引用本文:
张依, 刘育. 多电荷环糊精的超分子组装[J]. 有机化学,
2020, 40(11): 3802-3811.
doi:
10.6023/cjoc202004040

Citation: Zhang Yi, Liu Yu. Supramolecular Assemblies of Multi-Charged Cyclodextrins[J]. Chinese Journal of Organic Chemistry, 2020, 40(11): 3802-3811. doi: 10.6023/cjoc202004040
Citation: Zhang Yi, Liu Yu. Supramolecular Assemblies of Multi-Charged Cyclodextrins[J]. Chinese Journal of Organic Chemistry, 2020, 40(11): 3802-3811. doi: 10.6023/cjoc202004040
多电荷环糊精的超分子组装
English
Supramolecular Assemblies of Multi-Charged Cyclodextrins
Abstract:
As a class of macrocyclic host with excellent water solubility, low biotoxicity and high charge density, multi-charged cyclodextrins have been extensively studied because they can interact with organic/inorganic/biological molecules through multiple synergistic forces such as the hydrophobic cavities and electrostatic interactions to construct the smart supramolecular assemblies with stable topology, functional diversity and stimulus responsiveness. The latest research progress of pH-, photo-, enzyme-, redox-, magnetic-and multi-stimulus responsive smart supramolecular assemblies, which are constructed by typical positively/negatively charged and amphiphilic multi-charged cyclodextrins including their application in the fields of drug delivery, controlled release and sensory detection is introduced, and the challenges and future developments of multi-charged cyclodextrin smart supramolecular assemblies are discussed.
-
环糊精(cyclodextrin, 简称CD)作为超分子化学研究的第二代大环主体化合物, 自从被发现以来就受到科研工作者的持续关注.其本质是由D-吡喃葡萄糖通过α-1, 4-糖苷键环化而成的环状低聚糖, 具有截锥状拓扑结构和“内外”两亲的特性.其中, 常见的α、β、γ-环糊精分别具有6、7、8个吡喃葡萄糖单元.环糊精因其具有良好的水溶性、生物相容性以及廉价易得等特点被广泛应用于日用化工、农业、催化、智能材料和生物医学等领域[1].然而, 较低的溶解度和有限的包结能力限制了天然环糊精的进一步应用.为了进一步提高分子的选择性和键合能力以及功能的多样性, 许多环糊精衍生物被设计和合成出来.众所周知, 环糊精大口端和小口端拥有的众多羟基可被多种功能基团修饰, 功能基团的类型、数目和修饰位置可以配合其空腔衍生出具有不同性能的大环主体.其中, 多电荷环糊精是一类带有多个正电荷或者负电荷基团的典型大环主体分子, 具有诸多优点: (1)带电基团的引入大大改善了环糊精的水溶性和生物相容性, 丰富了其在生物领域的应用[2]; (2)通过静电相互作用力能够显著地提高带相反电荷客体分子的识别能力, 增强分子选择性[3]; (3)修饰后的环糊精衍生物空腔变深, CD部分或者取代基部分与客体之间的相互作用增强, 可以实现超分子纳米组装体的高稳定性和紧密性[4].更重要的是多电荷环糊精的高电荷密度使其成为基于分子诱导聚集策略构筑超分子组装体的最理想主体之一.
智能体系是一类当受到外部信号刺激时, 可以给出特定反馈的新型功能体系, 具体的反馈主要来源于体系内部结构的变化.刺激信号也可能是多种多样的, 例如pH、氧化还原、酶、温度、光、磁力、超声和电压等.环糊精与客体分子的结合主要利用具有动态和可逆性质的非共价键作用力, 提供了一种有效的方式来模仿自然和生理的响应过程.通过在环糊精超分子体系中引入特定的响应基团, 合适地调整外部条件, 将基于CD的纳米体系结构破坏、重组、甚至降级为原始的构建模块, 这样的超分子体系具备智能材料的性能[5].基于多电荷环糊精的智能组装体对外部刺激的响应使主客体组装模式发生变化主要来源于三个方面: (1)主/客体分子上电荷数量或者电性的改变使主客体之间的静电相互作用受影响从而改变组装模式; (2)客体分子性质的变化使其从环糊精的空腔中逸出, 影响了主客体相互作用, 破坏了组装模式; (3)主/客体分子的降解或功能基团的离去导致组装体的解体.近些年来, 科研工作者利用非共价键自组装技术更快、更方便、更灵活地构建了许多结构新颖的功能化超分子组装体, 在纳米超分子领域取得一系列创新性突破, 极大地推进了超分子体系在智能识别与组装、新型生物医药功能材料等领域的应用.
多电荷环糊精智能组装体的设计理念是将智能化学方法赋予缺乏环境响应特性的离子大环受体体系中, 也就是说将刺激反应位点以共价或非共价方式引入CD.因此, 构筑的多电荷环糊精超分子组装体具有pH、光、酶、氧化还原和磁等多种刺激响应性, 从而提高组装体的生物相容性和多功能性,拓展超分子的应用领域.本综述将从典型的正/负电荷及两亲性多电荷环糊精构筑的超分子组装体出发, 根据组装体常见的刺激响应类型介绍多电荷环糊精智能超分子体系的最新进展; 同时, 关注了组装体结构和响应的对应关系以及其在药物传递、控制释放和传感检测领域的应用.
1. 基于多电荷环糊精的超分子组装
由于环糊精的多电荷衍生化赋予环糊精主体更加丰富的性质, 科研工作者会根据不同的研究目的来选择合适的带电基团, 并修饰到环糊精上以获得具有有序结构和独特功能的组装体.在以下部分, 根据组装体主体分子的不同, 选取典型的实例进行简要的讨论与介绍.
1.1 负电荷环糊精
典型的负电荷环糊精是以羧基、磺酸基等作为修饰基团.相比天然环糊精, 这类环糊精负电荷密度比较高, 在分子识别和分子组装方面对正电荷的客体分子具有优异的识别能力和较好诱导聚集能力, 可以通过非共价相互作用构筑功能性超分子组装体.
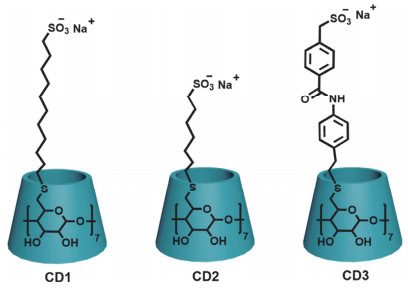
负电荷基团的修饰增强了环糊精与药物分子的相互作用, 早些年, Zhang等[6]发现羧基取代的γ-CD可以通过静电相互作用实现对神经肌肉阻断剂罗库溴铵选择性键合, 使该环糊精被广泛用于手术后去除罗库溴铵. Wenz等[4b]发现6位全磺化环糊精可以与抗癌药喜树碱通过非共价相互作用构筑成稳定的包合物, 大大增加了喜树碱的水溶性.这些研究为负电荷环糊精在生物医药领域的应用奠定了基础.此外, 在最近的研究中, Tapparel团队[7]发现磺化环糊精可以作为广谱抗病毒试剂.如图 1, 作者合成了三种类型的磺化环糊精CD1、CD2和CD3来模拟乙酰肝素硫酸盐用于抗病毒.实验结果表明, 这三种磺化环糊精均具有广谱抗病毒活性, 并揭示了柔性链和刚性链对抗病毒能力的影响.这个工作拓展了多电荷环糊精在抗病毒药物研发领域的应用.
图 1
静电作用的引入使负电荷环糊精在构筑功能性超分子组装体中也表现出优异的性质.我们课题组[8]报道了通过羧酸化环糊精(CCD)诱导含有季铵头基的正电荷两亲客体分子聚集形成的超分子组装体.作者合成了单羧酸取代β-CD、单羧酸取代全甲基化β-CD和全羧酸取代β-CD三种主体化合物, 研究了它们对正电荷客体的聚集诱导行为.其中, 只有全羧基取代β-CD展现出良好的诱导聚集能力, 突出了静电相互作用在分子诱导聚集中的重要作用.此外, 作者还利用这种组装机制实现了对模型药物分子的负载与缓慢释放.该组装体为新型纳米胶囊的构筑提供了理论基础.另一个典型例子是利用磺化环糊精通过静电相互作用诱导低聚对苯撑乙烯衍生物(OPV-I)聚集形成纳米粒子, 再将荧光染料尼罗红(NiR)负载到SCD/OPV-I纳米组装体的疏水层中构筑成新型高效的水相人工光捕获体系(图 2)[9].体系中SCD/OPV-I作为荧光给体, NiR作为荧光受体, 利用超分子组装策略有效地拉近了给体与受体之间的距离, 实现了高效的能量转移和超高的天线效应.利用该策略构筑的光捕获体系为人工模拟自然界的光合作用提供了新思路.
图 2

1.2 正电荷环糊精
典型的正电荷环糊精是以氨基、胍基和咪唑基等作为修饰基团.正电荷基团的引入不仅增强了环糊精与负电荷无机、有机客体的相互作用, 还促进其与生物大分子DNA和RNA相互作用并形成纳米体系.利用正电荷环糊精构筑的功能性纳米组装体是超分子领域的研究热点.
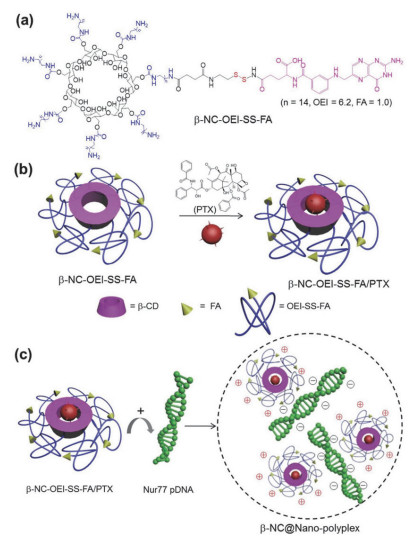
正电荷环糊精可以与负电荷表面活性剂相互作用形成两亲超分子聚集体用作纳米载体[10].另外, 还可以构筑基因载体纳米体系, Fernández等[11]报道了氨基环糊精适用于DNA传递, 实现了高转染效率和低毒性.此外, 有研究将多氨环糊精通过二硫键修饰上叶酸, 利用氨基环糊精对DNA的凝聚作用构筑成了靶向病变细胞的基因转染体系[12].如图 3所示, 通过体系中叶酸的靶向作用将DNA递送到特定肿瘤细胞内, 并利用细胞内的谷胱甘肽切断二硫键释放叶酸, 使叶酸受体循环, 保证了叶酸受体介导的内吞作用的持续性, 从而实现高效的基因表达.该超分子基因递送体系有望用于癌症的基因治疗.
图 3

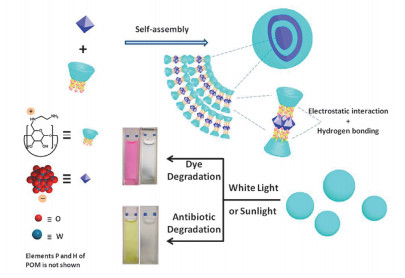
正电荷环糊精不仅可以与生物分子作用用于生物医药方面, 还可以与无机物相互作用构筑有机-无机杂化体系.如图 4所示, 利用全氨基取代的环糊精(EDA-CD)与磷钨酸(POM)相互作用构筑了超分子杂化组装体并用于污染物的光降解[13].研究结果显示在H2O2的存在下, 该组装体可以高效光解多种有机染料和抗生素.作者解释在光作用下, EDA-CD/POM促进H2O2产生的·OH, 而·OH可以通过环糊精空腔转移到溶液中, 在·OH的作用下污染物可以降解成无害物质.这种超分子杂化材料可以大大增加光降解效率.此外, 正电荷环糊精还可以用于构筑杂化凝胶材料.比如, 胍基β-环糊精和聚丙二醇(PPG)链通过非共价相互作用形成超分子准聚轮烷, 将准聚轮烷与锂皂石无机黏土相互作用就构筑了超分子水凝胶[3a].该水凝胶形成的主要驱动力是正电荷胍基与负电荷锂皂石纳米片的静电相互作用.研究结果显示该水凝胶具有自愈合性能和染料负载能力.这种简便构筑有机-无机杂化超分子水凝胶的方法为超分子软材料的实际应用奠定了基础.
图 4

1.3 两亲性环糊精
超分子囊泡一般由两亲性分子组装而成, 所以是一种能同时负载亲水性和疏水性两种活性成分的独特体系, 这种类似于天然生物膜的性质使其被广泛用于物质递送和生物领域[14].在2000年, Ravoo课题组[15]报道了首个完全由两亲环糊精组成的双层纳米囊泡体系, 该囊泡结合了脂质体和大环主体分子的特性, 为开发先进的主客体载体和递送系统奠定了研究基础.在之后的研究中发现, 两亲环糊精囊泡可以与具有多个键合位点的客体分子进行组装.这使得两亲环糊精构筑的软材料在纳米功能材料领域具有举足轻重的地位[16].这里我们主要介绍两亲性多电荷环糊精.
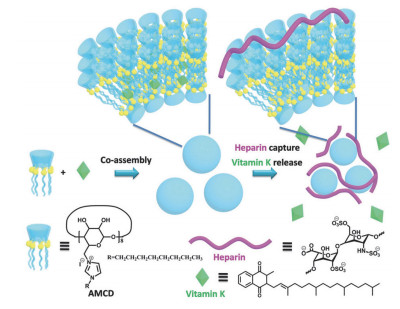
Nishimura等[17]在2002年报道了硫酸盐两亲环糊精形成的具有负电性的囊泡.紧接着, Ravoo课题组[18]报道了氨基两亲环糊精, 这是首次报道的在水溶液中形成具有正电性的双层囊泡, 开启了两亲性环糊精构筑功能超分子组装体的新篇章.得益于囊泡体系的负载能力, 这样的组装体常作为基因传递的非病毒载体[19].同时, 基于两亲性多电荷环糊精的新型功能性组装的不断发展.我们课题组[20]合成了烷基链修饰的多咪唑正电荷两亲性环糊精(AMCD)(图 5), 并构筑了用于抗肝素凝血剂的新型协同治疗超分子组装体.肝素是常用于血液透析手术中的抗凝血剂, 使用时会引起出血并发症和功能性维生素K (VK)缺乏症.肝素的结构决定了其具有负电荷多糖的特点, 可以与正电荷AMCD相互作用.为了测量AMCD和肝素未分级肝素(UFH)的键合常数, 作者利用主客体相互作用构筑了AMCD和8-羟基芘-1, 3, 6-三磺酸三钠盐(HPTS)的络合物, 通过荧光竞争滴定实验得出键合常数为2.4×106 L·mol-1, 也说明该体系可以用作UFH荧光传感的指示剂.活化部分促凝血酶原激酶时间(aPTT)凝血试验证实了AMCD对血浆中肝素的中和能力.在此研究基础上, 作者构筑了AMCD和VK的共组装体, 该组装体可以在中和肝素过程中实现对VK的释放.该研究为临床手术中实现肝素中和与VK补充协同治疗提供了新思路.
图 5

2. 多电荷环糊精的智能体系
多电荷环糊精超分子组装通过引入智能响应组分可衍生出许多具有独特拓扑特征和特定功能的纳米刺激响应结构, 这提供了一种有效的方式来构筑智能体系.超分子智能体系可以模拟多种自然和生理过程的刺激响应结构, 适应微环境变化并且动态响应, 可应用于各个领域.这部分主要根据常用的刺激敏感成分来介绍多电荷环糊精智能超分子组装体, 这些成分的研究可以了解刺激响应过程中组装体中组分的物理化学性质和分子间聚集模式的变化, 有助于精准地设计智能组装体结构.
2.1 pH响应
生物体内pH值在不同部位会有明显的不同, 例如, 正常组织pH为7.4, 肿瘤组织pH≈6.8, 感染和炎症部位pH≈6.5, 而溶酶体中pH为4~5.所以pH刺激响应水解或降解的体系对于选择性地将药物递送到器官、组织甚至亚细胞器至关重要, 可以实现更好的疗效并避免副作用.一般来说, 构筑pH刺激响应的体系是将pH响应性基团(羧基、亚胺基、硼酸基和乙酰基)或者聚合物(聚丙烯酰胺、甲基丙烯酸二乙氨基乙酯)共组装到超分子体系中, 这些基团或聚合物在酸性条件下可被质子化或分解, 从而导致组装体系的解离[21].多电荷环糊精组装体大多是利用静电相互作用和聚集诱导策略构筑而成, 可以方便地将上述基团或者聚合物引入到体系中从而成功构筑智能pH响应组装体.
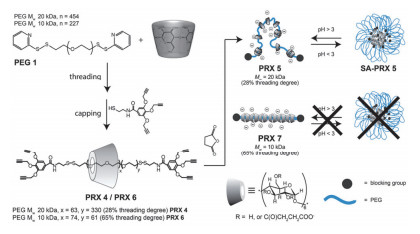
pH变化会影响主体分子的电荷性质的变化来影响组装体的组装模式, 从而实现控制释放.例如, 本课题组[22]构筑了羧酸化环糊精/抗癌药物包合物, 研究了环糊精主体与多种抗癌药物的键合行为.研究结果表明在不同pH条件下, 主体分子对抗癌药键合能力不同, 该体系实现了对药物分子pH响应释放.多羧酸环糊精与抗癌药物选择性键合和控制释放行为使其有望成为抗癌药物的新型传递体系.另外, 羧酸环糊精和季铵盐二茂铁衍生物构筑的pH响应超分子组装体[23], 也是由于主体在酸性条件下会质子化失去电性导致体系解组装从而实现抗癌药盐酸阿霉素(DOX)的有效释放.除此之外, 聚轮烷(PR)这种超分子结构也常用于构筑智能纳米组装体. Caruso等[24]报道了一种pH响应性的负电荷聚轮烷纳米粒子.如图 6, 选择一种由α-CD和聚乙二醇(PEG)构筑的低覆盖度聚轮烷(PRX 5)来确保CD在高分子链上高迁移率, 其中CD被响应性羧酸基团修饰后, 在酸性的条件下PRX5自组装成稳定的纳米粒子, 作者认为纳米颗粒的形成可能来源于CD的羧酸部分和羟基基团之间的分子内和分子间氢键.当pH上升时, 由于各个PRX上带负电荷的羧酸根之间的排斥导致纳米粒子发生解离, 且该响应过程具有一定的重复性.研究还证明了CD的迁移率为关键因素, 因为具有较高覆盖率PRX的酸化不会形成组装体.
图 6

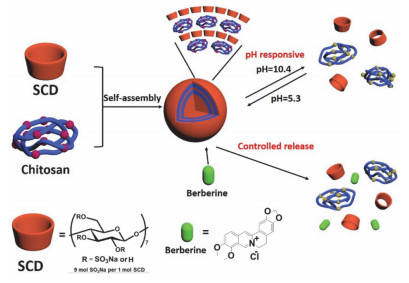
pH变化也会通过影响客体分子的电荷性质使组装体的组装模式发生改变.比如, 磺化环糊精和聚丙烯酰胺(PEI)构筑的pH响应ATP超分子智能载体[25], 由于pH影响PEI的质子化程度, 因此组装体表现出良好的pH响应组装/解组装行为以及对ATP的负载/持续释放能力.对于口服药物一般是希望在胃酸性环境中保持稳定而在肠碱性中释放.这就要求药物载体在不同pH下实现药物的可控释放, 而新型智能纳米粒子是解决这个问题的突破口.基于此, 本课题组[26]报道了磺化环糊精和壳聚糖(CS)构筑的pH响应智能纳米粒子(图 7).研究结果表明球形纳米粒子在室温下稳定存在不会发生解聚.由于构筑模块具有离子结构, pH的变化可以控制超分子组装体的组装与解组装, 当pH从5.3到10.4可以实现纳米粒子的解组装, 当pH调回到5.3时纳米粒子重新组装.在这个研究基础上, 作者利用SCD/CS体系负载了消化道的抑菌药物黄连素(BE), 研究表明当pH值从2(即胃的环境pH)增加到8(即肠的环境pH)时, 负载BE的释放效率明显增加.动物实验进一步证实了该纳米粒子负载药物在经过胃时保持稳定而在肠道中进行释放.该体系有望用于药物分子的精准释放.
图 7

2.2 酶响应
相比于其他刺激响应的智能体系, 酶响应体系具有良好的生物相容性、高度选择性和可控降解的优势, 一直是智能响应体系研究的热点.同时生物体内所有生命活动最终受酶的控制, 也包括特定的疾病.多电荷环糊精因其优异的生物相容性以及空腔特异性结合底物的性质, 常作为构筑酶响应体系的理想主体.在环糊精体系中引入酶响应位点或者引入特定疾病相关酶用于靶向治疗是构建酶响应超分子纳米组装体的主要思路[27].常见酶响应体系的构筑主要利用鱼精蛋白对胰蛋白酶、肉豆蔻酰胆碱和酯键对丁酰胆碱酯酶、透明质酸对透明质酸酶的特异性响应.
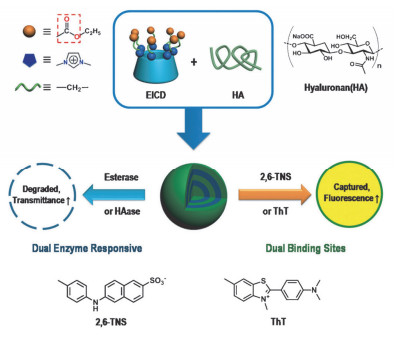
将酶响应酯键引入多电荷环糊精主体分子中是酶响应智能体系的一种构筑方法.如图 8所示, 以具有七个可裂解酯键的咪唑鎓β-CD衍生物(EICD)为主体分子, 通过EICD和透明质酸(HA)静电相互作用构筑了超分子组装体[28].该超分子组装体具有双重酶响应性, 人工羧酸酯对于乙酰胆碱酯酶的响应和透明质酸对透明质酸酶的响应, 并且透明质酸可以定向靶向癌细胞.同时由于EICD空腔可以结合负电荷底物, 而EICD/HA体系可以负载正电荷底物, 该体系具有正电性和负电性底物负载能力.双酶响应和双负载能力的结合使该体系具有靶向癌细胞和高效释放多种药物的能力.
图 8

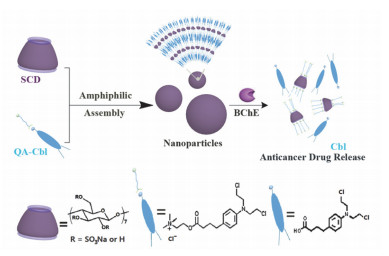
基于客体分子酶刺激响应的超分子组装体常被用于控制释放.我们课题组[29]利用带负电的磺化环糊精(SCD)与鱼精蛋白构筑了生物相容性优异的新型胰蛋白酶响应性的超分子组装体.由于鱼精蛋白可以特异性响应胰蛋白酶, 当向组装体溶液中加入胰蛋白酶可以破坏SCD/鱼精蛋白纳米粒子从而促进负载药物模型分子的释放.该超分子组装体提供了一种酶响应型药物控制释放的智能模型.此外, 羧酸β-CD诱导氯化肉豆蔻酰胆碱聚集形成的组装体[30].主客体通过静电相互作用形成球形纳米粒子, 并且在丁酰胆碱酯酶的作用下纳米粒子能够实现对药物模型分子的控制释放.另一个典型例子是磺化环糊精诱导聚集前药分子胆碱修饰的苯丁酸氮芥(QA-Cbl)形成的组装体[31](如图 9).客体分子可以被丁酰胆碱酯酶特异性切断并释放抗癌药物苯丁酸氮芥(Cbl).这种利用静电相互作用构筑的纳米粒子安全无毒, 而且前药直接作为客体分子大大提高了药物的负载率.在丁酰胆碱酯酶作用下组装体解组装并且释放活性抗癌药物.该高效酶响应的智能超分子组装体利用多电荷大环/前药的组装模式, 为解决癌症治疗中多药耐药性的问题提出新方法.
图 9

2.3 光响应
在多种类型的刺激响应中, 光因其远程可控、非侵入性、环保性和瞬时性等优势赋予光刺激响应体系巨大的应用价值.光响应体系的构筑离不开光敏基团, 如偶氮苯类、香豆素类和蒽等.一直以来, 基于特定光敏基团的光诱导异构化、降解和聚合等性质构筑了各种各样的光响应智能体系[16e, 32].因此, 将光敏基团引入到超分子纳米载体中就可以利用超分子策略实现精确地组装以及特定时间和特定空间的光刺激智能响应.
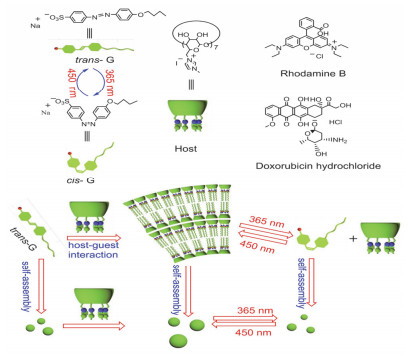
例如, 利用磺化环糊精和两亲性蒽衍生物通过静电作用构筑光响应降解的超分子组装体[33], 该组装体提供的疏水环境有利于蒽环的进一步堆积, 从而在光照条件下产生单线态氧, 加速蒽衍生物的光降解速率.本工作为利用超分子策略构筑多环芳烃的光解体系以及光敏材料体系提供了思路.我们课题组[34]报道了咪唑环糊精(ICD)与磺化偶氮苯衍生物(G)构筑的光响应超分子组装体(图 10).单独客体分子具有两亲性可以自组装成小的纳米粒子, 加入咪唑β-CD后被诱导聚集成较大的纳米粒子.而且, 该超分子组装体可以通过静电相互作用和π-π相互作用稳定地负载染料和药物.更重要的是, 在紫外光照射下, 偶氮苯分子由trans-G转变为cis-G, 导致组装体解体重组, 使纳米粒子体积变小.在可见光照射后, 又可以可逆地恢复到之前的组装模式.同时实验结果表明此纳米粒子光响应可逆变化具有良好的往复性.这种光控组装与解组装可以有效实现组装体中负载物阿霉素的可控释放.所以, 这种光响应的智能超分子组装体有望作为纳米载体应用于药物传递和可控释放, 减少抗癌药物的副作用并提高其生物利用度.
图 10

牛忠伟等[35]通过主体分子环糊精上氨基(NH2)和疏水性聚乳酸(PLA)末端羧基(COOH)之间的静电相互作用构筑了一种两亲超分子组装体.组装体在水中呈现的是以环糊精为壳的核壳结构.所以作者利用CD空腔包结光响应型分子偶氮苯衍生物, 赋予了组装体智能光刺激响应性质.实验结果证实了在紫外光和可见光的交替照射下, 组装体纳米结构的大小可以往复性改变.此外还研究了CD空腔对探针分子8-苯胺-1-萘磺酸盐(ANS)的包结以及组装体对阿霉素的负载和pH响应释放行为.
2.4 氧化还原响应
同时, 也有许多研究致力于探索具有氧化还原敏感性基团的纳米体系.最常用的方法是将还原敏感的二硫键或氧化-还原响应二茂铁基团引入到环糊精体系中, 为开发氧化还原响应纳米体系并应用于基因治疗提供了条件[36].
利用二硫键将靶向剂等功能基团连接到核心CD上形成的主体分子可以用于构筑具有氧化还原响应性的超分子基因载体.比如, 将靶向基团叶酸(FA)通过二硫键连接到多氨基取代的β-CD上作为主体化合物[37].如图 11所示, 通过主客体作用将抗癌药紫杉醇包封入CD腔中, 并与质粒DNA静电相互作用构筑了纳米粒子. FA诱导纳米粒子靶向肿瘤, 二硫键响应细胞内谷胱甘肽, 释放FA分子, 促进叶酸受体的循环, 进一步提高转染效率.结果表明, 该纳米超分子体系对肿瘤生长有明显的抑制作用.此外, 还有一种以CD为核心的星形阳离子聚合物与金刚烷改性离子聚合物通过主客体相互作用形成的多功能基因载体体系[38].体系中, 主体聚合物(β-CD-SS-P)是通过二硫键连接的聚(甲基丙烯酸2-二甲基氨基乙酯)(pDMAEMA)环糊精, 客体聚合物(Ad-pMPC)是用一个金刚烷封端的聚(2-甲基丙烯酰氧基乙基磷酸胆碱)(pMPC). Ad-pMPC赋予体系胞外稳定性和血清耐受性并且可以提高细胞摄取能力.同时, 在GSH存在的情况下, 环糊精上二硫键的还原响应使体系将逐渐解组装, 从而触发负载DNA的释放.该体系可以显著增强基因转染效率.作者认为该组装体优异的基因传递能力可用于癌症的治疗.
图 11

除此之外, 聚轮烷(PR)这种超分子结构也常用于构筑还原响应智能纳米组装体.在之前的报道中就曾研究过由正电荷环糊精构成的聚轮烷, 并评估了其与DNA络合以及体外转染能力[39].比较有代表性的是由含多个正电荷的α-环糊精和含二硫键的聚乙二醇(PEG)组成的可生物裂解的聚轮烷[40].该超分子组装体具有稳定的正电荷, 可作为非病毒基因载体, 而二硫键的引入为其提供了在细胞还原环境中的可降解性.作者研究发现聚轮烷的这种结构有助于增强基因传递效率.一个典型的例子是将由N, N-二甲基氨基乙基(DMAE)基团修饰的α-CD穿入PEG轴上并用二硫键引入封端基团构成正电荷聚轮烷[41], 该聚轮烷具有GSH还原响应性, 可用于siRNA的递送.研究发现聚轮烷上α-CD数量的不同会影响PR/siRNA复合物对聚阴离子竞争的抵抗力.结果表明, 具有52个α-CD的PR可以高度稳定地凝聚siRNA, 并在细胞内GSH的作用下将其释放.因此作者认为该超分子智能组装体可用于siRNA传递与治疗.
2.5 磁响应
除了上面提到几种比较典型的刺激响应类型, 磁响应材料也因其在自然现象和生理活动中的重要作用而受到研究者的广泛关注[42].将多电荷环糊精引入到以磁纳米粒子为核心的磁性材料中, 再利用非共价相互作用赋予磁纳米体系相应的功能, 就可以构筑具有特殊功能并可以高度靶向外部磁场部位的磁响应超分子组装体.磁性氧化铁纳米粒子作为磁性材料最常用的构筑原料有望应用于物质分离和生物治疗领域[43].
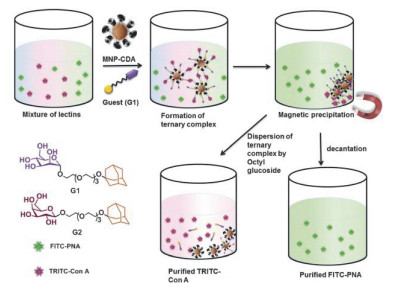
Ravoo课题组[44]报道了一个简单的动态超分子系统, 可以在磁场中选择性地从蛋白质混合物中捕获和分离所需的蛋白质.如图 12, 作者将羧酸化环糊精修饰到磁纳米粒子表面, 利用主客体相互作用将金刚烷糖类衍生物负载到超分子纳米粒子上, 从而功能化磁纳米粒子, 客体分子上的糖类可以特异性地与蛋白质结合.该磁响应纳米粒子为蛋白质的选择性吸附提供了非常大的有效表面积, 并且通过施加外部磁场使纳米粒子与溶液分离而且不会造成蛋白质变性, 实现了对混合蛋白质的高效分离.该研究促进了超分子纳米体系对特定蛋白质的捕获和分离.
图 12

全氟化合物(PFCs)和重金属是新兴污染物, 这些有害物质的清除也受到了极大关注.比如, Suri课题组[43a]报道了一种基于环糊精分子的多功能磁性吸附剂, 用于去除水溶液中的全氟辛烷磺酸(PFOS)、全氟辛酸(PFOA)和Cr(VI).环糊精的引入使纳米粒子可以利用疏水相互作用和静电相互作用吸附PFC和Cr(VI).此外, 该纳米粒子的回收和重复使用可以通过外加磁场进行控制, 重复使用次数高达10次且无任何明显的效率损失.
2.6 多重刺激响应
为了最大限度提高多电荷环糊精智能组装体的功能性和利用率, 研究者考虑将多种刺激集成到环糊精纳米平台体系中, 这样的体系更方便灵活和智能, 应用领域更加广泛, 可表现出协同效应和协同增强作用.
Singh等[45]研究了磺化β-环糊精诱导聚集染料金胺O (AuO)客体分子形成的聚集体.仔细研究了该聚集体的光物理行为, 发现在SCD的高电荷环境下, AuO显示强烈荧光发射和更长的荧光寿命, 并且最大发射红移至560 nm.该智能超分子聚集体对介质的温度和离子强度等外界刺激具有灵敏的响应性.最近他们[46]还报道了硫酸化的β-CD诱导有机荧光探针芘的氨基衍生物(PMA)自聚集的超分子体系.带负电荷的硫酸盐基团对带正电荷探针的电荷中和以及环糊精边缘的紧密接近, 诱导了PMA分子的基态缔合.并且这种单体-二聚体平衡体系对外部化学刺激(例如温度、pH、介质的离子强度和有机溶剂)具有极强的响应能力.研究表明该超分子组装体可检测精氨酸, 有望用于传感检测.
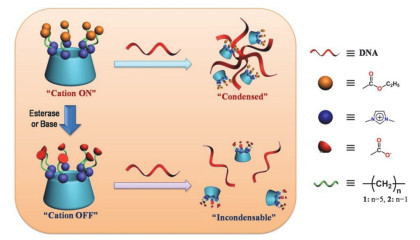
还有一个典型的例子是基于正电荷咪唑β-CD的超分子体系具有可控DNA凝聚能力[47].如图 13所示, 合成了由不同长度疏水链连接的咪唑鎓与羧酸酯作为修饰基团的全取代β-CD, 主体分子的正电性决定了其良好的DNA凝聚能力.研究表明在碱或酶的存在下, 由主体分子的酯水解产生了羧酸根, 此时电性变化导致DNA凝聚能力变弱.而且发现疏水链的长度影响DNA凝聚能力及环糊精/DNA体系的稳定性, 疏水链越长, CD对DNA凝聚能力越弱.随后研究了主体分子的细胞毒性, 结果证明该基因载体具有良好的生物相容性.该体系通过调节基因载体的电荷类型和分布来实现酶和pH双调控DNA的凝聚和释放, 有望作为高效的基因传递载体.
图 13

3. 总结与展望
综上所述, 通过一些典型的例子总结了多电荷环糊精智能组装体最新研究进展.从主体类型和智能组装体多种刺激响应模式的方面来进行介绍, 阐述了多电荷环糊精通过分子诱导聚集构筑超分子纳米结构带来的智能响应性的机理, 尤其是引入多种刺激响应基团之后体现了智能组装体优异的生物相容性、可控的生物降解性、高效的负载能力和精准的底物调节释放功能, 表明了该智能组装体可以应用于药物和基因传递、控制释放、癌症治疗和传感检测等领域.基于CD的智能体系已被经常用于多类型物质传递的纳米平台, 在精准递送领域存在巨大潜力.提高多电荷环糊精智能组装体环境的实时响应性和靶向的精准递送效率, 构建高效的纳米递送平台, 对于更好地拓展其智能应用的领域, 仍然充满了机遇和挑战.
目前主体分子修饰方法单一, 所以多电荷环糊精的类型也比较固定, 导致主体分子的识别能力比较有限, 超分子体系的应用相对局限.下一步应该致力于开发新的修饰方法, 对CD骨架进行区域选择性修饰, 通过修饰基的功能和环糊精的不同空腔尺寸来提高主体对客体分子的选择性键合, 开拓多电荷环糊精在化学、生物以及材料领域的应用.
环糊精中多电荷的引入可以通过静电相互作用极大促进智能纳米超分子体系的构筑.因此, 利用主体与多种客体共组装超分子体系可以增加多种非共价键作用力的协同效应, 提高多电荷环糊精超分子组装体的综合性能, 可为构筑多重刺激响应的超分子组装体提供新思路.
尽管天然环糊精和多电荷环糊精本身是无毒或低毒的, 然而其与客体分子共组装形成的环糊精智能体系在药物和基因传递以及疾病治疗方面仍存在安全性问题.因此, 低毒高效的纳米智能体系是未来发展的重要方向.
Dedicated to the 40th anniversary of Chinese Journal of Organic Chemistry
-
-
[1]
(a) Crini, G. Chem. Rev. 2014, 114, 10940.
(b) Chen, Y.; Zhang, Y. M.; Liu, Y. Chem. Commun. 2010, 46, 5622.
(c) Lai, W.-F.; Rogach, A. L.; Wong, W.-T. Chem. Soc. Rev. 2017, 46, 6379.
(d) Prochowicz, D.; Kornowicz, A.; Lewiński, J. Chem. Rev. 2017, 117, 13461.
(e) Chen, G.; Jiang, M. Chem. Soc. Rev. 2011, 40, 2254.
(f) Chen, H.; Liu, X.; Dou, Y.; He, B.; Liu, L.; Wei, Z.; Li, J.; Wang, C.; Mao, C.; Zhang, J.; Wang, G. Biomaterials 2013, 34, 4159.
(g) Schmidt, B.; Barner-Kowollik, C. Angew. Chem., Int. Ed. 2017, 56, 8350.
(h) Kolesnichenko, I. V.; Anslyn, E. V. Chem. Soc. Rev. 2017, 46, 2385.
(i) Chen, Y.; Liu, Y. Chem. Soc. Rev. 2010, 39, 495.
(j) Zhang, Y.; Liang, L.; Chen, Y.; Chen, X. M.; Liu, Y. Soft Matter 2018, 15, 73.
(k) Yu, Q.; Zhang, Y.-M.; Liu, Y.-H.; Xu, X.; Liu, Y. Sci. Adv. 2018, 4, eaat2297.
(l) Pinalli, R.; Pedrini, A.; Dalcanale, E. Chem. Soc. Rev. 2018, 47, 7006.
(m) Zhang, W.; Deng, W. Chin. J. Org. Chem. 2018, 38, 3002(in Chinese).
(张薇, 邓维, 有机化学, 2018, 38, 3002.)
(n) Chen, Y. Q.; Gui, X.; Duan, Z. B.; Zhu, L. J.; Xiang, Y. Z.; Xia, D. H. Chin. J. Org. Chem. 2019, 39, 1284(in Chinese).
(陈雅琪, 桂鑫, 段尊斌, 朱丽君, 项玉芝, 夏道宏, 有机化学, 2019, 39, 1284.)
(o) Qie, S. Y.; Hao, Y.; Liu, Z. J.; Wang, J.; Xi, J. N. Acta Chim. Sinica 2020, 78, 232(in Chinese).
(郄淑燕, 郝莹, 刘宗建, 王锦, 席家宁, 化学学报, 2020, 78, 232.) -
[2]
Eliseev, A. V.; Schneider, H.-J. J. Am. Chem. Soc. 1994, 116, 6081. doi: 10.1021/ja00093a004
-
[3]
(a) Li, Z.; Zhang, Y.-M.; Wang, H.-Y.; Li, H.; Liu, Y. Macromolecules 2017, 50, 1141.
(b) Zhang, Y.; Chen, Y.; Li, J.; Liang, L.; Liu, Y. Acta Chim. Sinica 2018, 76, 622(in Chinese).
(张依, 陈湧, 李晶晶, 梁璐, 刘育, 化学学报, 2018, 76, 622.)
(c) Niu, J.; Chen, Y.; Liu, Y. Chin. J. Org. Chem. 2019, 39, 151(in Chinese).
(牛杰, 陈湧, 刘育, 有机化学, 2019, 39, 151.) -
[4]
(a) Eliseev, A. V.; Schneider, H.-J. Angew. Chem., Int. Ed. 1993, 32, 1331.
(b) Steffen, A.; Thiele, C.; Tietze, S.; Strassnig, C.; Kämper, A.; Lengauer, T.; Wenz, G.; Apostolakis, J. Chem.-Eur. J. 2007, 13, 6801. -
[5]
(a) Zhang, Y. M.; Liu, Y. H.; Liu, Y. Adv. Mater. 2020, 32, e1806158.
(b) Liao, R.; Lv, P.; Wang, Q.; Zheng, J.; Feng, B.; Yang, B. Biomater. Sci. 2017, 5, 1736.
(c) Yao, X.; Mu, J.; Zeng, L.; Lin, J.; Nie, Z.; Jiang, X.; Huang, P. Mater. Horiz. 2019, 6, 846. -
[6]
(a) Bom, A.; Bradley, M.; Cameron, K.; Clark, J. K.; Van Egmond, J.; Feilden, H.; Maclean, E. J.; Muir, A. W.; Palin, R.; Rees, D. C.; Zhang, M.-Q. Angew. Chem., Int. Ed. 2002, 41, 265.
(b) Adam, J. M.; Bennett, D. J.; Bom, A.; Clark, J. K.; Feilden, H.; Hutchinson, E. J.; Palin, R.; Prosser, A.; Rees, D. C.; Rosair, G. M.; Stevenson, D.; Tarver, G. J.; Zhang, M.-Q. J. Med. Chem. 2002, 45, 1806. -
[7]
Jones, S. T.; Cagno, V.; Janeček, M.; Ortiz, D.; Gasilova, N.; Piret, J.; Gasbarri, M.; Constant, D. A.; Han, Y.; Vuković, L.; Král, P.; Kaiser, L.; Huang, S.; Constant, S.; Kirkegaard, K.; Boivin, G.; Stellacci, F.; Tapparel, C. Sci. Adv. 2020, 6, eaax9318. doi: 10.1126/sciadv.aax9318
-
[8]
Sun, H.-L.; Zhang, Y.-M.; Chen, Y.; Liu, Y. Sci. Rep. 2016, 6, 27. doi: 10.1038/s41598-016-0026-z
-
[9]
Li, J. J.; Chen, Y.; Yu, J.; Cheng, N.; Liu, Y. Adv. Mater. 2017, 29, 1701905. doi: 10.1002/adma.201701905
-
[10]
(a) Shi, R.-J.; Chen, Y.; Hou, X.-F.; Liu, Y. RSC Adv. 2016, 6, 15175.
(b) Zhao, D.; Chen, Y.; Liu, Y. Chin. Chem. Lett. 2015, 26, 829. -
[11]
Díaz-Moscoso, A.; Balbuena, P.; Gómez-García, M.; Ortiz Mellet, C.; Benito, J. M.; Le Gourriérec, L.; Di Giorgio, C.; Vierling, P.; Mazzaglia, A.; Micali, N.; Defaye, J.; García Fernández, J. M. Chem. Commun. 2008, 2001.
-
[12]
Zhao, F.; Yin, H.; Zhang, Z.; Li, J. Biomacromolecules 2013, 14, 476. doi: 10.1021/bm301718f
-
[13]
Wang, J.; Chen, Y.; Cheng, N.; Feng, L.; Gu, B.-H.; Liu, Y. ACS Appl. Bio Mater. 2019, 2, 5898. doi: 10.1021/acsabm.9b00845
-
[14]
(a) Lu, Y.; De Vries, W. C.; Overeem, N. J.; Duan, X.; Zhang, H.; Zhang, H.; Pang, W.; Ravoo, B. J.; Huskens, J. Angew. Chem., Int. Ed. 2019, 58, 159.
(b) Schibilla, F.; Holthenrich, A.; Song, B.; Linard Matos, A. L.; Grill, D.; Rota Martir, D.; Gerke, V.; Zysman-Colman, E.; Ravoo, B. J. Chem. Sci. 2018, 9, 7822.
(c) Himmelein, S.; Lewe, V.; Stuart, M. C. A.; Ravoo, B. J. Chem. Sci. 2014, 5, 1054. -
[15]
Ravoo, B. J.; Darcy, R. Angew. Chem., Int. Ed. 2000, 39, 4324. doi: 10.1002/1521-3773(20001201)39:23<4324::AID-ANIE4324>3.0.CO;2-O
-
[16]
(a) Mohan Nalluri, S. K.; Bultema, J. B.; Boekema, E. J.; Ravoo, B. J. Chem. Sci. 2011, 2, 2383.
(b) Samanta, A.; Ravoo, B. J. Chem.-Eur. J. 2014, 20, 4966.
(c) Samanta, A.; Stuart, M. C. A.; Ravoo, B. J. J. Am. Chem. Soc. 2012, 134, 19909.
(d) Nalluri, S. K. M.; Bultema, J. B.; Boekema, E. J.; Ravoo, B. J. Chem.-Eur. J. 2011, 17, 10297.
(e) Moratz, J.; Stricker, L.; Engel, S.; Ravoo, B. J. Macromol. Rapid Commun. 2018, 39, 1700256. -
[17]
Sukegawa, T.; Furuike, T.; Niikura, K.; Yamagishi, A.; Monde, K.; Nishimura, S.-I. Chem. Commun. 2002, 430.
-
[18]
Donohue, R.; Mazzaglia, A.; Ravoo, B. J.; Darcy, R. Chem. Commun. 2002, 2864.
-
[19]
(a) O'mahony, A. M.; Doyle, D.; Darcy, R.; Cryan, J. F.; O'driscoll, C. M. Eur. J. Pharm. Sci. 2012, 47, 896.
(b) Méndez-Ardoy, A.; Guilloteau, N.; Di Giorgio, C.; Vierling, P.; Santoyo-González, F.; Ortiz Mellet, C.; García Fernández, J. M. J. Org. Chem. 2011, 76, 5882. -
[20]
Li, P. Y.; Chen, Y.; Chen, C. H.; Liu, Y. Chem. Commun. 2019, 55, 11790. doi: 10.1039/C9CC06545H
-
[21]
(a) Zan, M.; Li, J.; Luo, S.; Ge, Z. Chem. Commun. 2014, 50, 7824.
(b) Zhao, D.; Yi, X.; Xu, J.; Yuan, G.; Zhuo, R.; Li, F. J. Mater. Chem. B 2017, 5, 2823.
(c) Durmaz, Y. Y.; Lin, Y.-L.; Elsayed, M. E. H. Adv. Funct. Mater. 2013, 23, 3885. -
[22]
Cheng, J. G.; Yu, H. J.; Chen, Y.; Liu, Y. Bioorg. Med. Chem. 2018, 26, 2287. doi: 10.1016/j.bmc.2018.03.013
-
[23]
Cheng, J.-G.; Zhang, Y.-M.; Liu, Y. ChemNanoMat 2018, 4, 758. doi: 10.1002/cnma.201800098
-
[24]
Tardy, B. L.; Tan, S.; Dam, H. H.; Ejima, H.; Blencowe, A.; Qiao, G. G.; Caruso, F. Nanoscale 2016, 8, 15589. doi: 10.1039/C6NR04841B
-
[25]
Liang, L.; Chen, Y.; Chen, X.-M.; Zhang, Y.; Liu, Y. Chin. Chem. Lett. 2018, 29, 989. doi: 10.1016/j.cclet.2017.12.022
-
[26]
Chen, X. M.; Chen, Y.; Hou, X. F.; Wu, X.; Gu, B. H.; Liu, Y. ACS Appl. Mater. Interfaces 2018, 10, 24987. doi: 10.1021/acsami.8b08651
-
[27]
(a) Gayam, S. R.; Venkatesan, P.; Sung, Y.-M.; Sung, S.-Y.; Hu, S.-H.; Hsu, H.-Y.; Wu, S.-P. Nanoscale 2016, 8, 12307.
(b) Lee, J.; Oh, E.-T.; Yoon, H.; Woo Kim, C.; Han, Y.; Song, J.; Jang, H.; Joo Park, H.; Kim, C. Nanoscale 2017, 9, 6901.
(c) Cheng, Y.-J.; Luo, G.-F.; Zhu, J.-Y.; Xu, X.-D.; Zeng, X.; Cheng, D.-B.; Li, Y.-M.; Wu, Y.; Zhang, X.-Z.; Zhuo, R.-X.; He, F. ACS Appl. Mater. Interfaces 2015, 7, 9078. -
[28]
Hu, P.; Chen, Y.; Li, J. J.; Liu, Y. Chem.-Asian J. 2016, 11, 505. doi: 10.1002/asia.201501029
-
[29]
Hou, X.-F.; Chen, Y.; Liu, Y. Soft Matter 2015, 11, 2488. doi: 10.1039/C4SM02896A
-
[30]
Han, X.; Chen, Y.; Sun, H.-L.; Liu, Y. Asian J. Org. Chem. 2018, 7, 870. doi: 10.1002/ajoc.201800076
-
[31]
Guan, X.; Chen, Y.; Wu, X.; Li, P.; Liu, Y. Chem. Commun. 2019, 55, 953. doi: 10.1039/C8CC09047E
-
[32]
(a) Zhao, Q.; Lian, Z.; Gao, X.; Yan, Y.; Huang, J. Langmuir 2016, 32, 11973.
(b) Mandl, G. A.; Rojas-Gutierrez, P. A.; Capobianco, J. A. Chem. Commun. 2018, 54, 5847.
(c) Stricker, L.; Fritz, E.-C.; Peterlechner, M.; Doltsinis, N. L.; Ravoo, B. J. J. Am. Chem. Soc. 2016, 138, 4547. -
[33]
Zhao, X.; Chen, Y.; Guan, X. R.; Li, P. Y.; Zhou, W. L.; Liu, Y. ChemistrySelect 2019, 4, 13241. doi: 10.1002/slct.201903889
-
[34]
Li, Z.-Y.; Chen, Y.; Wu, H.; Liu, Y. ChemistrySelect 2018, 3, 3203. doi: 10.1002/slct.201703091
-
[35]
Liu, Z.; Qiao, J.; Tian, Y.; Wu, M.; Niu, Z.; Huang, Y. Langmuir 2014, 30, 8938. doi: 10.1021/la501936a
-
[36]
Liu, J.; Xu, L.; Jin, Y.; Qi, C.; Li, Q.; Zhang, Y.; Jiang, X.; Wang, G.; Wang, Z.; Wang, L. ACS Appl. Mater. Interfaces 2016, 8, 14200. doi: 10.1021/acsami.6b04462
-
[37]
Chen, X.; Qiu, Y. K.; Owh, C.; Loh, X. J.; Wu, Y. L. Nanoscale 2016, 8, 18876. doi: 10.1039/C6NR08055C
-
[38]
Wen, Y.; Zhang, Z.; Li, J. Adv. Funct. Mater. 2014, 24, 3874. doi: 10.1002/adfm.201303687
-
[39]
(a) Yang, C.; Wang, X.; Li, H.; Goh, S. H.; Li, J. Biomacromolecules 2007, 8, 3365.
(b) Yamashita, A.; Kanda, D.; Katoono, R.; Yui, N.; Ooya, T.; Maruyama, A.; Akita, H.; Kogure, K.; Harashima, H. J. Control. Release 2008, 131, 137. -
[40]
Ooya, T.; Choi, H. S.; Yamashita, A.; Yui, N.; Sugaya, Y.; Kano, A.; Maruyama, A.; Akita, H.; Ito, R.; Kogure, K.; Harashima, H. J. Am. Chem. Soc. 2006, 128, 3852. doi: 10.1021/ja055868+
-
[41]
Tamura, A.; Yui, N. Biomaterials 2013, 34, 2480. doi: 10.1016/j.biomaterials.2012.12.006
-
[42]
Schroeder, T. B. H.; Houghtaling, J.; Wilts, B. D.; Mayer, M. Adv. Mater. 2018, 30, 1705322. doi: 10.1002/adma.201705322
-
[43]
(a) Badruddoza, A. Z. M.; Bhattarai, B.; Suri, R. P. S. ACS Sustainable Chem. Eng. 2017, 5, 9223.
(b) Lu, A.-H.; Salabas, E. L.; Schüth, F. Angew. Chem., Int. Ed. 2007, 46, 1222. -
[44]
Samanta, A.; Ravoo, B. J. Angew. Chem., Int. Ed. 2014, 53, 12946. doi: 10.1002/anie.201405849
-
[45]
Awasthi, A. A.; Singh, P. K. J. Phys. Chem. B 2017, 121, 6208. doi: 10.1021/acs.jpcb.7b03592
-
[46]
Singh, G.; Singh, P. K. Langmuir 2019, 35, 14628. doi: 10.1021/acs.langmuir.9b03083
-
[47]
Hu, P.; Chen, Y.; Liu, Y. Chem. Commun. 2015, 51, 10839. doi: 10.1039/C5CC03248B
-
[1]
-
-

 下载:
下载:



 下载:
下载:












 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 98
- 文章访问数: 10762
- HTML全文浏览量: 1862
