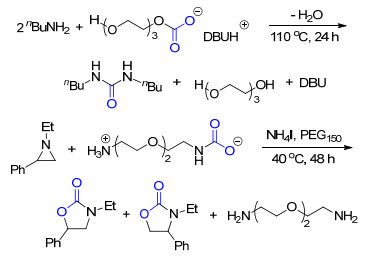
图式 1.
超强碱和聚乙二醇的催化体系用于CO2捕集与转化
Scheme 1.
CO2 capture and utilization by superbase/polyethyl- ene glycol catalyst system

世界范围内工业化的快速发展, 导致化石资源逐渐耗竭, 大气中CO2浓度不断升高, 并由此引发了一系列能源危机和环境问题.因此, 开发可再生资源、减少大气中CO2的累积尤为重要.将CO2作为C1合成子应用于有机合成与绿色化学化工, 既能在一定程度上减少化石资源的使用, 还起到了消耗和固定CO2的作用, 可谓一举两得.
CO2是一个典型的直线型分子, 具有两个C=O键.由于CO2分子上存在的两个三中心四电子的离域π键, 使得该分子的C=O键的键长缩短为116.3 pm, 键能更是升高到804.4 kJ/mol.并且, CO2上的碳原子处于最高氧化态, 为+4价, 所以第一电离能(13.8 eV)较高, 不易给出电子.虽然CO2的电子亲和能(38 eV)相对较高, 所以氧原子上较易接受电子, 但是这伴随着直线型的CO2分子发生弯曲, 而这通常都是耗能反应, 且需要的能量都比较高.可见, CO2具有热力学稳定性和动力学惰性, 要成功实现其化学转化, 不仅需要合理设计转化路线, 还需要开发高活性催化体系.为了使CO2化学转化反应在热力学上可行, 常需要高能底物参与或者采用剧烈的反应条件.近年来, 光、电促进的CO2转化兼储能过程也是CO2转化策略之一; 此外, 二氧化碳是高度对称的分子, CO2的动力学惰性还体现在低压、低浓度反应速率低.因此, 低压下二氧化碳的快速转化反应是本领域中最具挑战的课题之一.动力学上的惰性决定了二氧化碳转化的关键在于发展高效的捕集材料以及催化体系.二氧化碳分子中的C—O键的极性和π电子使其易于受亲核试剂的进攻或发生氧化加成反应, 这是二氧化碳化学转化的分子科学基础.
作为一种温室气体, CO2的捕集已经受到人们的广泛关注.醇胺水溶液是目前工业上常用的CO2吸收剂, 其对CO2吸收容量大.但是, 醇胺在重生过程中需要的高能耗, 大大地增加了CO2吸收的成本.与CO2脱附相比, 将捕集的CO2直接进行催化转化, 不仅能避免耗能的捕集试剂再生过程, 还能利用CO2这一种丰富、可再生的C1资源合成很多具有高附加值的化合物[1].我们[2]率先提出将CO2的捕集与转化相偶合具有重要的应用前景.基于此, 我们课题组设计了离子液体、含氮有机碱等CO2捕集试剂, 实现了CO2捕集和转化的偶合.
由于具有低蒸气压、宽液程和高稳定性等优点, 离子液体已被广泛地应用于CO2的捕集与转化中.我们[3]利用超强碱与醇在CO2存在下可以形成质子型离子液体的特性, 设计了含有超强碱和聚乙二醇(PEG)的二元催化体系.通过CO2的吸收实验, 发现超强碱的碱性越强, 二元催化体系的CO2捕集能力也越强.而聚乙二醇的链长则对体系吸收CO2的能力没有明显影响.这类二元催化体系捕集了CO2之后形成的离子液体可看作CO2的活化形式, 能够在温和的条件下与胺反应生成脲, 还能在碘化铵(NH4I)催化下与氮丙啶类化合物反应生成噁唑烷酮, 这两个反应都能以接近100%的收率得到相应的产物(Scheme 1).
使用聚乙二醇功能化的非质子型离子液体同样可以促进CO2与氮丙啶类化合物的反应(Scheme 2).我们课题组[4]设计了含有超强碱的聚乙二醇功能化的离子液体.这类离子液体可以有效地催化CO2与氮丙啶类化合物的反应, 其中BrDBNPEG150DBNBr具有适中的碱性和较小的位阻, 表现出了最好的活性.若是将该离子液体中的聚乙二醇基团替换为长链的烷基, 其催化效果会有明显的降低, 证明了聚乙二醇基团对该反应的促进作用.该离子液体还具有很好的循环利用性, 重复利用了5次其活性也没有明显降低.
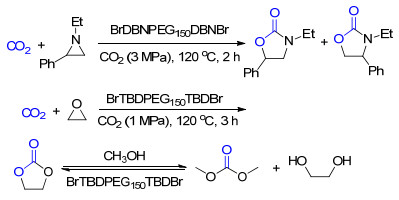
除了催化合成噁唑烷酮, 聚乙二醇功能化的离子液体在催化CO2与环氧类化合物的反应中也有很好的活性[5]. BrTBDPEG150TBDBr可以在101 kPa的CO2下高效地将CO2和环氧类化合物转化为环状碳酸酯, 并具有很好的循环利用性(Scheme 2).通过原位红外光谱表征, 发现催化剂的阳离子部分不仅能通过氢键活化环氧丙烷, 还能以形成氨基甲酸盐的形式捕集CO2.
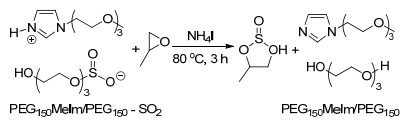
聚乙二醇功能化的离子液体还可以用于SO2的捕集与转化中(Scheme 3).研究发现, 弱碱与聚乙二醇混合形成的质子型离子液体可以快速、可逆、高选择性地吸收SO2, 并且其吸收容量和弱碱的碱性密切相关[6].使用PEG衍生化的碱作为阳离子可以通过PEG上的氧原子与SO2的物理作用进一步提高体系对SO2的吸收容量.此外, 被吸收(活化)的SO2可以在NH4I的催化下与环氧化合物反应生成环状亚硫酸酯.
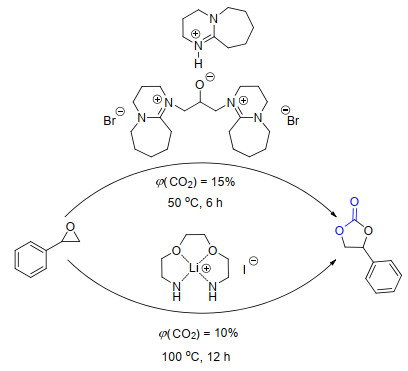
CO2捕集与转化的策略还被用于低浓度下的CO2利用反应中(Scheme 4).最近, 张香平课题组[7a]报道了一种1, 8-二氮杂二环十一碳-7-烯(DBU)功能化的质子型离子液体可以高效地催化CO2与环氧化合物的反应.即使在模拟烟气的CO2浓度[φ(CO2)=15%]下, 依然可以96%的收率得到相应的环状碳酸酯产物.通过密度泛函理论(DFT)计算和红外光谱(FT-IR)证实, 这类离子液体是因为可以先将CO2进行捕集并活化再用于反应中, 因而在低浓度的CO2条件下同样具有比较高的反应活性.利用多氨基螯合型离子液体, 王从敏课题组[7b]也实现了模拟烟气条件下将CO2与环氧化合物转化为环状碳酸酯.离子液体阳离子上的氨基具有捕集CO2的能力, 阴离子(碘离子)可以活化底物, 这种阴阳离子的协同作用是其具有高催化活性的主要原因.
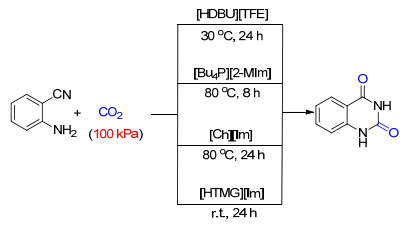
以CO2为原料之一合成广泛存在于生物、医药分子中的喹唑啉-2, 4(1H, 3H)-二酮及其衍生物是100%原子经济反应, 因而开发高效催化剂, 实现CO2的捕集和转化尤其值得研究(Scheme 5).刘志敏课题组[8a]以1, 8-二氮杂二环十一碳-7-烯三氟乙酸盐([DBU][TFA])为催化剂, 实现了2-氨基苯甲腈与101 kPa下的CO2反应生成喹唑啉-2, 4(1H, 3H)-二酮.通过1H NMR和13C NMR的表征, 他们认为[DBU][TFA]可以同时捕集CO2并活化底物, 因此能够在较低的CO2压力下实现该反应.随后, 该课题组[8b]还发展了以2-甲基咪唑(2-Methylimidazole, 2-MeIm)为阴离子的非质子型离子液体, 用于CO2的捕集和转化反应中.因为2-MeIm阴离子都具有很好的捕集CO2的性能, 所以这些反应在常压的CO2下同样具有较好的活性.同样使用含唑类阴离子的离子液体, 王从敏等[8c]发现胆碱咪唑盐([Ch][Im])在2-氨基苯甲腈与CO2的反应在中也有较高的催化活性.其中, 离子液体的阳离子部分的碱性被发现对其催化活性有明显的影响, 并且阳离子羟基上的氢可以通过与反应中间体形成氢键来降低反应能垒.我们课题组[8d]发展了基于1, 1, 3, 3-四甲基胍(TMG)与咪唑的质子型离子液体, 用于催化邻氨基苯腈与CO2反应生成喹唑啉-2, 4(1H, 3H)-二酮的反应(Scheme 5):在120 ℃ 100 kPa CO2的条件下能够以96%的收率得到产物.通过1H NMR、13C NMR和FT-IR的测试, 我们认为CO2是先被[HTMG][Im]中的阴离子捕集到反应体系中, 然后再与邻氨基苯腈进行反应.
含氨基的化合物是目前工业上常用的碳捕集剂, 它们一般是通过与CO2形成氨基甲酸盐的形式来进行碳捕集的.虽然氨基被广泛地应用于碳捕集中, 但由于在吸收CO2的同时, 会发生质子向另一个胺分子的转移, 因而它们的CO2吸收容量一般为50 mol% (CO2与吸附剂之比)左右.为提高该类CO2捕集试剂的吸收容量, 我们课题组[9]采用在氨基酸的N上修饰一个具有大位阻的基团(比如叔丁基、异丙基等)来抑制氨基甲酸盐的形成, 使得这些氨基酸盐能够吸收等物质的量的CO2.重要的是, 这些被捕集的CO2还能在催化剂的存在下分别与炔丙胺或者氮杂环丙烷反应(Scheme 6).
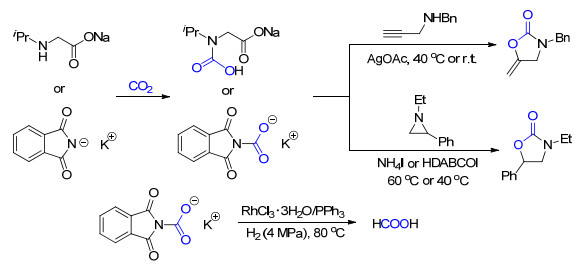
随后, 我们[10]发现邻苯二甲酰亚胺钾同样可以等物质的量地吸收CO2, 而且其吸收焓仅有-10.6 kJ/mol.通过对实验结果、NMR谱图、原位FT-IR测试以及DFT计算的分析, 我们发现阴离子的碱性和亲核性都对CO2的吸收容量有至关重要的影响.该捕集试剂不仅利于CO2脱附, 吸收了CO2之后的产物还能与炔丙胺或者氮杂环丙烷反应, 都能以较好的收率得到相应产物(Scheme 6).当在体系中加入H2, 并以三氯化铑作为催化剂, 可以将被吸收的CO2还原为重要化工产品与能源分子甲酸.
此外, 我们[11a]还利用聚乙烯亚胺(PEI)作为CO2吸收剂和氢化反应的促进剂, 高效地实现了将被捕集的CO2原位催化氢化为甲酸(Scheme 7).该反应以铑为催化剂并加入单齿膦配体, 产生甲酸的转化数可以达到852.通过反应机理的研究, 证明催化活性组分为原位形成的铑氢化合物, 而PEI则是通过与铑的配位作用来促进反应的进行.利用CO2原位催化氢化的策略, Olah和Prakash课题组[11b-11c]先用聚胺的水溶液吸收空气中的CO2, 再使用金属钌配合物催化CO2原位氢化为甲酸和甲醇等高附加值产物.
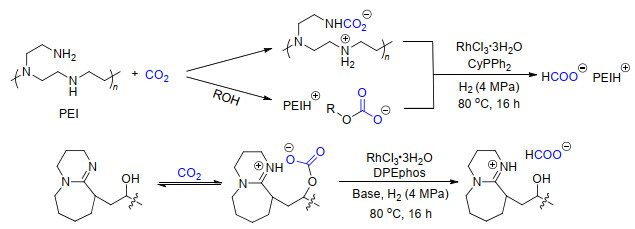
为了进一步提高CO2捕集并原位转化的效率, 我们[12]借鉴了超强碱功能化的质子型离子液体体系, 设计了以脒类衍生物作为CO2捕集剂的催化体系(Scheme 7).在该体系下, 使用双齿膦配体的铑催化剂能够将99%的被吸收的CO2转化为甲酸盐.并且, 使用二氧化硅负载的羟基脒同样具有高的反应活性, 在使用三次之后仍然能够保持80%以上的反应活性.
2012年, Cantat课题组[13]首次提出了“CO2还原功能化”的概念:将CO2的官能团化和CO2的还原相结合.他们使用1, 5, 7-三氮杂二环[4.4.0]癸-5-烯(TBD)为催化剂, PhSiH3为还原剂, 将CO2还原并同时与二级脂肪胺反应, 得到了一系列的甲酰化产物.这种策略不仅可以扩大从CO2直接获取的化学品的利用范围, 还能实现能量的积累, 将会在一定程度上代替现有的石油化学工业, 具有巨大的发展潜力.为此, 我们课题组建立了通过改变还原剂类型、反应条件、配体结构等实现CO2分级可控还原功能化的策略.
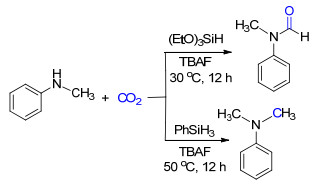
基于氟的亲硅性, 我们[14a]以廉价的有机小分子四正丁基氟化铵为催化剂活化氢硅烷, 高效地实现了胺参与的CO2还原功能化反应.通过改变还原剂——氢硅烷的类型, 能够选择性地得到CO2的2电子或者6电子还原产物(Scheme 8):使用三乙氧基硅烷为还原剂可以得到甲酰胺类产物, 而用苯硅烷时则是得到甲胺类化合物.基于反应过程检测到甲酸硅脂这一关键的中间体的事实, 提出了甲酰胺是经由甲酸硅酯与苯胺的反应生成的, 在较强还原剂存在的条件下, 甲酰胺可进一步被还原为甲胺的反应机理.相似地, 傅尧等[14b]也通过调节硅烷的种类, 在Cs2CO3的催化下, 分别经过CO2的N-甲酰化和N-甲基化反应得到了这两类产物.
含氧的亲核物种同样可以通过与硅相互作用来活化氢硅烷.基于此, 我们[15a]以碳酸盐为催化剂、二苯基硅烷为还原剂, 实现了CO2的6电子还原并与C—N键的形成相偶合(Scheme 9).通过对控制实验的分析, 认为甲酰胺并非该反应的中间体, CO2是先还原至C0物种缩醛胺再进一步被还原至N-甲基化的.除了碳酸盐, 常用的有机溶剂二甲基甲酰胺(DMF)由于含有亲核性的酰胺氧原子且其周围位阻较小, 因而也能够促进CO2的2/6电子还原功能化(Scheme 9)[15b].其中, DMF既是反应溶剂也是催化剂.
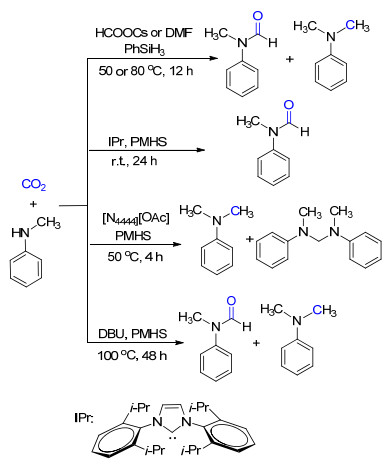
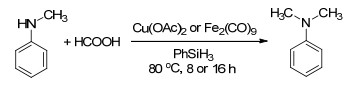
相比于其他的氢硅烷, 聚甲基氢硅氧烷(Poly- methylhydrosiloxane, PMHS)更稳定, 能够长时间地储存而不损失其活性; 而且PMHS是硅工业的副产物, 是一种廉价易得的还原剂.但是PMHS的反应活性很低.为了活化PMHS, Cantat等[16a]以活性更高的氮杂卡宾(N-heterocyclic carbene, NHC)作为催化剂, 用于CO2的N-甲酰化反应.我们[16b]设计了含醋酸阴离子的离子液体, 通过其较强的亲核性来活化PMHS从而实现CO2的还原功能化(Scheme 9).此外, 通过调节CO2的用量可以有选择性地得到4电子或者6电子的还原产物.这是首例离子液体催化的CO2选择性4/6电子还原功能化反应.随后, 夏纪宝课题组[16c]通过改变DBU催化的CO2还原功能化反应的温度, 也使用PMHS作为还原剂可以分别得到N-甲酰化和N-甲基化的产物.
最近, 我们[17a]发现直接调节CO2的压力也能够改变CO2在还原功能化反应中的选择性(Scheme 10).以廉价易得的钨酸钾为催化剂, 苯硅烷为还原剂, 在2 MPa压力下的CO2中可得到2电子还原的甲酰胺产物, 而在100 kPa的CO2下则主要得到6电子还原的甲胺产物.
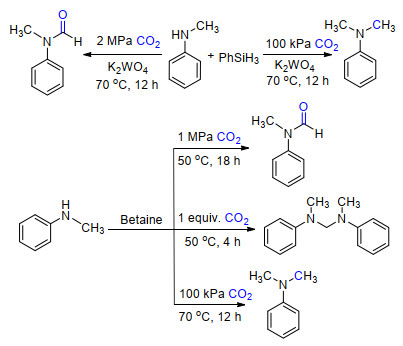
但是, 在胺和还原剂存在下将CO2分级可控还原至甲酰胺、缩醛胺和甲胺仍面临着巨大的挑战.我们[17b]利用甜菜碱作为催化剂, 通过调节CO2的压力与用量便能够高效高选择性地分别得到CO2不同还原水平的产物(Scheme 10).由于在反应过程中可以检测到C0硅基缩醛和缩醛胺, 因此认为它们是反应的中间体.这是首次采用一种催化剂实现了分级可控地将CO2进行2/4/6电子的还原功能化反应.
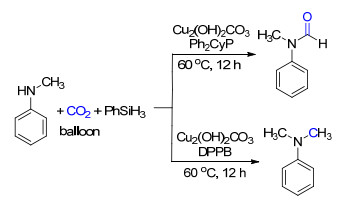
在使用过渡金属铜作为催化剂时, 不同的配体同样也可以调控CO2的还原水平[18].同样是使用碱式碳酸铜作为催化剂, 苯硅烷为还原剂, 以双齿的1, 4-双(二苯基膦)丁烷(DPPB)作为配体可以得到6电子还原的胺甲基化产物, 将配体换为单齿的Ph2CyP配体则是得到2电子还原的胺甲酰化产物(Scheme 11)[18a].最近报道, 通过膦配体调控铁催化的CO2选择性地还原官能化到甲酰胺和甲胺的成功实例, 表明了这一方法的通用性[18b].
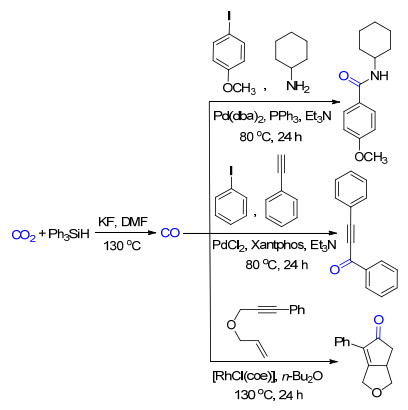
CO作为一种常见的羰基源被广泛用于有机反应中, 并且在工业上作为费托合成的原料之一也有重要的应用.将CO2还原为CO再进行功能化反应可以极大地提高CO2的应用范围.基于此, 我们[19]发展了一种两步的合成法(Scheme 12):先在一个反应釜中以氟化铯(CsF)为催化剂, 用苯硅烷将CO2还原成CO; 然后通过另一个与之相连的反应釜将得到的CO直接用于胺羰基化反应、Sonogashira偶联反应或者Pauson-Khand反应.其中, 所得到的炔酮类化合物还能用于合成吡唑类杂环化合物, 进一步拓展了反应的应用范围.
甲酸可以由CO2催化氢化得到, 在催化剂的作用下也可以重新释放出CO2, 因此甲酸参与的反应可以被认为是CO2的一种间接利用.我们[20]以铜或者铁作为催化剂发展了一种利用甲酸作为C1资源的N-甲基化反应(Scheme 13).该反应具有很好的底物适用性, 对一级胺、二级胺都能以较好的收率得到相应产物.通过控制实验的研究, 我们认为该反应是经过了缩醛中间体的历程.该方法提供了一种二氧化碳间接利用途径.
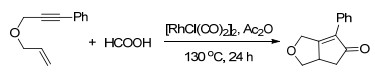
此外, 甲酸也可以作为一种安全、环保的CO替代物来进行利用.以铑配合物为催化剂, 乙酸酐为添加剂可以高效地实现以甲酸作为CO前体的Pauson-Khand反应(Scheme 14)[21].通过对一系列甲酸盐的筛选, 可以证实甲酸是反应产物中羰基的来源.并且, 该策略也适用于杂环的Pauson-Khand反应, 能以较好的收率得到相应的杂环化合物, 进一步表现了该方法在生物医药方面的应用前景.
由于CO2分子具有热力学稳定性, 多数CO2参与的化学反应是热力学不利的过程.因此, 开发热力学可行的CO2转化新途径是面向可持续发展的CO2化学领域具有挑战的课题之一.以环碳酸酯和噁唑啉酮的合成为例, 采用邻二醇或2-乙醇胺和CO2为原料进行合成虽然原子经济性好, 但面临热力学限制, 即使采用脱水剂和苛刻的反应条件, 产物的收率仍然有限[22].基于此, 我们设计了CO2与炔丙醇、亲核试剂的三组分串联反应, 为CO2“变废为宝”的高值化利用提供新途径.即, 发展绿色、高效催化技术, 成功地以邻二醇或氨基醇为原料分别合成环状碳酸酯和2-噁唑啉酮; 建立了CO2热力学可行的高效转化新方法, 具有步骤经济性、原子经济性、实用性等特点.在三组分级联反应中, 将高能化合物炔丙醇引入邻二醇或2-乙醇胺和CO2的反应体系, 使CO2首先与炔丙醇发生反应, 得到的α-亚甲基环状碳酸酯进一步与亲核试剂邻二醇或2-乙醇胺发生亲核开环和闭环反应, 从而生成环碳酸酯或噁唑啉酮并联产α-羟基酮类化合物.以此为基础, 我们进一步将亲核试剂扩展到胺、一元醇和水, 从而合成出了β-羰基氨基甲酸酯、碳酸酯和β-羟基酮[23].根据催化机理, 能够促进炔丙醇与CO2羧化环化反应并能亲电活化碳基的Lewis酸以及能活化醇羟基和胺基的Lewis碱都能够对该反应起到促进作用.基于早期对炔丙胺和CO2羧化环化反应的认识, 我们开发了一系列催化剂, 实现了环碳酸酯和噁唑啉酮的高效合成.
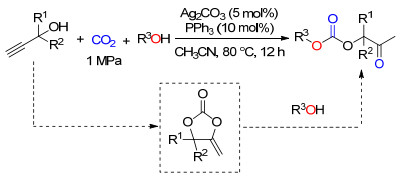
银催化剂能够促进炔丙醇和CO2的羧化环化反应, 同时银离子作为Lewis酸, 还能活化羰基, 促进α-亚甲基环碳酸酯与邻二醇反应(Scheme 15)[24].通过对一系列银盐和配体的筛选, 发现含有碱性阴离子的银盐AgOAc、Ag2WO4、Ag2CO3以及Ag2O与配体PPh3的组合对该反应具有很好的促进作用.其中以Ag2CO3和Xantphos配体的效果最好.控制实验表明, 反应经历了炔丙醇的羧化环化反应以及α-亚甲基与邻二醇的亲核开环和合环反应, Ag2CO3可作为双功能催化剂, 分别起到Lewis酸和碱的作用.该催化体系对多种结构的邻二醇(或间二醇)和炔丙醇化合物均具有较好的底物适用性, 所得环碳酸酯和α-羟基酮化合物的收率为38%~97%.
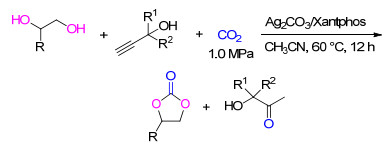
当三组分反应中的亲核试剂为2-乙醇胺时, 炔丙醇、CO2和2-乙醇胺经历级联反应, 可生成噁唑啉酮类化合物.对于该反应体系, 银盐以及银的氧化物、铜盐和离子液体均表现出优良的催化活性.
其中含有碱性阴离子的银盐可作为双功能催化剂, 在适宜配体的作用下, 银的配合物可作为Lewis酸, 而游离出的碱性阴离子可活化胺基和醇羟基.其中Ag2CO3/Xantphos组成的催化体系对不同结构的炔丙醇和2-乙醇胺化合物与CO2的级联反应均有促进作用, 噁唑啉酮和α-羟基酮化合物的收率为33%~92% (Scheme 16)[25].产物的收率受到2-乙醇胺中胺基N原子亲核能力的影响, N原子上所连基团的电负性增大会使目标产品的收率降低.
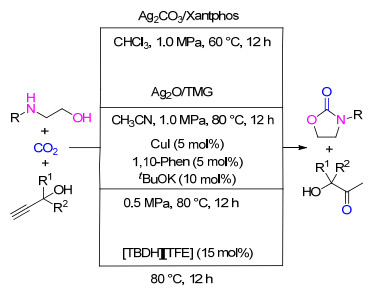
除了含有碱性阴离子的银盐, 其它银盐以及Ag2O在有机碱存在的条件下, 也能促进炔丙醇、CO2和2-乙醇胺的级联反应并生成目标产物(Scheme 16)[26].其中以Ag2O和有机碱TMG效果最好, 所得目标产品的收率为24%~99%.在反应过程中, 具有Lewis酸性的银活化炔键以及羰基, 有机碱则活化胺基和醇羟基.
除了银盐以及银的氧化物, 具有Lewis酸性的铜盐也能够活化炔键和羰基, 因而也可以作为炔丙醇、2-乙醇胺和CO2级联反应的催化剂(Scheme 16).其中, 在强碱tBuOK存在条件下, CuI和邻菲罗林组成的催化体系对该三组分级联反应表现出较好的催化效果.在0.5 MPa的CO2压力条件下, 可得到与银催化体系相当的收率[27].
上述基于银和铜的催化体系虽然能够促进炔丙醇、2-乙醇胺和CO2三组分级联反应, 但所需的CO2压力仍然较高.为实现常压下CO2参与三组分级联反应, 我们课题组[28]进一步对离子液体对该反应的催化性能进行了研究.结果表明, 含有碱性阴离子的质子型离子液体在常压CO2下对该反应具有催化活性, 其中以[TBDH][TFE]的活性最好, 能使目标产品的收率达到与金属催化体系相当的水平(Scheme 16).另外, 离子液体催化剂具有方便回收和可重复利用的优点.推测其反应机理与金属催化剂类似, 质子型阳离子起到活化炔键和羰基的作用, 而碱性阴离子活化胺基和醇羟基.
除了邻二醇和2-乙醇胺, 单元胺也可以作为亲核试剂与炔丙醇和CO2构成三组分级联反应.当采用伯胺作亲核试剂时, 产物为α-亚甲基噁唑啉酮类化合物; 而当采用仲胺时, 则生成氨基甲酸酯类化合物.基于该反应路线, 我们课题组开发了基于Ag和Cu的催化体系, 实现了噁唑啉酮和氨基甲酸酯化合物的高效合成.
含有碱性阴离子的Ag2WO4在配体PPh3的作用下, 能够促进炔丙醇与CO2的羧化环化反应, 同样, 该催化体系也能用于炔丙醇、CO2与胺的三组分级联反应(Scheme 17)[29].在无溶剂的条件下, 伯胺参与的三组分反应能以较高的收率得到噁唑啉酮类化合物.值得一提的是, 当苯胺作为亲核试剂时, 会得到非脱水的噁唑啉酮化合物, 而乙醇胺作为亲核试剂时, 能以91%的收率得到分子内脱水的五元并环化合物.当亲核试剂是仲胺时, 能以较高的收率得到氨基甲酸酯类化合物.
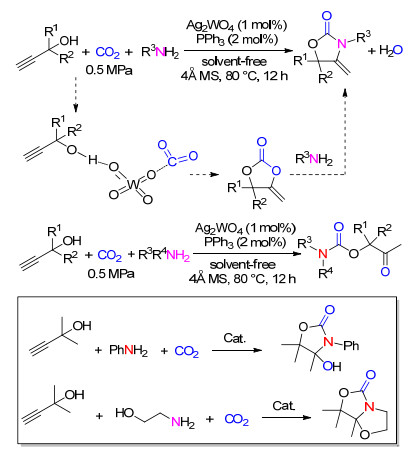
随后, 我们[30]进一步以伯胺吸收CO2后形成的氨基甲酸盐作为反应物与炔丙胺反应, 开发了无气体参与的氨基甲酸酯生成工艺(Scheme 18).当以Ag2O/PPh3为催化剂时, 能以37%~96%的收率得到氨基甲酸酯类化合物.在反应中, CO2被仲胺吸收并被活化, 氨基甲酸盐首先与炔丙醇发生转羧化, 释放出仲胺, 并在银盐的作用下形成环碳酸酯; 被释放出的仲胺进一步亲核进攻环碳酸酯中的羰基碳, 从而形成氨基甲酸酯. CO2和仲胺以氨基甲酸盐的形式参与反应, 便于反应过程的计量, 也避免了复杂的操作过程, 更重要的是反应可以在常压下达到满意的收率.
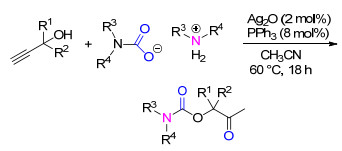
此外, 铜盐CuI在仲胺、炔丙醇和CO2三组分反应中也表现出优异的催化性能, 在100 kPa CO2以及无溶剂的条件下, 能够以满意的收率得到氨基甲酸酯产品(Scheme 19)[31].需要说明的是, 该催化体系不适用于内炔型的底物炔丙醇, 另外, 氧的存在会导致部分端炔偶联副产物的形成, 因而反应过程应避免氧对反应结果的影响.该反应还可以以仲胺与CO2反应生成的氨基甲酸为原料与炔丙醇反应, 能得到与三组分级联反应接近的目标产品的收率, 因而该催化体系也能用于无气体参与的三组分级联反应.
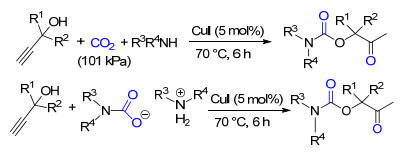
以单元醇作为亲核试剂时, 炔丙醇、CO2和醇的三组分级联反应会生成不对称碳酸酯类化合物. Ag2CO3/ PPh3组成的催化体系对该三元体系的反应具有良好的催化作用, 该催化体系能适用于多种结构的炔丙醇和醇类化合物, 且目标产物的收率在36%~97%之间(Scheme 20)[32].由于该反应中仍然是以α-亚甲基环碳酸酯为中间体, 故该催化体系不适用于非端炔型的炔丙醇.
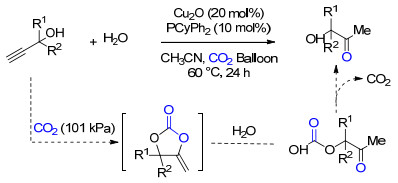
水作为亲核试剂, 也可以与炔丙醇、CO2构成三组分级联反应.在反应中, 炔丙醇与CO2首先发生羧化环化反应, 生成环碳酸酯, 之后水作为亲核试剂与环碳酸酯反应, 生成的烷基碳酸不稳定, 重新释放出CO2而得到α-羟基酮类化合物.其中CO2通过改变反应路径, 促进了炔丙醇的水解.基于铜的催化体系Cu2O/PCyPh2可有效促进该反应的进行, 对于R1和R2为多种取代基的端炔型炔丙醇, 均具有良好的底物适用性, 但对于内炔型的炔丙醇类化合物, 该催化体系不适用(Scheme 21)[33].
基于炔丙醇、CO2和亲核试剂的三组分级联反应的策略, 克服了亲核试剂与CO2反应热力学不利的问题, 我们发展的基于Ag、Cu以及离子液体的催化剂, 能够高效催化三组分级联反应并以较高的收率得到环碳酸酯、噁唑啉酮、氨基甲酸酯、碳酸酯以及α-羟基酮等重要的化学品.此外, 在以胺为亲核试剂时, 还可采用氨基甲酸盐为原料代替胺与CO2, 实现无气体参与的反应过程.值得指出的是, 对于上述所提到的反应, 只有端炔型的炔丙醇能够与CO2和亲核试剂构成级联反应, 内炔型的炔丙醇可能由于位阻效应, 使炔丙醇与CO2的羧化环化反应难以进行, 因而不能得到目标产物.
光促反应是CO2转化的一个重要策略, 光促进的CO2还原能将太阳能转化为化学能, 合成一系列能源化学品, 因而为广大研究者所关注[34].除了光催化的CO2还原, 通过精心的底物设计, 光催化CO2转化可得到结构各异的化合物, 目前已有多个课题组对光促进的CO2多样转化进行了广泛的研究[35].我们课题组也在基于光生自由基促进的CO2转化以及光促进的CO2还原及转化方面进行了相关研究.
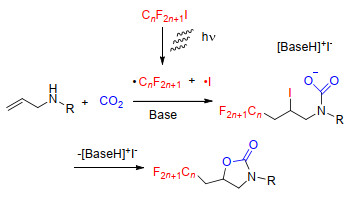
在烯丙胺与CO2的羧化环化反应中, 我们课题组[36]利用全氟碘代烷烃在可见光激发下产生的自由基, 对烯丙胺中双键进行亲电活化, 在等物质的量的碱的存在下实现了烯丙胺与CO2的羧化环化反应, 合成了含氟取代基的噁唑啉酮类化合物(Scheme 22).该方法适用于多种全氟碘代烷烃以及烯丙胺化合物, 具有较好的底物耐受性, 且该方法也同样适用于烯丙醇化合物.在该反应中, 碱可以活化胺基, 促进氨基甲酸盐负离子的形成; 而光生自由基使底物形成中间体碘和全氟烷基取代的氨基甲酸盐, 随后I-离去, 促进噁唑啉酮环的形成.自由基的反应历程使难以被具有Lewis酸性的金属离子活化的烯键与氨基甲酸跟或烷基碳酸根成环成为了可能, 从而使烯丙胺和烯丙醇类化合物也能与CO2完成羧化环化反应, 得到重要化学品环碳酸酯和噁唑啉酮类化合物.
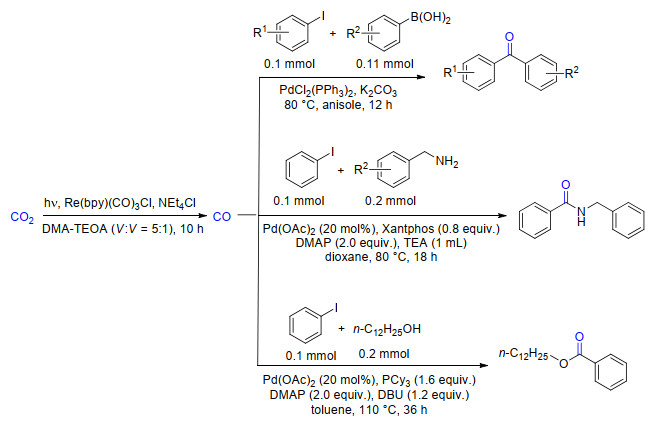
光驱动的CO2还原也是CO2高值化利用的一个重要手段, 在电子牺牲剂以及适宜光敏剂和光催化剂存在的作用下, CO2还可以被还原为CO、甲酸、甲醇或甲烷等能源化学品.为了使光还原CO2的产物更为丰富, 我们在采用Re(bpy)(CO)3Cl还原CO2至CO的基础上, 将生成的CO进一步与碘代芳烃进行Pd催化的Suzuki羰基化、氨羰基化以及烷氧羰基化反应, 构筑C—C、C—N和C—O键, 在相互联通的两个反应器内实现了CO2还原和复杂分子的构建, 这是光催化的二氧化碳还原功能化的第一例(Scheme 23)[37].
有效利用可再生碳资源、保护环境、发展绿色化学已成为大势所趋. CO2是一种主要的温室气体, 也是来源丰富、廉价易得的可再生碳资源.将CO2“变废为宝, 高值化利用”, 不仅可以实现CO2的固定, 还可以获得高附加值的材料、精细化学品和大宗化工产品, 以部分替代化石资源, 满足可持续发展的需求[38].目前, CO2已成功应用于尿素、无机碳酸盐和燃料、甲醇、水杨酸以及环碳酸酯的合成; 近年来, 为进一步推动CO2的大量固定和高值化利用, 多个课题组对CO2制备聚碳酸酯[39]、碳酸二甲酯[40]、异氰酸酯[41]等大宗化学品的工艺进行探索并取得了一定的成果; 另一方面, CO2参与的复杂化学分子构建也被逐步开发出来, 通过精心的底物设计, 在引入CO2分子的同时, 构筑多个化学键、形成有用的精细化学结构单元, 为精细化学品的合成提供了绿色方案[42].
当今世界正处于能源供给从过度依赖化石能源向基于太阳燃料转型的过渡时期, 利用太阳能以及可再生能源将CO2还原以生产碳氢燃料(CO2作为能量载体), 已成为本领域有巨大应用潜力及挑战的热点课题.在CO2催化还原中, 廉价易得的还原剂, 如氢气[43]、生物质[44]等的使用, 不断推动CO2还原向产业化方向发展; 而在电、光还原中, 研究者们则在廉价电子供体的使用[45]、高效催化剂以及光电转化效率的提高方面不断取得突破[46]; 同时, CO2还原可获得能源产品的范围也在进一步扩大, 目前CO2还原产物不仅包括C1化合物, 如甲酸、甲醇和甲烷, 但科学家们已经开始探索将CO2还原为长链酸、醇和烃, 以增加CO2还原产品的能量密度.
另外, 在温和条件下实现低浓度CO2的转化, 对与工业废气乃至大气中CO2的经济利用尤为重要[47].基于CO2活化方式的多位点活化催化体系的设计[48], 光、电催化体系的开发, 有助于使CO2在温和的反应条件下进行转化; 捕集与转化偶合的策略, 使得低浓度CO2的化学转化成为可能[49]; 开发能够耐受杂质气体的催化剂是实现低浓度CO2转化的另一途径.而适用于工业生产的非均相催化体系的开发将有助于推动CO2转化的工业化.
虽然CO2的利用已经发展了很多方法, 但是大规模工业化应用仍然面临经济性的巨大挑战.通过对反应机理的深刻理解, 设计合理、高效的反应路线具有重要的意义.并且, 如何对CO2进行高值化利用, 如构筑具有生物活性的复杂分子、用于合成能源分子(比如甲烷、甲醇、乙烯、乙烷等)或作为弱氧化剂来合成大宗化学品(比如苯乙烯、苯酚等)是一个具有广阔前景的发展方向.此外, 利用可再生、清洁的光能或电能作为CO2转化反应的驱动力, 减少反应能耗也是未来发展的趋势.
作为绿色化学的重要热点领域之一, CO2化学转化已进入一个快速发展的阶段, 在与CO2转化反应相关的活化机理、高活性催化剂开发、转化的新方法、新途径与新策略等方面不断取得了突破, 获得的产品范围不断扩大, 反应条件也越来越温和, 低浓度CO2的利用, 甚至是直接利用空气中的CO2在一些化学品的合成中也成为可能.随着以化学为中心的多学科协同研究的发展, CO2化学转化将会迎来新的发展机遇.
(a) He, M., Sun, Y.; Han, B. Angew. Chem., Int. Ed. 2013, 52, 9620.
(b) Song, Q.-W.; Zhou, Z.-H.; He, L.-N. Green Chem. 2017, 19, 3707.
(c) Yu, B.; He, L.-N. ChemSusChem 2015, 8, 52.
(d) Wang, S.; Xi, C. Chem. Soc. Rev. 2019, 48, 382.
(e) Yao, X.-Y.; Zhang, Y.; Gao, S.; He, L.-N. J. Huazhong Norm. Univ., Nat. Sci. 2019, 53, 834(in Chinese).
(姚向阳, 张彦, 高嵩, 何良年, 华中师范大学学报(自然科学版), 2019, 53, 834.)
(f) Yang, Z.-Z.; Zhao, Y.-N.; He, L.-N. RSC Adv. 2011, 1, 545.
(g) Yu, B.; Diao, Z.-F.; Guo, C.-X.; He, L.-N. J. CO2 Util. 2013, 1, 60.
(h) Li, Y.-N.; Ma, R.; He, L.-N.; Diao, Z.-F. Catal. Sci. Technol. 2014, 4, 1478.
(i) Cao, Y.; He, X.; Wang, N.; Li, H.-R.; He, L.-N. Chin. J. Chem. 2018, 36, 644.
(a) Yang, Z.-Z.; He, L.-N.; Gao, J.; Liu, A.-H.; Yu, B. Energy Environ. Sci. 2012, 5, 6602.
(b) Fu, H.-C.; You, F.; Li, H.-R.; He, L.-N. Front. Chem. 2019, 7, 525.
(c) Kar, S.; Goeppert, A.; Prakash, G. K. S. Acc. Chem. Res. 2019, 52, 2892.
Yang, Z.-Z.; He, L.-N.; Zhao, Y.-N.; Li, B.; Yu, B. Energy Environ. Sci. 2011, 4, 3971. doi: 10.1039/c1ee02156g
Zhao, Y.-N.; Yang, Z.-Z.; Luo, S.-H.; He, L.-N. Catal. Today 2013, 200, 2. doi: 10.1016/j.cattod.2012.04.006
Yang, Z.-Z.; Zhao, Y.-N.; He, L.-N.; Gao, J.; Yin, Z.-S. Green Chem. 2012, 14, 519. doi: 10.1039/c2gc16039k
Yang, Z.-Z.; He, L.-N.; Zhao, Y.-N.; Yu, B. Environ. Sci. Technol. 2013, 47, 1598. doi: 10.1021/es304147q
(a) Meng, X.; Ju, Z.; Zhang, S.; Liang, X.; Solms, N.; Zhang, X.; Zhang, X. Green Chem. 2019, 21, 3456.
(b) Luo, X.; Chen, K.; Li, H.; Wang, C. Int. J. Hydrogen Energy 2016, 41, 9175.
(a) Zhao, Y.; Yu, B.; Yang, Z.; Zhang, H.; Hao, L.; Gao, X.; Liu, Z. Angew. Chem., Int. Ed. 2014, 53, 5922.
(b) Liu, A.-H.; Yu, N.; He, L.-N. Greenhouse Gases: Sci. Technol. 2015, 5, 17.
(c) Shi, G.; Chen, K.; Wang, Y.; Li, H.; Wang, C. ACS Sustainable Chem. Eng. 2018, 6, 5760.
(d) Lang, X.-D.; Yu, Y.-C.; Li, Z.-M.; He, L.-N. J. CO2 Util. 2016, 15, 115.
Liu, A.-H.; Ma, R.; Song, C, ; Yang, Z.-Z.; Yu, A.; Cai, Y.; He, L.-N.; Zhao, Y.-N.; Yu, B.; Song, Q.-W. Angew. Chem., Int. Ed. 2012, 51, 11306. doi: 10.1002/anie.201205362
Zhang, S.; Li, Y.-N.; Zhang, Y.-W.; He, L.-N.; Yu, B.; Song, Q.-W.; Lang, X.-D. ChemSusChem 2014, 7, 1484. doi: 10.1002/cssc.201400133
(a) Li, Y.-N.; He, L.-N.; Liu, A.-H.; Lang, X.-D.; Yang, Z.-Z.; Yu, B.; Luan, C.-R. Green Chem. 2013, 15, 2825.
(b) Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G. A.; Prakash, G. K. S. J. Am. Chem. Soc. 2016, 138, 778.
(c) Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G. A.; Prakash, G. K. S. Green Chem. 2016, 18, 5831.
Li, Y.-N.; He, L.-N.; Lang, X.-D.; Liu, X.-F.; Zhang, S. RSC Adv. 2014, 4, 49995. doi: 10.1039/C4RA08740B
Das Neves Gomes, C.; Jacquet, O.; Villiers, C.; Thuery, P.; Ephritikhine, M.; Cantat, T. Angew. Chem., Int. Ed. 2012, 51, 187. doi: 10.1002/anie.201105516
(a) Liu, X.-F.; Ma, R.; Qiao, C.; Cao, H.; He, L.-N. Chem.-Eur. J. 2016, 22, 16489.
(b) Fang, C.; Lu, C.; Liu, M.; Zhu, Y.; Fu, Y.; Liu, B.-L. ACS Catal. 2016, 6, 7876.
(a) Liu, X.-F.; Qiao, C.; Li, X.-Y.; He, L.-N. Green Chem. 2017, 19, 1726.
(b) Liu, X.-F.; Qiao, C.; Li, X.-Y.; He, L.-N. Pure Appl. Chem. 2018, 90, 1099.
(c) Liu, X.-F.; Li, X.-Y.; Qiao, C.; He, L.-N. Synlett 2018, 28, 548.
(d) Liu, X.-F.; Li, X.-Y.; He, L.-N. Eur. J. Org. Chem. 2019, 2019, 2347.
(a) Jacquet, O.; Das Neves Gomes, C.; Ephritikhine, M.; Cantat, T. J. Am. Chem. Soc. 2012, 134, 2934.
(b) Li, X.-Y.; Zheng, S.-S.; Liu, X.-F.; Yang, Z.-W.; Tan, T.-Y.; Yu, A.; He, L.-N. ACS Sustainable Chem. Eng. 2018, 6, 8130.
(c) Li, G.; Chen, J.; Zhu, D.-Y.; Chen, Y.; Xia, J.-B. Adv. Synth. Catal. 2018, 360, 2364.
(a) Wang, M.-Y.; Wang, N.; Liu, X.-F.; Qiao, C.; He, L.-N. Green Chem. 2018, 20, 1564.
(b) Liu, X.-F.; Li, X.-Y.; Qiao, C.; Fu, H.-C.; He, L.-N. Angew. Chem., Int. Ed. 2017, 56, 7425.
(c) Cao, Y.; Wang, N.; He, X.; Li, H.-R.; He, L.-N. ACS Sustainable Chem. Eng. 2018, 6, 15032.
(a) Li, X.-D.; Xia, S.-M.; Chen, K.-H.; Liu, X.-F.; Li, H.-R.; He, L.-N. Green Chem. 2018, 20, 4853.
(b) Li, W.-D.; Zhu, D.-Y.; Li, G.; Chen, J.; Xia, J.-B. Adv. Synth. Catal. 2019, 361, 5098.
Lang, X.-D.; He, L.-N. ChemSusChem 2018, 11, 2062. doi: 10.1002/cssc.201800902
(a) Qiao, C.; Liu, X.-F.; Liu, X.; He, L.-N. Org. Lett. 2017, 19, 1490.
(b) Qiao, C.; Yao, X.-Y.; Liu, X.-F.; Li, H.-R.; He, L.-N. Asian J. Org. Chem. 2018, 7, 1815.
Lang, X.-D.; You, F.; He, X.; Yu, Y.-C.; He, L.-N. Green Chem. 2019, 21, 509. doi: 10.1039/C8GC03933J
(a) Diao, Z.-F.; Zhou, Z.-H.; Guo, C.-X.; Yu, B.; He, L.-N. RSC Adv. 2016, 6, 32400.
(b) Du, Y.; Kong, D.-L.; Wang, H.-Y.; Cai, F.; Tian, J.-S.; Wang, J.-Q.; He, L.-N. J. Mol. Catal. A: Chem. 2005, 241, 233.
(c) Tamura, M.; Honda, M.; Nakagawa, Y.; Tomishige, K. J. Chem. Technol. Biotechnol. 2014, 89, 19.
(d) Liu, A.-H.; Li, Y.-N.; He, L.-N. Pure Appl. Chem. 2012, 84, 581.
(e) Lang, X.-D.; He, L.-N. Chem. Rec. 2016, 16, 1337.
(f) Li, X.-D.; He, X.; Liu, X.-F.; He, L.-N. Sci. China: Chem. 2017, 60, 841.
(g) Wang, M.-Y.; He, L.-N. Sci. China: Chem. 2016, 59, 507.
Zhou Z.-H.; Xia S.-M.; He, L.-N. Acta Phys.-Chim. Sin. 2018, 34, 838. doi: 10.3866/PKU.WHXB201712271
Zhou, Z.-H.; Song, Q.-W.; He, L.-N. ACS Omega 2017, 2, 337. doi: 10.1021/acsomega.6b00407
Song, Q.-W.; Zhou, Z.-H.; Wang, M.-Y.; Zhang, K.; Liu, P.; Xun, J.-Y.; He, L.-N. ChemSusChem 2016, 9, 2054. doi: 10.1002/cssc.201600470
Li, X.-D.; Song, Q.-W.; Lang, X.-D.; Chang, Y.; He, L.-N. ChemPhysChem 2017, 18, 3182. doi: 10.1002/cphc.201700297
Li, X.-D.; Cao, Y.; Ma, R.; He, L.-N. J. CO2 Util. 2018, 25, 338. doi: 10.1016/j.jcou.2018.01.022
Xia, S.-M.; Song, Y.; Li, X.-D.; Li, H.-R.; He, L.-N. Molecules 2018, 23, 3033. doi: 10.3390/molecules23113033
Song, Q.-W.; Yu, B.; Li, X.-D.; Ma, R.; Diao, Z.-F.; Li, R.-G.; Li, W.; He, L.-N. Green Chem. 2014, 16, 1633. doi: 10.1039/c3gc42406e
Song, Q.-W.; Zhou, Z.-H.; Yin, H.; He, L.-N. ChemSusChem 2015, 8, 3967. doi: 10.1002/cssc.201501176
李雪冬, 郎咸东, 宋清文, 郭亚坤, 何良年, 有机化学, 2016, 36, 744. doi: 10.6023/cjoc201512037Li, X.-D.; Lang, X.-D.; Song, Q.-W.; Guo, Y.-K.; He, L.-N. Chin. J. Org. Chem. 2016, 36, 744(in Chinese). doi: 10.6023/cjoc201512037
(a) Song, Q.-W.; Chen, W.-Q.; Ma, R.; Yu, A.; Li, Q.-Y.; Chang, Y.; He, L.-N. ChemSusChem 2015, 8, 821.
(b) Zhou, Z.-H.; Guo, C.-X.; Xie, J.-N.; Liu, K.-X.; He, L.-N. Curr. Org. Synth. 2017, 14, 1185.
(c) He, L.-N. Curr. Org. Synth. 2020, 17, 2.
(d) He, L.-N. Mini-Rev. Org. Chem. 2019, 16, 409.
(a) Zhou, Z.-H.; Zhang, X.; Huang, Y.-F.; Chen, K.-H.; He, L.-N. Chin. J. Catal. 2019, 40, 1345.
(b) Zhou, Z.-H.; Chen, K.-H.; He, L.-N. Chin. J. Chem. 2019, 37, 1223.
(a) Bonin, J.; Maurin, A.; Robert, M. Coord. Chem. Rev. 2017, 334, 184.
(b) Tamaki, Y.; Ishitani, O. ACS Catal. 2017, 7, 3394.
(c) Chang, X.; Wang, T.; Yang, P.; Zhang, G.; Gong, J. Adv. Mater. 2018, 1804710.
(d) Wu, J.; Huang, Y.; Ye, W.; Li, Y. Adv. Sci. 2017, 4, 1700194.
(e) Zhao, Y.; Waterhouse, G. I. N.; Chen G.; Xiong, X.; Wu, L. Z.; Tung, C. H.; Zhang, T. Chem. Soc. Rev. 2019, 48, 1972.
(a) Ye, J. H.; Miao, M.; Huang, H.; Yan, S. S.; Yin, Z. B.; Zhou, W. J.; Yu, D. G. Angew. Chem., Int. Ed. 2017, 56, 15416.
(b) Yin, Z. B.; Ye, J. H.; Zhou, W. J.; Zhang, Y. H.; Ding, L.; Gui, Y. Y.; Yan, S. S.; Li, J.; Yu, D. G. Org. Lett. 2018, 20, 190.
(c) Sun, L.; Ye, J. H.; Zhou, W. J.; Zeng, X.; Yu, D. G. Org. Lett. 2018, 20, 3049.
(d) Ju, T.; Fu, Q.; Ye, J. H.; Zhang, Z.; Liao, L. L.; Yan, S. S.; Tian, X. Y.; Luo, S. P.; Li, J.; Yu, D. G. Angew. Chem., Int. Ed. 2018, 57, 13897.
(e) Liao, L. L.; Cao, G. M.; Ye, J. H.; Sun, G. Q.; Zhou, W. J.; Gui, Y. Y.; Yan, S. S.; Shen, G.; Yu, D. G. J. Am. Chem. Soc. 2018, 140, 17338.
(f) Fan, X.; Gong, X.; Ma, M. Nat. Commun. 2018, 9, 4936.
(g) Murata, K.; Numasawa, N.; Shimomaki, K.; Chem. Commun. 2017, 53, 3098.
(h) Yeung, C. S. Angew. Chem., Int. Ed. 2019, 58, 5491.
Wang, M.-Y.; Cao, Y.; Liu, X.; Wang, N.; He, L.-N.; Li, S.-H. Green Chem. 2017, 19, 1240. doi: 10.1039/C6GC03200A
He, X.; Cao, Y.; Lang, X.-D.; Wang, N.; He, L.-N. ChemSusChem 2018, 11, 3382. doi: 10.1002/cssc.201801621
(a) He, L. N.; Yang, Z. Z.; Liu, A. H.; Gao, J. In Advances in CO2 Conversion and Utilization, ACS Symposium Series, Vol. 1056, Ed.: Hu, Y. H., American Chemical Society, Washington DC, 2010, p. 77.
(b) He, L. N. Carbon Dioxide Chemistry, Science Press, Beijing, 2013(in Chinese).
(何良年, 二氧化碳化学, 科学出版社, 北京, 2013.
(a) Poland, S. I.; Darensbourg, D. J. Green Chem. 2017, 19, 4990.
(b) Wang, Y.; Darensbourg, D. J. Coord. Chem. Rev. 2018, 372, 85.
(c) Lu, X. B.; Darensbourg, D. J. Chem. Soc. Rev. 2012, 41, 1462.
(d) Kember, M. R.; Buchard, A.; Williams, C. K. Chem. Commun. 2011, 47, 141.
(e) Lu, X. B.; Ren, W. M.; Wu, G. P. Acc. Chem. Res. 2012, 45, 1721.
(a) Leino, E.; Maki-Arvela, P.; Eta, V. Appl. Catal., A 2010, 383, 1.
(b) Shukla, K.; Srivastava, V. C. Catal. Rev.: Sci. Eng. 2017, 59, 1.
(c) Dai, W. L.; Luo, S. L.; Yin, S. F. Appl. Catal., A 2009, 366, 2.
(d) Sakakura, T.; Kohno, K. Chem. Commun. 2009, 1312.
(e) Tamboli, A. H.; Chaugule, A. A.; Kim, H. Chem. Eng. J. 2017, 323, 530.
(a) Broere, D. L. J.; Mercado, B. Q.; Holland, P. L. Angew. Chem., Int. Ed. 2018, 57, 6507.
(b) Waldman, T. E.; Mcghee, W. D. J. Chem. Soc., Chem. Commun. 1994, 8, 957.
(c) Camp, C.; Chatelain, L.; Kefalidis, C. E. Chem. Commun. 2015, 51, 15454.
(d) Maria, L.; Bandeira, N. A. G.; Marcalo, J. Chem. Commun. 2020, 56, 431.
(e) Keane, A. J.; Farrell, W. S.; Yonke, B. L. Angew. Chem., Int. Ed. 2015, 54, 10220.
(a) Yoo, W. J.; Nguyen, T. V. Q.; Kobayashi, S. Angew. Chem., Int. Ed. 2014, 53, 10213.
(b) Ye, J. H.; Zhu, L.; Yan, S. S.; Miao, M.; Zhang, X. C.; Zhou, W. J.; Li, J.; Lan, Y.; Yu, D. G. ACS Catal. 2017, 7, 8324.
(c) Zhang, W. Z.; Yang, M. W.; Yang, X. T.; Shi, L. L.; Wang, H. B.; Lu, X. B. Org. Chem. Front. 2016, 3, 217.
(d) Cheng, L.; Xie, J. Chin. J. Org. Chem. 2020, 40, 247(in Chinese).
(程磊, 谢建华, 有机化学, 2020, 40, 247.)
(e) Xu, P., Wang, S.; Fang, Y.; Ji, S. J. Chin. J. Org. Chem. 2018, 38, 1626(in Chinese).
(徐佩, 汪顺义, 方毅, 纪顺俊, 有机化学, 2018, 38, 1626.)
(f) Zhang, W.; Lü, X. Chin. J. Catal. 2012, 33, 745(in Chinese).
(张文珍, 吕小兵, 催化学报, 2012, 33, 745.)
(g) Zhu, Q.; Wang, L.; Xia, C.; Liu, C. Chin. J. Org. Chem. 2016, 36, 2813(in Chinese).
(朱庆, 王露, 夏春谷, 刘超, 有机化学, 2016, 36, 2813.)
(h) Fan Q.; Liu, J.; Chen J.; Xia, C. Chin. J. Catal. 2012, 33, 1435(in Chinese).
(樊启佳, 刘建华, 陈静, 夏春谷, 催化学报, 2012, 33, 1435.)
(i) Jia, X.; Wang, Z.; Xia, C.; Ding, K. Chin. J. Org. Chem. 2013, 33, 1369(in Chinese).
(贾肖飞, 王正, 夏春谷, 丁奎岭, 有机化学, 2013, 33, 1369.)
(j) Liu, Q.; Shi, L.; Liu, N. Chin. J. Org. Chem. 2019, 39, 2882(in Chinese).
(刘铨瑶, 石磊, 刘宁, 有机化学, 2019, 39, 2882.)
(k) Jiang, H.; Zhang, Y.; Xiong, W.; Cen, J.; Wang, L.; Cheng, R.; Qi, C.; Wu, W. Org. Lett. 2019, 21, 345;
(l) Zhang, Y.; Xiong, W.; Cen, J.; Yan, W.; Wu, Y.; Qi, C.; Wu, W.; Jiang, H. Chem. Commun. 2019, 55, 12304.
(m) Yu, B.; Xie, J. N.; Zhong, C. L.; Li, W.; He, L. N. ACS Catal. 2015, 5, 3940;
(n) Qiu, J.; Gao, S.; Li, C.; Zhang, L.; Wang, Z.; Wang, X.; Ding, K. Chem.-Eur. J. 2019, 25, 13874.
(a) Xie, C.; Chen, C.; Yu, Y. Nano Lett. 2017, 17, 3798.
(b) Ramirez, A.; Ould-Chikh, S.; Gevers, L. ChemCatChem 2019, 11, 2879.
(c) Yang, H.; Zhang, C.; Gao, P. Catal. Sci. Technol. 2017, 7, 4580.
(d) Prieto, G. ChemSusChem 2017, 10, 1056.
(e) Aitbekova, A.; Goodman, E. D.; Wu, L. Angew. Chem., Int. Ed. 2019, 58, 17451.
(f) Saeidi, S.; Amin, N. A. S.; Rahimpour, M. R. J. CO2 Util. 2014, 5, 66.
(g) Rezayee, N. M.; Huff, C. A.; Sanford, M. S. J. Am. Chem. Soc. 2015, 137, 1028.
(h) Li, Y.; Wang, Z.; Liu, Q. Chin. J. Org. Chem. 2017, 37, 1978(in Chinese).
(李勇, 王征, 刘庆彬, 有机化学, 2017, 37, 1978.)
(i) Zhang, L.; Han, Z.; Zhang, L.; Li, M.; Ding, K. Chin. J. Org. Chem. 2016, 36, 1824(in Chinese).
(张琳莉, 韩召斌, 张磊, 李明星, 丁奎岭, 有机化学, 2016, 36, 1824.)
(j) Han, Z.; Rong, L.; Wu, J.; Zhang, L.; Wang, Z.; Ding, K. Angew. Chem., Int. Ed. 2012, 51, 13041.
(k) Zhang, L.; Han, Z.; Zhao, X.; Wang, Z.; Ding, K. Angew. Chem., Int. Ed. 2015, 54, 6186.
(l) Dong, K.; Razzaq, R.; Hu, Y.; Ding, K. Top Curr. Chem. 2017, 375, 203.
Wang, H.; Zhao, Y.; Ke, Z.; Yu, B.; Li, R.; Wu, Y.; Wang, Z.; Han, J.; Liu, Z. Chem. Commun. 2019, 55, 3069. doi: 10.1039/C9CC00819E
(a) Akatsuka, M.; Kawaguchi, Y.; Itoh, R.; Ozawa, A.; Yamamoto, M.; Tanabe, T.; Yoshida, T. Appl. Catal., B 2020, 262, 118247.
(b) Teramura, K.; Hori, K.; Terao, Y.; Huang, Z.; Iguchi, S.; Wang, Z.; Asakura, H.; Hosokawa, S.; Tanaka, T. J. Phys. Chem. C 2017, 121, 8711.
(c) Nakada, A.; Ishitani, O. ACS Catal. 2018, 8, 354.
(d) Takayama, T.; Sato, K.; Fujimura, T.; Kojima, Y.; Iwase A.; Kudo, A. Faraday Discuss. 2017, 198, 397.
(e) Barman, S.; Das, S.; Sreejith S. S.; Garai, S.; Pochamoni, R.; Roy, S. Chem. Commun. 2018, 54, 2369.
(f) Nakanishi, H.; Iizuka, K.; Takayama, T.; Iwase, A.; Kudo, A. ChemSusChem 2017, 10, 112.
(g) Yin, G.; Sako, H.; Gubbala, R. V.; Ueda, S.; Yamaguchi, A.; Abe, H.; Miyauchi, M. Chem. Commun. 2018, 54, 3947.
(a) Xu, S.; Carter, E. A. Chem. Rev. 2019, 119, 6631;
(b) Yuan, Y. P.; Ruan, L. W.; Barber, J. Energy Environ. Sci. 2014, 7, 3934.
(c) Daiyan, R.; Lu, X.; Ng, Y. H. ChemSusChem 2017, 10, 4342.
He, L. N.; Wang, J. Q.; Wang, J. L. Pure Appl. Chem. 2009, 81, 2069. doi: 10.1351/PAC-CON-08-10-22
(a) Lang, X. D.; Yu, Y. C.; He, L. N. J. Mol. Catal. A: Chem. 2016, 420, 208.
(b) Xu, H.; Liu, X. F.; Cao, C. S.; Zhao, B.; Cheng, P.; He, L. N. Adv. Sci. 2016, 3, 1600048.
(c) Cao, C. S.; Xia, S. M.; Song, Z. J.; Xu, H.; Shi, Y.; He, L. N.; Cheng, P.; Zhao, B. Angew. Chem., Int. Ed. 2020, 59, 8586.
(a) Li, Y. N.; He, L. N. Chin. Sci. Bull. 2015, 60, 1465(in Chinese).
(李雨浓, 何良年, 科学通报, 2015, 60, 1465.)
(b) Fu, H. C.; Fei, Y.; Li, H. R.; He, L. N. Front. Chem. 2019, 7, 525.

图式 1 超强碱和聚乙二醇的催化体系用于CO2捕集与转化
Scheme 1 CO2 capture and utilization by superbase/polyethyl- ene glycol catalyst system

图式 2 聚乙二醇功能化的离子液体用于CO2捕集与转化
Scheme 2 CO2 capture and utilization by polyethylene glycol functionalized ionic liquid

图式 3 PEG150MeIm/PEG150吸收后的SO2与环氧丙烷的反应
Scheme 3 Reactions of epoxides with SO2 absorbed by PEG150- MeIm/PEG150

图式 5 离子液体催化的CO2与2-氨基苯腈反应
Scheme 5 Cyclization of 2-aminobenzonitriles with CO2 catalyzed by ionic liquids

图式 6 氨基酸盐以及邻苯二甲酰亚胺盐用于催化CO2的捕集与转化
Scheme 6 Amino acid salts and phthalimide salts used in CO2 capture and utilization

图式 8 氢硅烷类型控制的CO2的还原功能化
Scheme 8 CO2 reduction functionalization controlled by the type of hydrosilanes

图式 9 不同条件下的CO2的还原功能化
Scheme 9 CO2 reduction functionalization under different conditions

图式 10 CO2压力控制的CO2的还原功能化
Scheme 10 CO2 reduction functionalization controlled by the pressure of CO2

图式 12 CO2通过还原为CO的还原功能化反应
Scheme 12 CO2 reduction functionalization through the formation of CO

图式 14 甲酸用于Pauson-Khand反应
Scheme 14 Pauson-Khand reaction with formic acid as substrate

图式 15 银催化的炔丙醇、邻二醇和CO2合成环碳酸酯
Scheme 15 Silver-catalyzed cyclic carbonates synthesis from propargylic alcohols, diols and CO2

图式 16 炔丙醇、CO2和2-乙醇胺类化合物合成2-噁唑啉酮的催化体系
Scheme 16 Catalytic systems for 2-oxazolidinones synthesis from propargylic alcohols, CO2 and 2-aminoethanols

图式 17 Ag2WO4/PPh3促进的炔丙醇、CO2和胺三组分级联反应
Scheme 17 Ag2WO4/PPh3 promoted the tandem reaction of propargylic alcohols, CO2 and amines

图式 18 Ag2O/PPh3催化的炔丙醇与氨基甲酸盐合成β-羰基氨基甲酸酯的反应
Scheme 18 Ag2O/PPh3 facilitated β-oxopropylcarbamates synthesis from propargylic alcohols and ammonium carbamates

图式 19 CuI催化炔丙醇、CO2和二级胺合成β-羰基氨基甲酸酯的反应
Scheme 19 CuI cayalyzed β-oxopropylcarbamates synthesis from propargylic alcohols, CO2 and secondary amines

图式 20 Ag2CO3/PPh3催化炔丙醇、CO2和一元醇生成β-羰基碳酸酯的反应
Scheme 20 Ag2CO3/PPh3 promoted β-oxopropylcarbonates synthesis from propargylic alcohols, CO2 and monohydric alcohols

图式 21 CO2作为助催化剂实现的炔丙醇水解合成β-羟基酮的反应
Scheme 21 Copper(I)-catalyzed hydration of propargylic alcohols to β-hydroxy ketones with CO2 as a cocatalyst

图式 22 光致自由基驱动的烯丙胺与CO2的羧化环化反应
Scheme 22 Photoinduced radical-initiated carboxylative cyclization of allyl amines with CO2

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:




