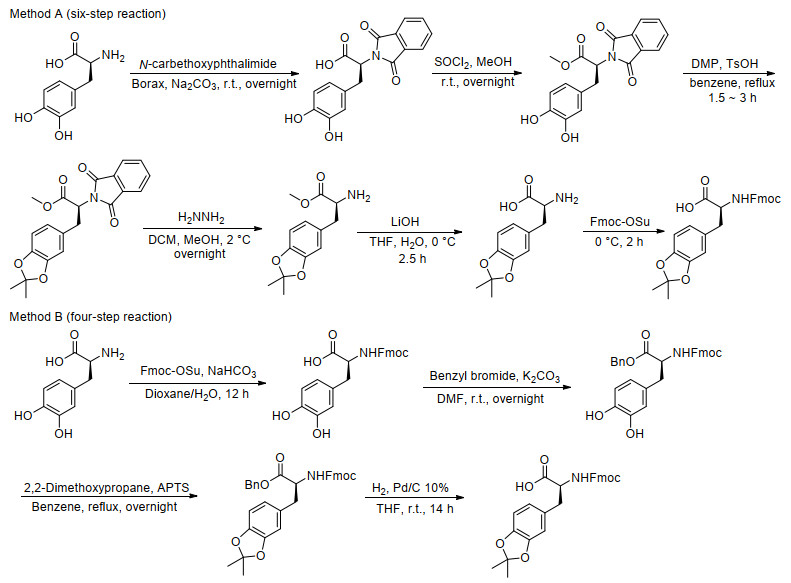
图式 1.
Fmoc-DOPA(acetonide)-OH已有的合成方法
Scheme 1.
Previous synthesis methods to obtain Fmoc-DOPA(acetonide)-OH

海洋生物贻贝分泌粘附蛋白, 使其能在复杂环境中紧密地附着于各种表面, 如岩石、金属和高分子材料等[1].贻贝粘附蛋白中高含量的3, 4-二羟基苯丙氨酸(DOPA)在贻贝粘附行为中起到关键作用[2], 贻贝粘附蛋白的优异粘合性能引起了人们对开发DOPA衍生物以增强生物材料的界面粘合[3]、表面改性[4]、自组装[5]等领域的广泛兴趣.
Fmoc-DOPA(acetonide)-OH广泛用作粘附蛋白和多肽的固相合成原料[6].然而, 由于其较高的商业价格和繁琐的合成步骤, 使其在实际应用中受到极大的阻碍.关于Fmoc-DOPA(acetonide)-OH的合成方法仅有少数报道, 其中重要的突破包括(Scheme 1): Messersmith等[7]设计了六步反应合成Fmoc-DOPA(acetonide)-OH, 并发表了专利涵盖了该化合物类似的多步合成路线[8]; Marsault等[9]简化了反应路线, 通过四步反应得到Fmoc-DOPA(acetonide)-OH.然而, 这两种方法都有各自的缺点, 前一种六步合成法步骤繁琐耗时、涉及反应试剂较多, 后一种四步合成法需要使用易燃易爆的氢气.因此, 迫切需要开发一种简单、经济高效的方法来合成Fmoc-DOPA(acetonide)-OH.本工作报告了一种简单、经济的Fmoc-DOPA(acetonide)-OH两步合成方法.
Wilker等[10]尝试以Fmoc-DOPA为原料, 在不同的温度、溶剂和催化剂条件下, 将其与丙酮回流, 虽然能够获得缩酮化产物, 但收率并不高.在此基础上, Messersmith等[7]认为充分保护L-DOPA的氨基和羧基基团是将邻苯二酚成功缩酮化的关键前提, 因此分别先用邻苯二甲酰亚胺和甲酯保护氨基和羧基, 再进行邻苯二酚基团的保护, 但是多步的保护与脱保护步骤导致反应路线复杂繁琐.我们考虑到在缩酮化的过程中会产生水分子, 这些少量的水分子会阻碍反应进程, 使得反应的收率不理想.我们以L-DOPA为起始原料, 第一步将氨基通过Fmoc保护得到化合物Fmoc-DOPA, 第二步在催化剂对甲苯磺酸和脱水剂无水硫酸镁的作用下通过与丙酮一步反应将邻苯二酚基团直接转化为缩丙酮基团得到最终化合物Fmoc-DOPA(acetonide)-OH, 整个过程不需要对羧基进行保护.该方法具有原料及试剂便宜易得、反应条件温和、操作简单、成本低的优点.
在之前的六步合成方法中[7], 需要先用邻苯二甲酰亚胺基团保护氨基, 甲酯保护羧基, 再进行邻苯二酚基团的保护, 虽然这种方法可以得到最终的产物, 但是邻苯二甲酰亚胺和甲酯基团的保护与脱除增加了反应的步骤, 使得合成路线变得繁琐.在之后的实验中, Marsault等[9]优化了合成路线, 用四步法得到了最终的产物, 并将Fmoc-DOPA上的羧基和苄基溴反应得到Fmoc-DOPA-OBn, 但是苄基酯的脱除需要用到氢气, 这使得这个合成路线存在安全隐患.基于以上相关文献的调阅与分析, 我们设计了如Scheme 2所示的新的合成路线.在得到Fmoc-DOPA的基础上, 用一步反应直接得到所需要的最终产物, 使得反应过程变得非常简单, 同时避免易燃易爆炸的氢气的使用.
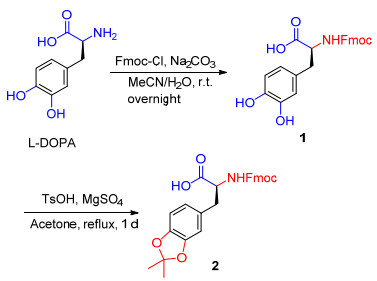
在Scheme 2所示的合成路线中, 使用9-芴基甲基氯甲酸酯(Fmoc-Cl)对化合物L-DOPA上的氨基进行保护, 乙腈和水作为溶液, Na2CO3提供碱性环境, 室温下反应过夜, 以88%的收率得到了化合物Fmoc-DOPA (1).第二步将化合物1在过量的丙酮中回流, 加入无水硫酸镁作为脱水剂, 对甲苯磺酸作为催化剂, 得到1.1 g的化合物Fmoc-DOPA(acetonide)-OH (2), 收率为49%.在这个反应中, 丙酮同时充当试剂和溶剂, 两步反应的总收率为43%.此外, 通过计算, 上述两种合成路线获得的最终产品的总成本远低于Sigma-Aldrich公司的价格和先前报道的两种方法的合成成本(表 1).
 下载:
导出CSV
下载:
导出CSV
| Source | Price ($•g-1) |
| Sigma-Aldrich | 563 |
| The six-step reaction method a | ≈6 |
| The four-step reaction method b | ≈20 |
| Our method c | ≈2 |
| a The cost of laboratory synthesis using method developed by Messersmith et al.[7]. b The cost of laboratory synthesis using method developed by Marsault et al.[9]. c The cost of laboratory synthesis using both strategies in the article. | |
为了确定Fmoc-DOPA(acetonide)-OH的手性结构, 用含有25%哌啶的二氯甲烷和体积比为TFA/TIS/H2O (V:V:V=95:2.5:2.5)溶液处理Fmoc-DOPA(aceto- nide)-OH, 脱除Fmoc和缩丙酮保护基团, 从而得到DOPA的粗产物进行手性HPLC分析[11], 同时使用市售的L-DOPA和D-/L-DOPA作为参考.结果显示, 由合成的Fmoc-DOPA(acetonide)-OH脱保护后得到的DOPA的手性HPLC谱图中, 仅有L-DOPA峰, 证实了合成的Fmoc-DOPA(acetonide)-OH的光学纯度ee值大于99%.在整个合成路线中, L-DOPA保留了其手性结构.
本工作的Fmoc-DOPA(acetonide)-OH合成路线首先选择氨基的Fmoc保护, 随后再进行邻苯二酚的缩酮化.之所以这样设计, 是因为如果改变这两步反应的顺序会有以下缺点: (1) DOPA很难直接缩酮化, 必须用DOPA盐酸盐作为原料以提高在丙酮中的溶解性才能实现[12], 而DOPA盐酸盐商业不可得, 必须经过一步制备, 增加了实验的步骤; (2)先进行氨基Fmoc保护, 得到的产物Fmoc-DOPA易溶于多种有机溶剂, 可以通过萃取、进一步简单的重结晶或快速柱层析提纯得到产物; 而如果先进行邻苯二酚的缩酮化反应, 由于原料和产物都是极性很大的氨基酸, 不仅很难通过薄层色谱监测反应, 而且很难实现产物的提纯.基于以上原因, 可以推断, 本工作采用的Fmoc-DOPA(acetonide)-OH合成路线为理论上最简单高效的方法.
开发了一种简单、高效的Fmoc-DOPA(acetonide)- OH合成方法, 并具有良好的收率, 这极大地促进了其在贻贝粘附蛋白和多肽固相合成中的应用, 有望推动更多新型粘合剂材料的合成开发及其在生物材料和医疗设备表面改性中的应用.
核磁共振谱图在AVANCE Ⅲ (400 MHz)超导傅立叶变换核磁共振波谱仪上测定, 以四甲基硅烷(TMS)为内参, 使用CDCl3或DMSO-d6为溶剂; 质谱仪为XEVO G2 TOF型(Waters), 采用电喷雾离子化(ESI)离子源; 使用配备有常州三泰科技有限公司生产的Sepaflash色谱柱的SepaBean机器纯化合成的化合物; 使用配有Daicel crownpak cr(+)色谱柱的液相色谱仪来进行手性分析, 流动相为等梯度的高氯酸水溶液(0.01 mol/L), 流速为0.5 mL/min; 所有试剂均为国产分析纯, 不经处理直接使用.
将DOPA (15 g, 76.1 mmol)和9-芴基甲基氯甲酸酯(23.6 g, 91.3 mmol)置于1000 mL的圆底烧瓶中, 并加入160 mL乙腈搅拌, 将其在冰浴中冷却.将碳酸钠(32.3 g, 304.3 mmol)溶于120 mL去离子水中, 逐滴加入到上述的反应体系中.恢复至室温, 并将混合物搅拌过夜, 将混合物在室温搅拌过夜, 然后用2 mol/L HCl调节pH到2~3.通过减压蒸馏除去乙腈后, 将粗产物萃取到二氯甲烷(400 mL×3)中, 3次萃取结束后将有机相收集在一起, 用饱和食盐水(200 mL)洗涤, 无水硫酸镁干燥有机相后过滤, 通过减压蒸馏得到浓缩液.通过硅胶柱层析, 用二氯甲烷和甲醇作为流动相得到28.1 g的纯产物1 [10], 收率为88%. 1H NMR (400 MHz, DMSO-d6) δ: 8.85 (br, 1H), 7.88 (d, J=7.2 Hz, 2H), 7.69 (d, J=8.0 Hz, 2H), 7.66 (d, J=7.6 Hz, 1H), 7.42 (td, J=7.6, 3.2 Hz, 2H), 7.37~7.22 (m, 2H), 6.77~6.64 (m, 2H), 6.56 (dd, J=8.0, 1.6 Hz, 1H), 4.31~4.07 (m, 4H), 2.94 (dd, J=13.6, 4.4 Hz, 1H), 2.74 (dd, J=13.7, 10.4 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 173.7, 156.1, 145.0, 143.9, 143.9, 143.9, 140.8, 128.8, 127.7, 127.2, 125.5, 125.4, 120.2, 120.0, 116.6, 115.4, 65.8, 56.1, 46.7, 36.1; ESI-MS m/z: 837.3 [2M+H]+.
将化合物(1) (2 g, 4.8 mmol)置于50 mL的圆底烧瓶中, 加入20 mL丙酮搅拌溶解, 然后向反应混合物中加入对甲苯磺酸(0.4 g, 2.4 mmol)和无水硫酸镁(2.3 g, 19.1 mmol).将反应混合物在氮气氛围下回流搅拌1 d.冷却至室温后, 通过减压蒸馏除去丙酮.将粗产物萃取到乙酸乙酯(30 mL×3)中, 3次萃取结束后将有机相收集在一起并用饱和食盐水(20 mL)洗涤, 用无水硫酸镁干燥有机相后过滤, 通过减压蒸馏得到浓缩液.通过硅胶柱层析, 用二氯甲烷和甲醇作为流动相, 得到1.1 g的纯产物2[7], 收率为49%. 1H NMR (400 MHz, CDCl3) δ: 7.77 (d, J=7.6 Hz, 2H), 7.62~7.48 (m, 2H), 7.40 (t, J=7.2 Hz, 2H), 7.31 (td, J=7.2, 2.8 Hz, 2H), 6.66 (d, J=7.7 Hz, 1H), 6.59~6.43 (m, 2H), 5.26 (d, J=8.1 Hz, 1H), 4.66 (dd, J=13.2, 5.6 Hz, 1H), 4.46 (dd, J=10.8, 7.2 Hz, 1H), 4.37 (dd, J=10.8, 7.2 Hz, 1H), 4.22 (t, J=7.2 Hz, 1H), 3.18~2.98 (m, 2H), 1.65 (s, 6H). 13C NMR (100 MHz, CDCl3) δ: 176.3, 156.0, 147.8, 146.7, 143.9, 143.8, 141.4, 128.6, 127.8, 127.2, 125.2, 125.2, 122.0, 120.1, 118.1, 109.5, 108.4, 67.2, 54.9, 47.2, 37.5, 29.8, 26.0; ESI-MS m/z: 482.3 [M+Na]+.
辅助材料(Supporting Information) 化合物1和2的1H NMR和13C NMR谱图以及手性HPLC谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(a) Waite, J. H.; Qin, X. Biochemistry 2001, 40, 2887.
(b) Kord Forooshani, P.; Lee, B. P. J. Polym. Sci., Part A: Polym. Chem. 2016, 55, 9.
(c) Lee, B. P.; Messersmith, P. B.; Israelachvili, J. N.; Waite, J. H. Annu. Rev. Mater. Res. 2011, 41, 99.
(a) Lee, H.; Dellatore, S. M.; Miller, W. M.; Messersmith, P. B. Science 2007, 318, 426.
(b) Lee, H. A.; Ma, Y. F.; Zhou, F.; Hong, S.; Lee, H. Acc. Chem. Res. 2019, 52, 704.
(c) Ye, Q.; Zhou, F.; Liu, W. M. Chem. Soc. Rev. 2011, 40, 4244.
(a) Liu, Y.; Ai, K.; Lu, L. Chem. Rev. 2014, 114, 5057.
(b) Wu, R.; Ding, X. K.; Qi, Y.; Zeng, Q.; Wu, Y. W.; Yu, B. R.; Xu, F. J. Small 2018, 14, 11.
(c) Xu, M.; Khan, A.; Wang, T.; Song, Q.; Han, C.; Wang, Q.; Gao, L.; Huang, X.; Li, P.; Huang, W. ACS Appl. Bio Mater. 2019, 2, 3329.
(d) Li, X. Y.; Gao, P.; Tan, J. Y.; Xiong, K. Q.; Maitz, M. F.; Pan, C. J.; Wu, H. K.; Chen, Y.; Yang, Z. L.; Huang, N. ACS Appl. Mater. Interfaces 2018, 10, 40844.
(e) Pandey, N.; Hakamivala, A.; Xu, C. C.; Hariharan, P.; Radionov, B.; Huang, Z.; Liao, J.; Tang, L. P.; Zimmern, P.; Nguyen, K. T.; Hong, Y. Adv. Healthcare Mater. 2018, 7, 1701069.
(a) Barclay, T. G.; Hegab, H. M.; Clarke, S. R.; Ginic-Markovic, M. Adv. Mater. Interfaces 2017, 4, 1601192.
(b) Liu, X. S.; Cao, J. M.; Li, H.; Li, J. Y.; Jin, Q.; Ren, K. F.; Ji, J. ACS Nano 2013, 7, 9384.
(c) Yang, C.; Ding, X.; Ono, R. J.; Lee, H.; Hsu, L. Y.; Tong, Y. W.; Hedrick, J.; Yang, Y. Y. Chem. Sci. 2014, 26, 7346.
(d) Sheng, W. B.; Li, B.; Wang, X. L.; Dai, B.; Yu, B.; Jia, X.; Zhou, F. Chem. Sci. 2015, 6, 2068.
(e) Zhang, S. X.; Liu, W. Y.; Dong, Y. S.; Wei, T.; Wu, Z. Q.; Chen, H. Langmuir 2019, 35, 3470.
(f) Liu, L.; Tian, X. H.; Ma, Y.; Duan, Y. Q.; Zhao, X.; Pan, G. Q. Angew. Chem., Int. Ed. 2018, 57, 7878.
(g) Wu, J. X.; Zheng, Y. J.; Jiang, S. B.; Qu, Y. C.; Wei, T.; Zhan, W. J.; Wang, L.; Yu, Q.; Chen, H. ACS Appl. Mater. Interfaces 2019, 11, 12357.
(a) Zhao, Y. H.; Wu, Y.; Wang, L.; Zhang, M. M.; Chen, X.; Liu, M. J.; Fan, J.; Liu, J. Q.; Zhou, F.; Wang, Z. K. Nat. Commun. 2017, 8, 8.
(b) Sheng, W. B.; Li, W.; Yu, B.; Li, B.; Jordan, R.; Jia, X.; Zhou, F. Angew. Chem., Int. Ed. 2019, 58, 12018.
(c) Cong, Z. Q.; Zhang, L.; Ma, S. Q.; Lam, K. S.; Yang, F. F.; Liao, Y. H. ACS Nano 2020, 14, 1958.
(a) Liu, L.; Tian, X. H.; Ma, Y.; Duan, Y. Q.; Zhao, X.; Pan, G. Q. Angew. Chem., Int. Ed. 2018, 57, 7878.
(b) Kuang, J.; Messersmith, P. B. Langmuir 2012, 28, 7258.
(c) Statz, A. R.; Meagher, R. J.; Barron, A. E.; Messersmith, P. B. J. Am. Chem. Soc. 2005, 127, 7972.
Liu, Z.; Hu, B. H.; Messersmith, P. B. Tetrahedron Lett. 2008, 49, 5519. doi: 10.1016/j.tetlet.2008.07.052
Messersmith, P. B.; Hu, B.-H.; Liu, Z. US 0087622, 2010.
St-Georges, C.; Desilets, A.; Beliveau, F.; Ghinet, M.; Dion, S. P.; Colombo, E.; Boudreault, P. L.; Najmanovich, R. J.; Leduc, R.; Marsault, E. Eur. J. Med. Chem. 2017, 129, 110. doi: 10.1016/j.ejmech.2017.02.006
Sever, M. J.; Wilker, J. J. Tetrahedron 2001, 57, 6139. doi: 10.1016/S0040-4020(01)00601-9
(a) Tredwell, M.; Preshlock, S. M.; Taylor, N. J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. Angew. Chem., Int. Ed. 2014, 53, 7751.
(b) Liu, Z.; Hu, B. H.; Messersmith, P. B. Tetrahedron Lett. 2008, 49, 5519.
oloshonok, V. A.; Ueki, H. Synthesis 2008, 693.

图式 1 Fmoc-DOPA(acetonide)-OH已有的合成方法
Scheme 1 Previous synthesis methods to obtain Fmoc-DOPA(acetonide)-OH

图式 2 Fmoc-DOPA(acetonide)-OH的合成路线
Scheme 2 Synthesis method of Fmoc-DOPA(acetonide)-OH
表 1 不同方法制备Fmoc-DOPA(acetonide)-OH的成本比较
Table 1. Comparison of cost to obtain Fmoc-DOPA(acetonide)- OH from different ways
| Source | Price ($•g-1) |
| Sigma-Aldrich | 563 |
| The six-step reaction method a | ≈6 |
| The four-step reaction method b | ≈20 |
| Our method c | ≈2 |
| a The cost of laboratory synthesis using method developed by Messersmith et al.[7]. b The cost of laboratory synthesis using method developed by Marsault et al.[9]. c The cost of laboratory synthesis using both strategies in the article. | |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们