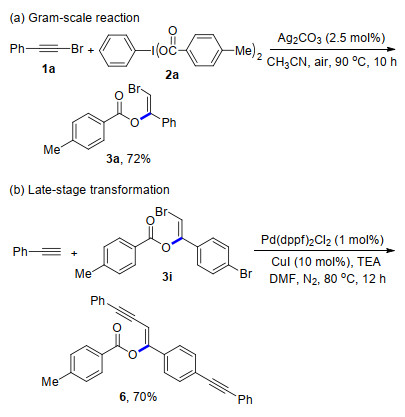
Furthermore, methyl 3-phenylpropiolate was employed to react with HIRs under the modified reaction conditions, and which delivered the vinyl esters in excellent yields (4i~4l). Then, a gram-scale reaction of 1a with 2a was performed under standard conditions, which generated 3a in 72% yield (Scheme 3a). Finally, a transformation of 3i with 1-phenylethyne by Sonogashira reaction was realized, and produced the product 6 in 70% yield (Scheme 3b).
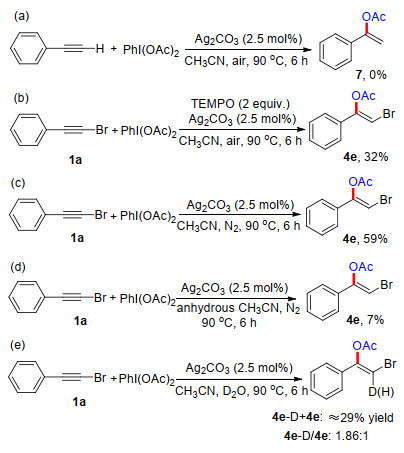
To reveal the reaction mechanism, several control experiments were subsequently designed and performed under given conditions (Scheme 4). It was found that the reaction of 1-phenylethyne with DIB under air condition did not give the desired product 7, which demonstrated the critical role of halogens (Scheme 4a).[12] The addition of a radical quencher, 2, 2, 6, 6-tetramethyl-1-piperidinyloxy (TEMPO), into the reaction of 1a with DIB did not completely inhibit the process under air atmosphere, generating 4e in 32% yield, which indicated a radical pathway was not involved therein (Scheme 4b). The model reaction was carried out in CH3CN at 90 ℃ under a N2 atmosphere for 6 h, and generated 4e in 59% yield, showing the presence of oxygen did not affect the addition event (Scheme 4c). Further, it was found that the selected reaction almost did not proceed in anhydrous CH3CN (Scheme 4, d). We then treated this reaction with heavy water, and a 1.86:1 ratio of (vinylic) D/H was observed by the 1H NMR analysis (Scheme 4e). The above result demonstrates that the vinylic hydrogen comes from H2O and a protonation step also takes part in this catalytic system.
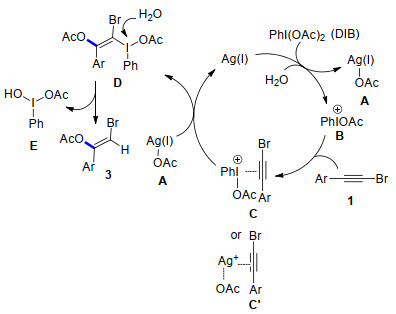
Based on the above results and previous report, [4-7] a possible reaction mechanism for the silver-catalyzed stereoselective addition reaction of functionalized alkynes with DIB is depicted in Scheme 5. Initially, in the presence of H2O, DIB interacts with silver(I) via a I—O bond cleavage to generate species A and cationic iodine intermediate B.[12] Then bromoalkyne 1 is activated by weak coordination with A or B to afford the complex C or C', [4, 6b, 13] followed by a nucleophilic attack of OAc- on the carbon- carbon triple bond to give intermediate D.[13] Finally, the hydrolysis of D releases a hypervalent iodine compound E and desired product 3.
In summary, a silver-catalyzed stereoselective addition reactions of functionalized alkynes with hypervalent iodine(III) reagents via I—O bond cleavage has been developed, which provides an efficient access to vinyl esters in high yields. The control experiment supports that the presence of aryl group considerably affects the cleavage model of I—O bond within hypervalent iodine(III) reagents. The further exploration of this strategy using HIRs and transition metal catalysis in synthetic chemistry and mechanistic studies is underway in our laboratory.
All 1H NMR and 13C NMR spectra were recorded on 400, 600 MHz Bruker FT-NMR spectrometers (400 MHz and 100 MHz, 600 MHz and 150 MHz, respectively). All chemical shifts are given with reference to tetramethylsilane (TMS) as an internal standard. High resolution mass spectroscopy data of the products were collected on a Thermo Fisher Scientific LTQ FTICR-MS instrument.
The starting materials, such as (bromoethynyl)benzene and hypervalent iodine(III) reagents, are prepared according to the reported methods, respectively.[1-2] All chemical reagents used in this work were purchased from commercial suppliers, Huawei Ruike Chemical company, Energy Chemical company, or Shanghai Chemical Company of China and Aldrich of USA. All solvents were dried and freshly distilled prior to use. Products were purified by flash chromatography on silica gels with petroleum ether/ethyl acetate (V : V=10:1 to 20:1) as eluent.
A 10 mL oven-dried reaction tube equipped with a magnetic stirrer bar was charged with alkyne 1 (0.20 mmol), HIRs 2 (0.20 mmol), silver carbonate (2.5 mol%), and freshly distilled CH3CN (1.0 mL) under air atmosphere. The reaction tube was placed in an oil bath and stirred at 90 ℃ for 6 h. After the reaction was completed, the mixture was cooled to room temperature, detected by thin-layer chromatography (TLC), and extracted with ethyl acetate (5 mL×3). The organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to yield the crude product, which was further purified by flash chromatography [silica gel, V (petroleum ether): V (ethyl acetate)=10:1 to 20:1], affording the desired product 3.
(Z)-2-Bromo-1-phenylvinyl 4-methylbenzoate (3a): Yellow liquid, 51.8 mg, 82% yield. 1H NMR (600 MHz, CDCl3) δ: 8.13 (d, J=8.0 Hz, 2H), 7.49~7.48 (m, 2H), 7.36~7.32 (m, 5H), 6.66 (s, 1H), 2.47 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 163.17, 150.65, 144.81, 133.41, 130.40, 129.39, 129.34, 128.78, 125.96, 124.92, 96.65, 21.77; HRMS (ESI) calcd for C16H13BrNaO2 [M+Na]+ 338.9991, found 338.9991.
(Z)-2-Bromo-1-(p-tolyl)vinyl 4-methylbenzoate (3b): Pale yellow solid, 52.7 mg, 80% yield. m.p. 46.8~47.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.13 (t, J=8.4 Hz, 2H), 7.36 (dd, J=16.0, 8.0 Hz, 4H), 7.16 (d, J=8.0 Hz, 2H), 6.60 (s, 1H), 2.47 (s, 3H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.18, 150.69, 144.72, 139.46, 130.62, 130.37, 129.45, 129.36, 126.02, 124.82, 95.61, 21.75, 21.25; HRMS (ESI) calcd for C17H14BrNaO2 [M+Na]+ 353.0148, found 353.0150.
(Z)-2-Bromo-1-(4-propylphenyl)vinyl 4-methylbenzoate (3c): Pale yellow solid, 60.2 mg, 84% yield. m.p. 98.7~100.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.13 (d, J=8.0 Hz, 2H), 7.38 (d, J=8.0 Hz, 2H), 7.32 (d, J=8.0 Hz, 2H), 7.15 (d, J=8.0 Hz, 2H), 6.61 (s, 1H), 2.59~2.55 (m, 2H), 2.47 (s, 3H), 1.65~1.59 (m, 2H), 0.93 (t, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.19, 150.71, 144.72, 144.24, 130.82, 130.39, 129.36, 128.86, 126.04, 124.82, 95.66, 37.75, 24.25, 21.75, 13.73; HRMS (ESI) calcd for C19H19BrNaO2 [M+Na]+ 381.0461, found 381.0460.
(Z)-2-Bromo-1-(4-butylphenyl)vinyl 4-methylbenzoate (3d): Pale yellow solid, 61.8 mg, 83% yield. m.p. 89.2~91.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.13 (d, J=8.4 Hz, 2H), 7.39 (d, J=8.0 Hz, 2H), 7.33 (d, J=8.0 Hz, 2H), 7.16 (d, J=8.0 Hz, 2H), 6.61 (s, 1H), 2.59 (t, J=7.6 Hz, 2H), 2.46 (s, 3H), 1.58 (t, J=8.0 Hz, 2H), 1.34 (dd, J=15.2, 7.6 Hz, 2H), 0.92 (t, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.19, 150.73, 144.72, 144.48, 130.79, 130.39, 129.36, 128.82, 126.06, 124.84, 95.65, 35.37, 33.30, 22.27, 21.76, 13.87; HRMS (ESI) calcd for C20H21- BrNaO2 [M+Na]+ 395.0617, found 395.0617.
(Z)-2-Bromo-1-(4-pentylphenyl)vinyl 4-methylbenzoate (3e): Pale yellow solid, 65.6 mg, 85% yield. m.p. 101.4~103.2 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.14 (d, J=8.0 Hz, 2H), 7.39 (d, J=8.0 Hz, 2H), 7.34 (d, J=8.0 Hz, 2H), 7.16 (d, J=8.0 Hz, 2H), 6.61 (s, 1H), 2.59 (t, J=7.6 Hz, 2H), 2.47 (s, 3H), 1.61~1.58 (m, 2H), 1.36~1.30 (m, 4H), 0.90 (t, J=6.9 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 163.25, 150.78, 144.77, 144.56, 130.84, 130.45, 129.42, 128.87, 126.12, 124.90, 95.71, 35.71, 31.45, 30.90, 22.53, 21.81, 14.02; HRMS (ESI) calcd for C21H23BrNaO2 [M+Na]+ 409.0774, found 409.0774.
(Z)-2-Bromo-1-(4-methoxyphenyl)vinyl 4-methyl-benzoate (3f): Pale yellow solid, 57.5 mg, 83% yield. m.p. 101.6~102.6 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.13 (d, J=8.0 Hz, 2H), 7.42 (d, J=8.0 Hz, 2H), 7.33 (d, J=7.6 Hz, 2H), 6.87 (d, J=7.6 Hz, 2H), 6.51 (s, 1H), 3.80 (s, 3H); 2.46 (s, 3H); 13C NMR (100 MHz, CDCl3) δ:164.45, 161.73, 151.68, 145.97, 131.63, 130.62, 127.68, 127.34, 115.44, 95.72, 56.56, 23.00; HRMS (ESI) calcd for C17H16BrNaO3 [M+Na]+ 369.0097, found 369.0097.
(Z)-2-Bromo-1-(4-ethoxyphenyl)vinyl 4-methylbenzoate (3g): Pale yellow solid, 61.9 mg, 86% yield. m.p. 102.7~103.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.13 (d, J=8.0 Hz, 2H), 7.40 (d, J=8.8 Hz, 2H), 7.33 (d, J=8.0 Hz, 2H), 6.85 (d, J=8.8 Hz, 2H), 6.51 (s, 1H), 4.02 (dd, J=13.0, 8.4 Hz, 2H), 2.46 (s, 3H), 1.40 (t, J=6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.19, 159.85, 150.45, 144.70, 130.38, 129.36, 126.39, 126.08, 125.86, 114.67, 94.35, 63.53, 21.76, 14.70; HRMS (ESI) calcd for C18H17- BrNaO3 [M+Na]+ 383.0253, found 383.0253.
(Z)-2-Bromo-1-(4-fluorophenyl)vinyl 4-methylbenzoate (3h): Pale yellow solid, 59.5 mg, 89% yield. m.p. 76.2~77.5 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.12 (d, J=8.0 Hz, 2H), 7.48~7.45 (m, 2H), 7.33 (d, J=7.8 Hz, 2H), 7.04 (t, J=9.0 Hz, 2H), 6.59 (s, 1H), 2.46 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 163.25 (d, J=249.9 Hz), 162.42, 149.81, 144.95, 130.40, 129.75 (d, J=3.2 Hz), 129.43, 126.94 (d, J=8.1 Hz), 125.80, 115.89 (d, J=21.8 Hz), 96.38, 21.77; 19F NMR (600 MHz, CDCl3) δ: -111.2; HRMS (ESI) calcd for C16H12BrFNaO2 [M+ Na]+ 356.9897, found 356.9899.
(Z)-2-Bromo-1-(4-bromophenyl)vinyl 4-methylbenzoate (3i): Pale yellow solid, 67.7 mg, 86% yield. m.p. 83.3~84.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.11 (d, J=8.0 Hz, 2H), 7.48 (d, J=8.8 Hz, 2H), 7.34 (dd, J=8.5, 2.5 Hz, 4H), 6.67 (s, 1H), 2.47 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.08, 149.81, 144.99, 132.45, 131.98, 130.40, 129.44, 126.44, 125.73, 123.53, 97.33, 21.77; HRMS (ESI) calcd for C16H12Br2NaO2 [M+Na]+ 416.9096, found 416.9097.
(Z)-2-Bromo-1-(4-cyanophenyl)vinyl 4-methylbenzoate (3j): Pale yellow solid, 61.4 mg, 90% yield. m.p. 156.3~157.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.11 (d, J=8.0 Hz, 2H), 7.64 (d, J=8.4 Hz, 2H), 7.57 (d, J=8.4 Hz, 2H), 7.34 (d, J=8.0 Hz, 2H), 6.84 (s, 1H), 2.47 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.02, 149.14, 145.30, 137.62, 132.62, 130.44, 129.53, 125.38, 118.24, 112.80, 100.42, 21.78; HRMS (ESI) calcd for C17H12BrNNaO2 [M+Na]+ 363.9944, found 363.9942.
(Z)-2-Bromo-1-(4-nitrophenyl)vinyl 4-methylbenzoate (3k): Pale yellow solid, 66.4 mg, 92% yield. m.p. 148.0~149.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.21 (d, J=8.8 Hz, 2H), 8.12 (d, J=8.4 Hz, 2H), 7.64 (d, J=9.2 Hz, 2H), 7.35 (d, J=8.0 Hz, 2H), 6.91 (s, 1H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.00, 148.91, 147.92, 145.35, 139.34, 130.43, 129.54, 125.58, 125.30, 124.12, 101.10, 21.78; HRMS (ESI) calcd for C16H12BrNNaO4 [M+Na]+ 383.9842, found 383.9842.
(Z)-2-Bromo-1-(4-(trifluoromethyl)phenyl)vinyl 4-methylbenzoate (3l): Pale yellow solid, 66.8 mg, 87% yield. m.p. 75.3~77.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.12 (d, J=8.0 Hz, 2H), 7.63~7.57 (m, 4H), 7.35 (d, J=8.0 Hz, 2H), 6.79 (s, 1H), 2.47 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 163.11, 149.53, 145.17, 136.83, 131.12 (q, J=32.4 Hz), 130.78, 130.44, 129.50, 123.79 (q, J=270.4 Hz), 125.85 (q, J=3.0 Hz), 125.188, 99.15, 21.79; 19F NMR (600 MHz, CDCl3) δ: -62.8; HRMS (ESI) calcd for C17H13BrF3NaO2 [M+Na]+ 406.9865, found 406.9865.
(Z)-1-([1, 1'-Biphenyl]-4-yl)-2-bromovinyl 4-methyl-benzoate (3m): Pale yellow solid, 59.9 mg, 76% yield. m.p. 123.3~124.6 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.16 (d, J=8.4 Hz, 2H), 7.60~7.54 (m, 6H), 7.44 (t, J=7.2 Hz, 2H), 7.36 (t, J=8.8 Hz, 3H), 6.72 (s, 1H), 2.48 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.24, 150.42, 144.86, 142.16, 140.18, 132.26, 130.44, 129.43, 128.84, 127.68, 127.49, 127.00, 125.96, 125.31, 96.62, 21.78; HRMS (ESI) calcd for C22H17BrNaO2 [M+Na]+ 415.0304, found 415.0306.
(Z)-2-Bromo-1-(m-tolyl)vinyl 4-methylbenzoate (3n): Pale yellow solid, 52.1 mg, 79% yield. m.p. 45.8~47.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.18 (d, J=8.4 Hz, 2H), 7.38 (d, J=8.0 Hz, 2H), 7.33 (d, J=6.4 Hz, 2H), 7.29 (d, J=7.6 Hz, 1H), 7.21 (d, J=7.2 Hz, 1H), 6.68 (s, 1H), 2.51 (s, 3H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.19, 150.79, 144.75, 138.47, 133.36, 130.39, 130.18, 129.37, 128.66, 126.01, 125.53, 122.09, 96.38, 21.74, 21.39; HRMS (ESI) calcd for C17H15BrNaO2 [M+Na]+ 353.0148, found 353.0148.
(Z)-2-Bromo-1-(3-fluorophenyl)vinyl 4-methylbenzoate (3o): pale yellow solid, 54.1 mg, 81% yield. m.p. 78.0~79.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.13 (d, J=8.0 Hz, 2H), 7.42 (td, J=7.7, 1.6 Hz, 1H), 7.36~7.29 (m, 3H), 7.14~7.09 (m, 2H), 6.91 (s, 1H), 2.46 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.08, 159.48 (d, J=250.3 Hz), 145.07 (d, J=4.3 Hz), 144.86, 130.58 (d, J=8.7 Hz), 130.40, 129.39, 127.72 (d, J=1.7 Hz), 125.80, 124.36 (d, J=3.5 Hz), 121.26 (d, J=11.4 Hz), 116.39 (d, J=22.5 Hz), 101.81 (d, J=13.7 Hz), 21.75; 19F NMR (600 MHz, CDCl3) δ: -111.9; HRMS (ESI) calcd for C16H12Br- FNaO2 [M+Na]+ 356.9897, found 356.9897.
(Z)-2-Bromo-1-(thiophen-2-yl)vinyl 4-methylbenzoate (3p): Pale yellow solid, 43.8 mg, 68% yield. m.p. 86.1~87.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.12 (d, J=8.4 Hz, 2H), 7.33 (d, J=8.0 Hz, 2H), 7.28 (dd, J=4.8, 0.8 Hz, 1H), 7.15 (dd, J=4.0, 0.8 Hz, 1H), 6.98 (dd, J=5.2, 4.0 Hz, 1H), 6.59 (s, 1H), 2.46 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 162.87, 145.65, 144.91, 136.56, 130.44, 129.39, 127.57, 126.36, 125.74, 125.15, 95.51, 21.77; HRMS (ESI) calcd for C14H11Br NaO2S [M+Na]+ 344.9555, found 344.9556.
(Z)-2-Bromo-1-(thiophen-3-yl)vinyl 4-methylbenzoate (3q): Pale yellow solid, 39.3 mg, 61% yield. m.p. 88.6~89.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.12 (d, J=8.4 Hz, 2H), 7.34~7.31 (m, 4H), 7.19 (dd, J=4.8, 1.2 Hz, 1H), 6.61 (s, 1H), 2.47 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.03, 146.88, 144.85, 135.01, 130.39, 129.39, 126.93, 125.87, 124.20, 122.36, 96.27, 21.76; HRMS (ESI) calcd for C14H11BrNaO2S [M+Na]+ 344.9555, found 344.9555.
(Z)-1-Bromohept-1-en-2-yl 4-methylbenzoate (3r): Pale yellow liquid, 46.5 mg, 75% yield. 1H NMR (600 MHz, CDCl3) δ: 8.03 (d, J=8.4 Hz, 2H), 7.29 (d, J=7.8 Hz, 2H), 5.91 (s, 1H), 2.44 (s, 3H), 2.42~2.39 (m, 2H), 1.53~1.51 (m, 2H), 1.33~1.31 (m, 4H), 0.90~0.87 (m, 3H); 13C NMR (150 MHz, CDCl3) δ: 163.19, 153.27, 144.48, 130.20, 129.26, 126.31, 93.70, 33.66, 31.09, 25.85, 22.30, 21.73, 13.91; HRMS (ESI) calcd for C15H19BrNaO2 [M+Na]+ 333.0461, found 333.0463.
For the synthesis of compound 4, see the general procedure for compound 3.
(Z)-2-Bromo-1-phenylvinyl benzoate (4a):[14] Pale yellow solid, 44.7 mg, 74% yield. m.p. 45.8~47.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.25 (dd, J=8.0, 0.8 Hz, 2H), 7.67 (t, J=7.6 Hz, 1H), 7.54 (t, J=7.8 Hz, 2H), 7.51~7.48 (m, 2H), 7.38~7.36 (m, 3H), 6.68 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 163.10, 150.63, 133.86, 133.31, 130.33, 129.39, 128.79, 128.74, 128.67, 124.92, 96.71.
(Z)-2-Bromo-1-phenylvinyl 4-(tert-butyl)benzoate (4b): White solid, 58.0 mg, 81% yield. m.p. 82.4~83.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.17 (d, J=8.8 Hz, 2H), 7.55 (d, J=8.4 Hz, 2H), 7.50~7.47 (m, 2H), 7.36~7.33 (m, 3H), 6.66 (s, 1H), 1.38 (s, 9H); 13C NMR (100 MHz, CDCl3) δ: 163.09, 157.76, 150.68, 133.46, 130.29, 129.35, 128.78, 125.95, 125.70, 124.95, 96.64, 35.24, 31.09; HRMS (ESI) calcd for C19H19BrNaO2 [M+Na]+ 381.0461, found 381.0461.
(Z)-2-Bromo-1-phenylvinyl 3-chlorobenzoate (4c): Pale yellow solid, 51.7 mg, 77% yield. m.p. 79.9~80.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.23 (t, J=1.6 Hz, 1H), 8.13 (dt, J=8.0 Hz, 1.2 Hz, 1H), 7.66~7.63 (m, 1H), 7.50~7.46 (m, 3H), 7.37 (dd, J=6.0, 2.4 Hz, 3H), 6.69 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 161.91, 150.47, 134.87, 133.90, 133.01, 130.44, 130.26, 130.01, 129.51, 128.83, 128.42, 124.89, 96.89; HRMS (ESI) calcd for C15H10Br- ClNaO2 [M+Na]+ 358.9445, found 358.9445.
(Z)-2-Bromo-1-phenylvinyl 2-methylbenzoate (4d): Pale yellow solid, 50.5 mg, 80% yield. m.p. 47.8~48.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.29 (d, J=8.0 Hz, 1H), 7.54~7.50 (m, 3H), 7.38~7.33 (m, 5H), 6.67 (s, 1H), 2.68 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.50, 150.68, 141.66, 133.51, 132.97, 131.98, 131.33, 129.34, 128.80, 127.75, 125.96, 124.92, 96.63, 21.86; HRMS (ESI) calcd for C16H13BrNaO2 [M+Na]+ 338.9991, found 338.9990.
(Z)-2-Bromo-1-phenylvinyl acetate (4e)[4a]: Yellow liquid, 25.4 mg, 53% yield. 1H NMR (400 MHz, CDCl3) δ: 7.44~7.40 (m, 2H), 7.38~7.35 (m, 3H), 6.55 (s, 1H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 167.23, 150.50, 133.29, 129.39, 128.77, 124.91, 96.55, 20.57.
(Z)-2-Bromo-1-(p-tolyl)vinyl heptanoate (4f): Yellow liquid, 23.9 mg, 37% yield. 1H NMR (400 MHz, CDCl3) δ: 7.32 (d, J=8.8 Hz, 2H), 6.85 (d, J=8.8 Hz, 2H), 6.39 (s, 1H), 4.03 (q, J=7.2 Hz, 2H), 2.59 (t, J=7.6 Hz, 2H), 1.78 (q, J=7.2 Hz, 2H), 1.44~1.34 (m, 7H), 0.93 (t, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 170.11, 159.83, 150.28, 126.37, 125.89, 114.62, 94.17, 63.53, 33.99, 31.25, 24.52, 22.27, 14.70, 13.87; HRMS (ESI) calcd for C16H21BrNaO2 [M+Na]+ 347.0617, found 347.0616.
(Z)-2-Chloro-1-phenylvinyl acetate (4g):[4a] Pale yellow liquid, 21.5 mg, 55% yield. 1H NMR (600 MHz, CDCl3) δ: 7.39~7.36 (m, 5H), 6.45 (s, 1H), 2.34 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 167.33, 148.41, 132.76, 129.35, 128.76, 124.70, 107.98, 20.46.
(Z)-2-Iodo-1-phenylvinyl acetate (4h):[4a] Pale yellow liquid, 35.7 mg, 62% yield. 1H NMR (600 MHz, CDCl3) δ: 7.42 (s, 2H), 7.35 (s, 3H), 6.63 (s, 1H), 2.35 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 167.33, 155.04, 133.66, 129.39, 128.73, 125.15, 68.28, 20.98.
(Z)-Methyl-3-acetoxy-3-phenylacrylate (4i): Pale yellow liquid, 41.8 mg, 95% yield. 1H NMR (600 MHz, CDCl3) δ: 7.59 (d, J=7.8 Hz, 2H), 7.44 (d, J=6.6 Hz, 1H), 7.41 (t, J=7.2 Hz, 2H), 6.28 (s, 1H), 3.74 (s, 3H), 2.40 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 168.12, 164.60, 158.31, 133.20, 131.06, 128.80, 125.90, 105.57, 51.49, 20.92; HRMS (ESI) calcd for C12H13O4 [M+H]+ 221.0808, found 221.0808.
(Z)-3-Methoxy-3-oxo-1-phenylprop-1-en-1-yl butyrate (4j): Pale yellow liquid, 45.6 mg, 92% yield. 1H NMR (600 MHz, CDCl3) δ: 7.58 (d, J=7.2 Hz, 2H), 7.45~7.39 (m, 3H), 6.28 (s, 1H), 3.74 (s, 3H), 2.68 (t, J=7.8 Hz, 2H), 1.86~1.80 (m, 2H), 1.06 (t, J=7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 170.64, 164.56, 158.29, 133.46, 130.95, 128.77, 125.89, 105.73, 51.43, 36.01, 18.04, 13.70; HRMS (ESI) calcd for C14H17O4 [M+H]+ 249.1121, found 249.1120.
(Z)-3-Methoxy-3-oxo-1-phenylprop-1-en-1-yl 3-chloro-benzoate (4k): White solid, 60.7 mg, 96% yield. m.p. 146.1~147.4 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.74 (s, 1H), 7.73 (d, J=7.8 Hz, 1H), 7.25~7.22 (m, 2H), 7.10~7.00 (m, 5H), 6.02 (s, 1H), 3.30 (s, 3H); 13C NMR (150 MHz, CDCl3)δ: 164.29, 162.61, 158.03, 134.73, 133.69, 132.91, 131.15, 130.71, 130.24, 129.95, 128.85, 128.41, 125.86, 106.00, 51.51; HRMS (ESI) calcd for C17H14ClO4 [M+H]+ 317.0575, found 317.0576.
(Z)-3-Methoxy-3-oxo-1-phenylprop-1-en-1-yl 4-(tert-butyl)benzoate (4l): White solid, 60.8 mg, 90% yield. m.p. 149.1~149.8 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.18 (d, J=7.8 Hz, 2H), 7.66 (d, J=7.8 Hz, 2H), 7.55 (d, J=7.8 Hz, 2H), 7.43 (d, J=6.6 Hz, 1H), 7.40 (t, J=7.8 Hz, 2H), 6.41 (s, 1H), 3.68 (s, 3H), 1.37 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 164.46, 163.80, 158.38, 157.44, 133.41, 130.97, 130.24, 128.79, 126.17, 125.95, 125.68, 106.04, 51.49, 35.16, 31.06; HRMS (ESI) calcd for C21H23O4 [M+H]+ 339.1591, found 339.1590.
A 10 mL oven-dried reaction tube equipped with a magnetic stirrer bar was charged with bromoalkyne 1a (0.20 mmol), BI-OAc (0.20 mmol), silver carbonate (5 mol%) and freshly distilled CH3CN (1.0 mL) under air atmosphere. The reaction tube was placed in an oil bath and stirred at 90 ℃ for 6 h. After the reaction was completed (detected by TLC), the mixture was cooled to room temperature, and extracted with ethyl acetate (5 mL×3). The organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to yield the crude product, which was further purified by flash chromatography [silica gel, V (petroleum ether):V (ethyl acetate)=10:1 to 20:1], affording the desired product (Z)-2-bromo-1-phenylvinyl 2-iodobenzoate (5) as white solid (65.0 mg, 76% yield). m.p. 61.4~62.8 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.26 (dd, J=7.8, 1.8 Hz, 1H), 8.11 (d, J=7.8 Hz, 1H), 7.54~7.52 (m, 3H), 7.41~7.39 (m, 3H), 7.28 (t, J=7.8 Hz, 1H), 6.70 (s, 1H); 13C NMR (150 MHz, CDCl3) δ: 162.40, 150.40, 141.91, 133.59, 132.92, 132.74, 131.85, 129.48, 128.81, 128.08, 124.99, 96.98, 95.11; HRMS (ESI) calcd for C15H10BrINaO2 [M+Na]+ 450.8801, found 450.8801.
A 25 mL oven-dried reaction tube equipped with a magnetic stirrer bar was charged with 1a (5.0 mmol), HIRs 2a (5.00 mmol), silver carbonate (2.5 mol%) and freshly distilled CH3CN (10 mL) under air atmosphere. The reaction tube was placed in an oil bath and stirred at 90 ℃ for 10 h. After the reaction was completed, the mixture was cooled to room temperature, and extracted with ethyl acetate (20 mL×3). The organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to yield the crude product, which was further purified by flash chromatography [silica gel, V (petroleum ether): V (ethyl acetate)=10:1 to 20:1], affording the desired product 3a as pale yellow liquid (1.14 g, 72%).
Under nitrogen atmosphere, a 10 mL oven-dried reaction tube equipped with a magnetic stirrer bar was charged with 3i (0.40 mmol), 1-phenylethyne (0.84 mmol), Pd(dppf)2Cl2 (1 mol %), CuI (10 mol%), triethylamine (2.0 mol) and anhydrous N, N-dimethylformamide (DMF, 1.0 mL). The reaction tube was placed in an oil bath and stirred at 80 ℃ for 12 h. After the reaction was completed, the mixture was cooled to room temperature, and extracted with ethyl acetate (5 mL×3). The organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to yield the crude product, which was further purified by flash chromatography [silica gel, V (petroleum ether): V (ethyl acetate)=9:1], affording the desired product (Z)-4-phenyl-1-(4-(phenyl- ethynyl)phenyl)but-1-en-3-yn-1-yl 4-methylbenzoate (6) as white solid (122.7 mg, 70%). m.p. 185.1~186.3 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.19 (d, J=7.8 Hz, 2H), 7.56~7.51 (m, 6H), 7.36~7.34 (m, 5H), 7.24~7.14 (m, 6H), 6.30 (s, 1H), 2.49 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 163.93, 155.42, 144.70, 133.22, 131.91, 131.80, 131.66, 131.41, 130.45, 129.42, 128.48, 128.37, 128.32, 128.16, 126.31, 124.49, 123.04, 123.00, 98.61, 98.04, 91.37, 89.05, 84.39, 21.80; HRMS (ESI) calcd for C32H23O2 [M+H]+ 439.1693, found 439.1693.
A 10 mL oven-dried reaction tube equipped with a magnetic stirrer bar was charged with 1a (0.20 mmol), (diacetoxyiodo)benzene (DIB, 0.20 mmol), silver carbonate (2.5 mol%), TEMPO (0.40 mmol) and freshly distilled CH3CN (1.0 mL) under air atmosphere. The reaction tube was placed in an oil bath and stirred at 90 ℃ for 6 h. After that, the reaction mixture was cooled to room temperature, and extracted with ethyl acetate (5 mL×3). The organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to yield the crude product, which was further purified by flash chromatography [silica gel, V (petroleum ether): V (ethyl acetate)=10:1 to 20:1], affording the desired product 4e in 32% yield. Therefore, a radical pathway can be ruled out for this silver-catalyzed system.
A 10 mL oven-dried reaction tube equipped with a magnetic stirrer bar was charged with 1a (0.20 mmol), (diacetoxyiodo)benzene (DIB, 0.20 mmol), silver carbonate (2.5 mol%), D2O (0.20 mmol) and freshly distilled CH3CN (1.0 mL) under N2 atmosphere. The reaction tube was placed in an oil bath and stirred at 90 ℃ for 6 h. After the reaction was completed, the mixture was cooled to room temperature, and extracted with ethyl acetate (5 mL×3). The organic layers were combined, dried over anhydrous MgSO4, and concentrated under reduced pressure to yield the crude product, which was further purified by flash chromatography [silica gel, V (petroleum ether): V (ethyl acetate)=10:1 to 20:1], affording the desired product 4e/[D]-4e (ca. 29% total yield). The ratio of mixture ([D]-4e/4e=1.86) was determined by 1H NMR analysis of the obtained mixture, which indicates the pronation step was involved therein.
Supporting Information 1H NMR and 13C NMR spectra of new compounds 3~6. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Haines, A. H. Methods for the Oxidation of Organic Compounds. Alkanes, Alkenes, Alkynes, and Arenes, Academic Press, New York, 1985.
(b) Meldal, M.; Tornoe, C. W. Chem. Rev. 2008, 108, 2952.
(c) Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302.
(d) Gilmore, K.; Alabugin, I. V. Chem. Rev. 2011, 111, 6513.
(a) Wu, W.; Jiang, H. Acc. Chem. Res. 2014, 47, 2483.
(b) Fang, G.; Bi, X. Chem. Soc. Rev. 2015, 44, 8124.
(c) Zhang, F.; Peng, X.; Ma, J. Chin. J. Org. Chem. 2019, 39, 109(in Chinese). (张发光, 彭星, 马军安, 有机化学, 2019, 39, 109.)
(d) Cheng, Z.; Guo, J.; Lu, Z. Chem.Commun.2020, 56, 2229.
(e) Ackermann, L. Acc. Chem. Res. 2020, 53, 84.
(a) Al-awar, R. S.; Joseph, S. P.; Comins, D. L. J. Org. Chem. 1993, 58, 7732.
(b) Kamei, K.; Maeda, N.; Tatsuoka, T. Tetrahedron Lett. 2005, 46, 229.
(c) Spaggiari, A.; Vaccari, D.; Davoli, P.; Torre, G.; Prati, F. J. Org. Chem. 2007, 72, 2216.
(d) Su, W.; Jin, C. Org. Lett. 2007, 9, 993.
(e) Iwai, T.; Fujihara, T.; Terao, J.; Tsuji, Y. J. Am. Chem. Soc. 2012, 134, 1268.
(f) Wu, X.-F.; Bezier, D.; Darcel, C. Adv. Synth. Catal. 2009, 351, 367.
(a) Chen, Z.; Li, J.; Jiang, H.; Zhu, S.; Li, Y.; Qi, C. Org. Lett. 2010, 12, 3262.
(b) Chen, Z.; Jiang, H.; Li, Y.; Qi, C. Chem. Commun. 2010, 46, 8049.
(c) Jiang, G.; Zhu, C.; Li, J.; Wu, W.; Jiang, H. Adv. Synth. Catal. 2017, 359, 1208.
Mo, D.-L.; Dai, L.-X.; Hou, X.-L. Tetrahedron Lett. 2009, 50, 5578. doi: 10.1016/j.tetlet.2009.07.081
(a) Wang, S.; Li, P.; Yu, L.; Wang, L. Org. Lett. 2011, 13, 5968.
(b) Hong, X.; Ma, F.; Zha, D.; Li, H. Asian J. Org. Chem. 2018, 7, 2552.
Zeng, X.; Liu, S.; Shi, Z.; Xu, B. Org. Lett. 2016, 18, 4770. doi: 10.1021/acs.orglett.6b02061
Lu, X.; Zhu, G.; Ma, S. Tetrahedron Lett. 1992, 33, 7205. doi: 10.1016/S0040-4039(00)60873-0
(a) Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299.
(b) Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650.
(c) Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328.
(d) Duan, Y.; Jiang, S.; Han, Y.; Sun, B.; Zhang, C. Chin. J. Org. Chem. 2016, 36, 1973(in Chinese).
(段亚南, 姜山, 韩永超, 孙博, 张弛, 有机化学, 2016, 36, 1973.)
(e) Zhang, H.; Tang, R.; Shi, X.; Xie, L.; Wu, J. Chin. J. Org. Chem. 2019, 39, 1837(in Chinese).
(张怀远, 唐蓉萍, 石星丽, 颉林, 伍家卫, 有机化学, 2019, 39, 1837.)
(f) Wang, X.; Studer, A. Acc. Chem. Res. 2017, 50, 1712. For recent examples, see: (g) Chen, Q.; Yang, Y.; Wang, X.; Zhang, Q.; Li, D. Chin. J. Org. Chem. 2020, 40, 454(in Chinese).
(陈倩雯, 杨耀成, 王霞, 张谦, 李栋, 有机化学, 2020, 40, 454.)
(h) Gao, P.; Fan, M.; Bai, Z.; Wei, Y. Chin. J. Chem. 2015, 33, 479.
(a) Frei, R.; Wodrich, M. D.; Hari, D.; Borin, P.; Chauvier, C.; Waser, J. J. Am. Chem. Soc. 2014, 136, 16563.
(b) Zhang, J.; Szabo, K, J.; Himo, F. ACS Catal. 2017, 7, 1093.
(c) Wang, S.; Gu, Q.; You, S. J. Org. Chem. 2017, 82, 11829.
(d) Xia, H.-D.; Zhang, Y.-D.; Wang, Y.-H.; Zhang, C. Org. Lett. 2018, 20, 4052.
(e) Li, J.; Liu, Z.; Wu, S.; Chen, Y. Org. Lett. 2019, 21, 2077.
(f) Li, G.-X.; Hu, X.; He, G.; Chen, G. Chem. Sci. 2019, 10, 688.
(g) Lan, T.; Zhang, Y.; Liu, W.; Xi, C.; Chen, C. Chin. J. Org Chem. 2019, 39, 2166(in Chinese).
(兰天磊, 张越, 刘伟, 席婵娟, 陈超, 有机化学, 2019, 39, 2166.)
(h) Liu, Q.-R.; Pan, C.-X.; Ma, X.-P.; Mo, D.-L.; Su, G.-F. J. Org. Chem. 2015, 80, 6496.
(i) Ma, X.-P.; Li, K.; Wu, S.-Y.; Liang, C.; Su, G.-F.; Mo, D.-L. Green Chem. 2017, 19, 5761.
(a) Tan, H.; Li, H.; Ji, W.; Wang, L. Angew. Chem., Int. Ed. 2015, 54, 8374.
(b) Zha, D.; Li, H.; Li, S.; Wang, L. Adv. Synth. Catal. 2017, 359, 467.
(c) Wang, L.; Li, H.; Wang, L. Org. Lett. 2018, 20, 1663.
(d) Yang, S.; Li, H.; Li, P.; Yang, J.; Wang, L. Org. Biomol. Chem 2020, 18, 715.
Deng's group reported a reaction of terminal alkynes with DIB using AgOAc as a catalyst, but which only forms α-acetoxy ketones as sole products. Under Deng's conditions, this stereoselective addition reaction of functionalized alkynes with hypervalent iodine(Ⅲ) reagents does not give the desired product (Entry 2, Table 1). Based on these experiment results, a different pathway for this silver-catalyzed addition is proposed in Scheme 5.
(a) Yamada, W.; Sugawara, Y.; Cheng, H. M.; Ikeno, T.; Yamada, T. Eur. J. Org. Chem. 2007, 16, 2604.
(b) Deng, G.; Luo, J. Tetrahedron 2013, 69, 5937.
Jiang, G.; Li, J. X.; Zhu, C.; Wu, W.; Jiang, H. Org. Lett. 2017, 19, 4440. doi: 10.1021/acs.orglett.7b01919
Table 1. Optimization of reaction conditionsa
|
|||
| Entry | Catalyst | Solvent | Yieldb/% |
| 1 | Ag2O | CH3CN | 21 |
| 2 | AgOAc | CH3CN | 0 |
| 3 | AgOTf | CH3CN | 0 |
| 4 | AgNO3 | CH3CN | 0 |
| 5 | Ag2CO3 | CH3CN | 86 |
| 6 | CuO | CH3CN | 0 |
| 7 | Cu2O | CH3CN | Trace |
| 8 | Fe(acac)2 | CH3CN | 0 |
| 9 | Fe(acac)3 | CH3CN | 0 |
| 10 | Pd(OAc)2 | CH3CN | 0 |
| 11 | — | CH3CN | 0 |
| 12 | Ag2CO3 | EtOH | 15 |
| 13 | Ag2CO3 | AcOH | 22 |
| 14 | Ag2CO3 | THF | Trace |
| 15 | Ag2CO3 | DMF | n.r. |
| 16 | Ag2CO3 | DMSO | 17 |
| 17 | Ag2CO3 | 1, 4-Dioxane | Trace |
| 18 | Ag2CO3 | PhMe | 0 |
| 19 | Ag2CO3 | CH3CN | 65c |
| 20 | Ag2CO3 | CH3CN | 83d |
| 21 | Ag2CO3 | CH3CN | 72e |
| 22 | Ag2CO3 | CH3CN | 58f |
| 23 | Ag2CO3 | CH3CN | 78g |
| 24 | Ag2CO3 | CH3CN | 64h |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol) and catalyst (2.5 mol%) in CH3CN (1 mL) at 90 ℃ under ambient air for 6 h.b Isolated yield.cAg2CO3 (1 mol%). dAg2CO3 (5 mol%). e 80 ℃. f100 ℃. g 8 h. h5 h. | |||
 下载: 导出CSV
下载: 导出CSV
Table 2. Scope of bromoalkynesa
|
|
|
|
|
|
| a Reaction conditions: 1a (0.2 mmol), 2 (0.2 mmol) and Ag2CO3 (2.5 mol%) in CH3CN (1 mL) at 90 ℃ under ambient air for 6 h. b Isolated yield. |
下载: 导出CSV
Table 3. Scope of HIRs and alkynesa
|
|
|
|
| a Reaction conditions: 1 (0.2 mmol), 2 (0.2 mmol) and Ag2CO3 (2.5 mol%) in CH3CN (1 mL) at 90 ℃ under ambient air for 6 h. bIsolated yield. c24 h. d10 h. |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们