


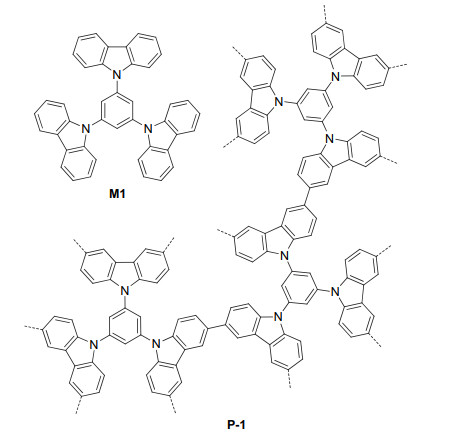
Figure 1.
Structures of M-1 and P-1
Porous Organic Polymers as Heterogeneous Catalysts for Visible Light-Induced Organic Transformations
Zi-Yue Xu , Yi Luo , Hui Wang , Dan-Wei Zhang , Zhan-Ting Li

Catalysis plays a key role in modern chemistry and chemical engineering. Homogeneous catalysis has many advantages, including the structural diversity of catalysts that work for wide ranges of reactions, high catalysis efficiency and good reaction controllability, [1] but typically suffers from the difficulty in the recovery and recyclable use of the catalysts. For a long time, heterogeneous catalysis through flow reactors has seized the majority of scaled industrial catalytic processes due to their engineering advantages.[2] In the past decades, heterogeneous catalysis in closed reactors for complicated organic transformations has also received increasing attention because repeated use of the catalysts can be readily realized.[3] However, for heterogeneous catalysis with solid or solid-supported catalysts, only catalytic sites on the surface can allow for the approach of reactant molecules, whereas the surface area of solid catalysts is generally limited. Porous architectures provide large platforms for the development of new heterogeneous catalysts that not only possess large surface areas for increased catalysis efficiency, but also allow for convenient recovery and repeated use of the catalysts. In this category, crystalline metal-organic frameworks and covalent-organic frameworks, which are constructed through coordination or reversible covalent bonding, have been vigorously investigated due to their large surface areas and well-defined pore sizes.[4-5] Porous organic polymers and related structures represent another family of frameworks that do not form regular pores, but can be synthesized and characterized more conveniently. The covalent frameworks themselves can contain catalytic sites for organic transformations. Their backbones can also be incorporated with or attached with additional transition metal complexes for the catalysis of organic transformations. In the past decade, studies along this line has become one of important topics for porous organic polymers and related structures.[6]
Visible light-induced organic transformations have attracted attention of chemists in the early 20th century.[7] In recent years, this bonding-forming strategy has come to the forefront in organic synthesis due to its efficient utilization of sunlight, mild reaction conditions and broad applicability for a large variety of organic transformations.[8] Compared with solid polymers, porous organic polymers usually exhibit increased surface areas and transparency that facilitate visible light excitation on the active sites in the interior. This review highlights the recent advance in the applications of POPs and related porous polymers as heterogeneous catalysts for visible light-induced organic transformations.
Porous organic polymers (POPs) are a class of three-dimensional (3D) porous network polymers which are formed through strong covalent linkages between organic components. Depending on their degree of long- range order, POPs can be generally divided into crystalline and amorphous ones. Crystalline POPs are the so-called COFs. Two-dimensional COFs (2D) are versatile platforms for the development of heterogeneous catalysts through loadings or postmodifications.[5] However, the characterization of the ordering of the structures has also been a challenge, [9] and the decomposition of the dynamic covalent bonds that enable the ordering of the structures may impede the accomplishment of high recyclability.[10] Amorphous POPs do not form regular pores, but usually do not introduce dynamic covalent bonding and thus generally have a high stability. Their synthesis, purification and characterizations are generally straightforward, whereas the pores can be large enough to allow designed catalytic reactions to take place in the interior. Many amorphous porous organic polymers are also termed as polymers of intrinsic micropores (PIMs), [11] conjugated microporous polymers (CMPs), [12] porous aromatic frameworks (PAFs), [13] or hyper-crosslinked polymers (HCPs), [14] etc. The key feature of these solid polymers is that they all produce pores for exploring different functions, including heterogeneous photocatalysis, which are more environmentally friendly and sustainable compared with traditional metal-based photosensitization. This review focuses on their intrinsic or loading- or postmodification-derived functions in visible light-induced catalysis of organic transformations.
Porous organic polymers are typically prepared from the coupling reactions of structurally complementary multi- armed molecular components. In 2012, Han et al.[15] reported that carbazole-connected porous polymer P-1 could be prepared from the oxidation coupling of compound M1 by FeCl3 (Figure 1). The Brunauer-Emmett-Teller (BET) specific surface area of P-1 was determined to be 2220 m2·g-1 and exhibited strong capacity of adsorbing gases. Loh et al.[16] further prepared P-1 from M1. This porous polymer had a BET surface area of 1137 m2·g-1 and was thermally stable to 600 ℃. Carbazole and its derivatives have been demonstrated as photoactive molecules that exhibit strong electron-donating capabilities.[17] P-1 prepared by Loh et al. was found to be a highly effective heterogeneous photocatalyst for visible light-induced molecular O2 oxidations of different organic compounds (Eqs. 1~3), such as the oxidative coupling of primary amines 1 to imines 2, aerobic dehydrogenation of nonactive secondary amines 3, including pharmaceutically relevant nitrogen heterocycles, to imines 4, and selective molecular O2 oxidation of sulfides 5 to sulfoxides 6 and sulfones 7 with a high selectivity for the sulfoxide products. The catalyst could be readily recycled by simple filtration and rinsing in acetonitrile. For the oxidation of thioanisole, P-1 could maintain a high photocatalytic activity for seven times of reuse.
|
|
(1) |
|
|
(2) |
|
|
(3) |
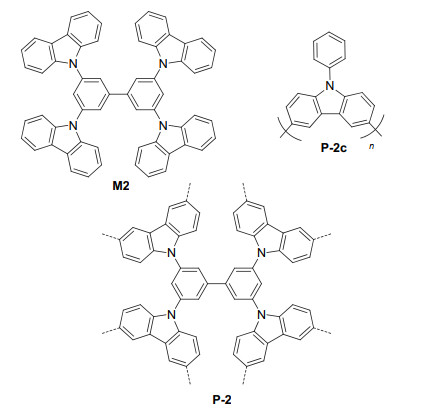
Zhang et al.[18] used FeCl3 to oxide M2 to prepare porous polymer P-2 (Figure 2). P-2 had a BET surface area of 2065 m2·g-1. The extended carbazole π-conjugation enhanced its visible light absorption. It was revealed that the large porosity of P-2 could accelerate the reaction rates of organic transformations under visible light irradiation. For the dehalogenation of phenacyl bromides 8 to afford the corresponding ketones 9 with Hantzsch ester as the reductant (Eq. 4).[19] The conversion of different bromides was quantitative, and the ketone products were obtained in 79%~98% yields. When a nonporous control polymer P-2c, which has a BET surface area of 95 m2·g-1, was used as the catalyst, the reaction proceeded with a slower reaction rate and lower yield, which showed the important effect of the porosity on the reaction rate. With NEt3 as a sacrificial reductant and molecular O2 in air as the green oxidant, P-2 also catalyzed the photooxidation of arylboronic acids 10 to the corresponding phenols 11 in 64%~97% yields (Eq. 5). In contrast, under the identical reaction conditions, control polymer P-2c-mediated reaction of phenylboronic acid afford phenol in only 28% yield, which was much lower than that (94%) obtained by P-2. For the visible light-induced α-alkylation of aldehydes 12 with electron-withdrawing group (EWG)-bearing bromides 13, P-2 also exhibited strong catalysis capacity and the corresponding alkylated aldehydes 14 could be obtained in 68%~92% yields (Eq. 6), using the reaction conditions developed for homogeneous photocatalysis. Again, nonporous control P-2c led to poor yield and selectivity even after longer reaction time. Polymer P-2 can also be easily recovered and reused via centrifugation. For the debromination of phenacyl bromide and the oxidative hydroxylation of phenylboronic acid, after 5 and 10 times of recycling, it did not show significantly decreasing reactivity, and Fourier Transform Infrared (FTIR) spectra of the recovered sample indicated no apparent structural change, which suggested a satisfactory stability.
|
|
(4) |
|
|
(5) |
|
|
(6) |
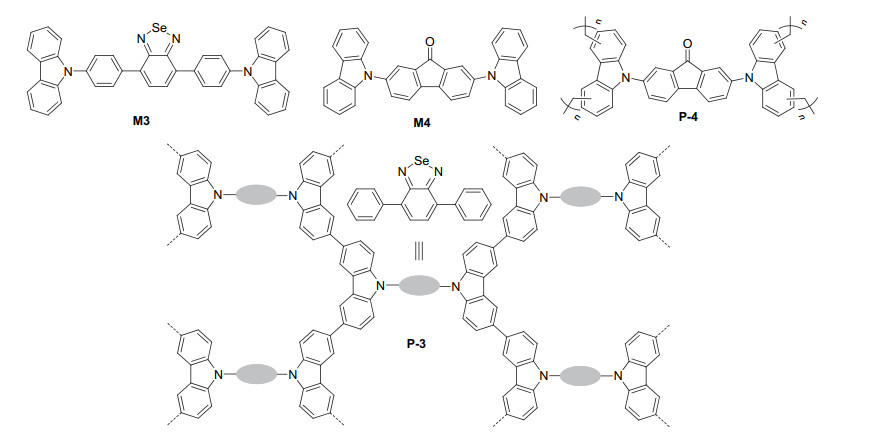
Several other carbazole-based POPs have also been synthesized for a variety of visible light-induced organic transformations. For example, Liu et al.[20] conducted the oxidation coupling of M3 with FeCl3 to prepare polymer P-3 (Figure 3), which has a BET surface area of 793 m2· g-1. This POP was found to efficiently catalyze the oxidation of mustard gas simulant 2-chloroethyl ethyl sulfide into nontoxic product 2-chloroethyl ethyl sulfoxide in methanol. After 1 h, the sulfoxide could be formed in 99% yield. The POP catalyst could be used for at least ten times with no observable reduction of the activity. P-3 could also catalyze visible light-induced oxidation of α-terpiene 15 by molecular oxygen, which afforded compounds 16~18 and others (Eq. 7). After 1 h of irradiation, ascaridole 16 was obtained as major product in 80.7% yield. Liu et al.[21] also prepared polymer P-4 from the cross-linking of M4 with dimethoxymethane under the catalysis of FeCl3. The BET surface area of P-4 was determined to be 1217 m2·g-1. The incorporation of the fluorenone unit as linker increased the absorption of the polymer for visible light and enriched its redox property. Similar to P-1, P-4 could also catalyze the O2 oxidation of primary and secondary amines to afford the corresponding imine products (Eqs. 1 and 2). For the oxidation of methylphenylsufane and its derivatives (Eq. 3), the corresponding sulfoxide products were exclusively formed and the further oxidation of the sulfoxides to sulfones was avoided.
|
|
(7) |
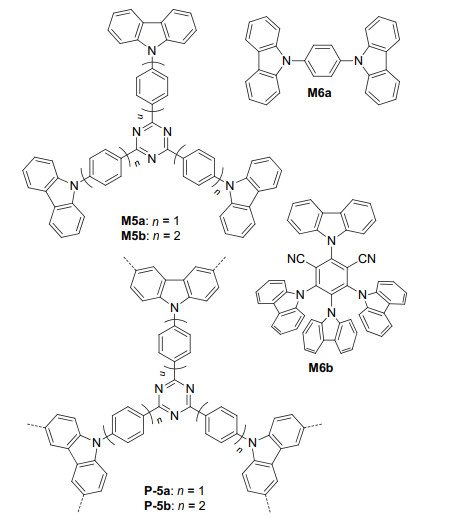
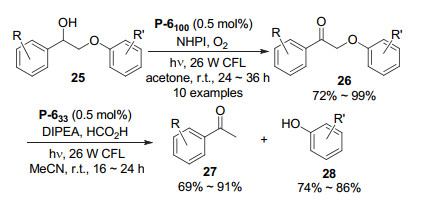
Zhang et al.[22] also prepared polymers P-5a and P-5b from the oxidative coupling of triangular monomers M5a and M5b by FeCl3 (Figure 4). These two polymers have a BET surface area of 1057 and 680 m2·g-1, respectively. Similar to P-2, these two polymers were also able to catalyze the oxidation of primary amines and sulfides (Eqs. 1~3). Both polymers catalyzed the O2 oxidation of 1, 3-diphenylisobenzofuran 19 to give rise to diketone 20 (Eq. 8). The reaction was also revealed to involve the engagement of O2·-. P-5a was also excellent catalyst for the well-studied photooxidative Mannich reaction. In the presence of L-proline, the reactions of tertiary amines 21 and acetone in MeCN afforded ketone products 22 in 86%~98% yields (Eq. 9). With all the substituents being the electron-donating methoxy groups, the corresponding ketone product was obtained in the lowest yield of 86%. The result was consistent with that of reported homogeneous Mannich reactions.[23] For all the oxidation reactions, the polymer catalysts could be used recyclably. After repeated use for six times, the catalysts still maintained their activity with no important loss. Zhang et al.[24] further prepared polymer series P-6n from FeCl3-induced, oxidative polymerization of M6a and M6b (Figure 4) at nine different ratios from 100:0 to 0:100 with 10% of gradual difference. The BET surface areas of these polymers were determined to range from 690 to 1280 m2·g-1. The benzene ring of M6a and the 4, 6-dicyanophenyl unit of M6b are electron-donating and withdrawing. Thus, the carbazole units of the two monomers formed donor-acceptor systems in the polymers, which tuned the photoredox potentials of the polymers. It is known that N-hydroxy- phthalimide (NHPI) serves as an effective hydrogen atom transfer (HAT) catalyst for the oxidation of benzyl alcohol.[25] Polymer P-6100, which was prepared from M6b, was studied to oxidize NHPI to generate the phthalimide- N-oxyl (PINO) radical for the aerobic oxidation of benzyl alcohol 23. Using such P-6100/NHPI photocatalytic system, the corresponding ketone product 24 could be formed in 99% yield (Eq. 10). This reaction was also extended to another ten alcohols 25 and the corresponding lignin ketones 26 were produced in 90%~99% yields (Scheme 1). P-633, which was prepared from M6a and M6b (molar ratio=2:1), was found to catalyze the reductive cleavage of ketones 26 with the highest activity, as compared with other polymers. For the studied ten reactions, the corresponding acetopheones 27 (69%~91%) and phenols 28 (74%~84%) were obtained in good to high yields.
|
|
(8) |
|
|
(9) |
|
|
(10) |
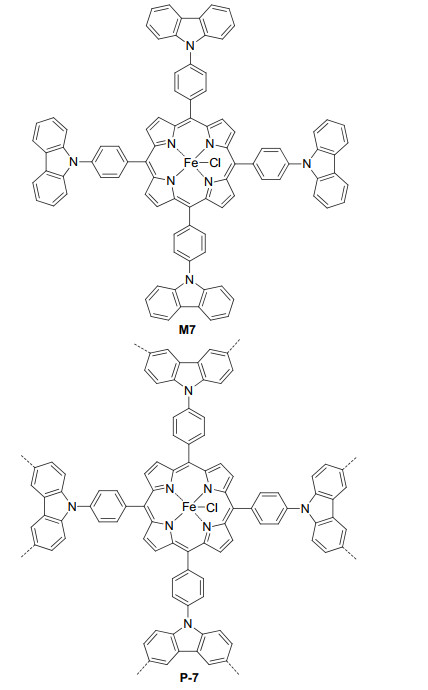
Han et al.[26] reported the preparation of porphyrin-cored polymer P-7 from the carbazole oxidative coupling polymerization of M-7 (Figure 5). The BET surface area of P-7 was 1320 m2·g-1. This polymer exhibited high catalytic activity for the hydrogen peroxide oxidation of sulfides 29 to sulfoxides 30 (Eq. 11), an important glycosyl donor in carbohydrate synthetic chemistry. Compared with M7, P-7 could realize higher yields for all four studied sulfides and could be reused in subsequent reactions without significant decrease in catalytic activity after three runs. The function of the porphyrin units in the catalytic activity was unclear. The oxidation reactions did not need the irradiation of a lamp. However, the results obtained in a closed, dark room were not available.
|
|
(11) |
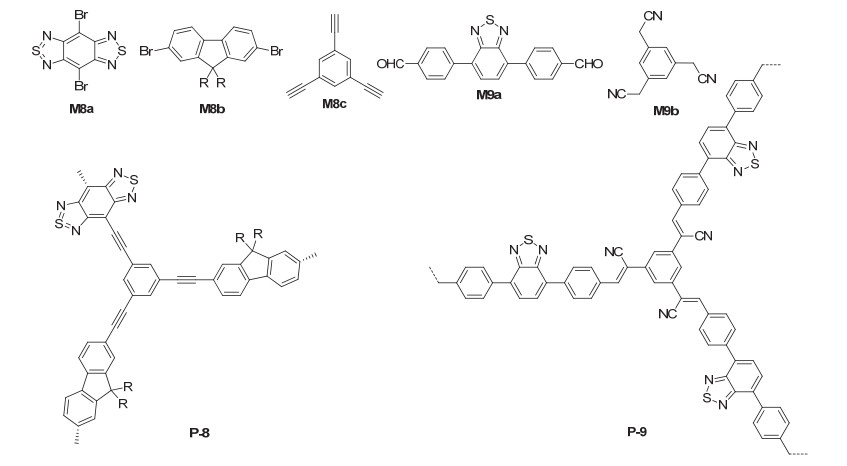
Benzothiadiazole and its derivatives have strong electron-withdrawing capacity, excellent stability, strong light absorption efficiency, tailorable band potentials and excited state lifetimes. Since the first report that the benzothiadiazole unit was utilized in conjugated microporous polymer networks as heterogeneous photocatalysts for visible light-induced singlet oxygen generation, [27] significant advance has been made for the development of poly(benzo- thiadiazole)-based photocatalysts for heterogeneous organic photoredox reactions.[28] Zhang et al.[29] reported the synthesis of polymer P-8 from Sonogashira reaction of monomers M8a and M8b with M8c (Figure 6). P-8 had a small BET surface area of 23 m2·g-1. Although the BET value was small, P-8 exhibited excellent heterogeneous photocatalytic activity for visible light-induced dehalogenation reactions of α-bromoacetophenones 31 (Eq. 12). For the seven studied reactions, acetophenones 32 were obtained in 89%~97% yields. Five additional repeating experiments substituent-free α-bromoacetophenone revealed that the catalyst was stable and maintained the catalytic activity after repeated use.[30] Zhang et al.[30] also prepared another benzothiadiazole-incorporated polymer P-9 from the Knoevenagel polycondensation of monomers M9a and M9b in tetrahydrofuran (THF) in the presence of potassium tert-butoxide. The obtained polymer exhibited a BET surface area of 130 m2·g-1 and could be applied as a robust and efficient heterogeneous photocatalyst for the oxidative hydroxylation of arylboronic acids under visible light irradiation (Eq. 5). Ten arylboronic acids 10 were studied and the corresponding aryl alcohols 11 were formed in 76%~96% yields. Repeating experiments for hydroxylation of phenylboronic acid showed a slight decrease with a generally high conversion of more than 80% during the whole five repeating cycles, which was attributed to the loss of the sample during the recovery processes.
|
|
(12) |
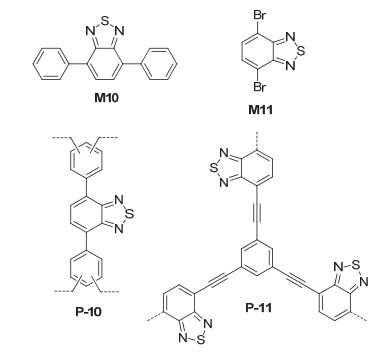
From FeCl3-promoted Friedel-Crafts alkylation reaction of M10 and formaldehyde dimethyl acetal, Zhang et al.[31] prepared polymer P-10 (Figure 7), which had a BET surface area of 586 m2·g-1. P-10 was an excellent heterogeneous photocatalyst for a visible light-induced, selective bromination reaction of electron-rich aromatic compounds (Eq. 13), using hydrogen bromide as a bromine source and molecular oxygen as clean oxidant. The reactions of studied eight aromatic ethers and thiophenes (33a~33k) all selectively afforded monobromides 34a~34k. Generally, substituents bearing two or three methoxy groups led to higher yields of the bromides. Thiophene and benzothiophene substrates also gave rise to bromination products with good yields. However, toluene did not afford the corresponding bromide. Instead, benzaldehyde was obtained in 4% yield. The method was also applied for thiophenes and benzothiophene. However, the reaction of toluene yielded benzaldehyde in 4% yield. Repeating experiments for 1, 3, 4-trimethoxybenzene were conducted, which revealed that P-10 could be used for at least six repeating cycles without significant change in the catalytic efficiency. Zhang et al.[32] also reported the visible light-induced heterogeneous photocatalysis of P-11 (Figure 7) for the [2+2] cycloaddition of alkenes. The polymer can be prepared from Sonogashira-Hagihara cross-coupling of monomers M8c and M11.[33] Its BET surface area was determined to be 120586 m2·g-1. P-11 was found to catalyze visible light-induced [2+2] cycloaddition of 35 and 36 to give cyclobutene derivative 37 in 86% yield (Eq. 14), together with 38 as minor product (6%). For another eight pairs of reactants, the unsymmetrical cycloaddition products were all produced as major products and nitromethane was found to be the most suitable solvent. When the solution of pure 35 in nitromethane was subjected to the same reaction conditions, 38 was formed in 80% yield. Thus, it was concluded that the formation of the heterocycloaddition product was energetically favorable than that of the homocycloaddition product.
|
|
(13) |
|
|
(14) |
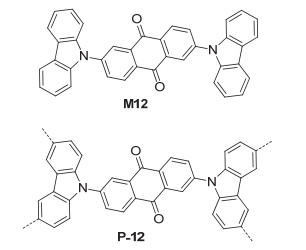
Donor-acceptor (D-A) fluorophores possess adjustable photoelectric properties for the enhancement of their photocatalytic performances.[34] Liu et al.[35] prepared polymer P-12 (Figure 8) from oxidative coupling polymerization reaction of monomer M12 with FeCl3. This polymer had a BET surface area of 758 m2·g-1. Under visible-light irradiation, compound 39 could oxidized by molecular oxygen to hemiaminal 40 in methanol in 97% yield under the catalysis of P-12 (Eq. 15). The reaction conditions were also applied for the oxidative C—H functionalization reactions of 39 with a variety of substrates 41a~41h (Eq. 16). The resulting C—C oxidative coupling products 42a~42h were produced in 70%~94% yields. With bromine, methoxy or nitro being introduced to the para-position of the benzene ring that connected the amino group, the corresponding C—C coupling products were also obtained in high yields. The reaction of 39 and 41a was studied for the recyclability of P-12. It was found that the polymer could be efficiently recycled and reused for at least ten times without obvious loss of photocatalytic activity and selectivity. The reaction of 39 and 41a had also been studied through an easy-to-build continuous flow reactor. Full conversion to the target product 42a could be obtained.
|
|
(15) |
|
|
(16) |
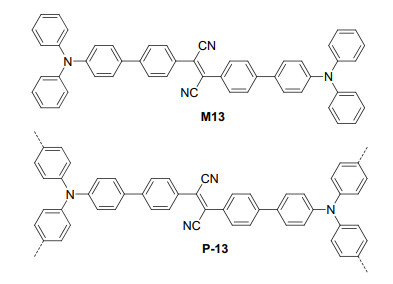
Liu et al.[36] prepared polymer P-13 from the oxidative coupling polymerization of M13 (Figure 9). Triphenylamine and fumaronitrile were used as the donor and acceptor in the monomer, which were separated by the biphenyl unit to promote their intersystem crossing process and favor 1O2 generation. As a result, P-13 showed high catalytic capacity for visible light-induced molecular oxygen oxidation of aromatic aldehydes 43, which might bear electron-donating and withdrawing substituent(s), to aromatic acids 44 (Eq. 17). The catalyst also worked well for the oxidation of 5-ethylthiophene-2-carbaldehyde to the corresponding acid (78%). However, the reaction of 4-diethyl- aminobenzaldehyde did not afford the corresponding acid. For liquid aldehydes, the photooxidations could take place in a solvent-free manner. With lowered amount of the catalyst, the acids were still obtained in comparable yields. For the reaction of benzaldehyde, after 10 cycles, the activity of P-13 could be maintained to be about 85% as that for the first run.
|
|
(17) |
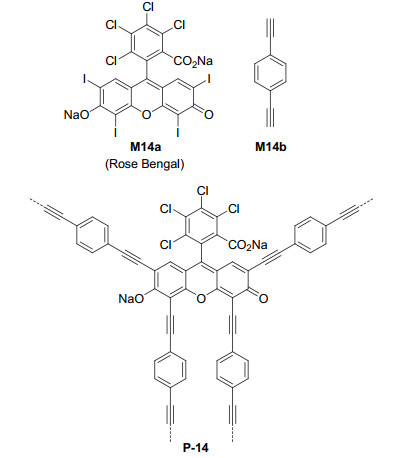
Xanthene dyes represent a family of photoredox organocatalysts that have been widely used in visible light induced organic transformations.[37] Rose Bengal M14a is one of the most studied xanthene dye that bears four iodine atoms. Jiang et al.[38] reported the synthesis of polymer P-14 from the palladium-catalyzed Sonogashira-Hagihara cross-coupling polycondensation of Rose Bengal M14a with M14b (Figure 10). This dye-cored polymer was stable with respect to aqueous solutions of acids and bases, such as HCl and NaOH and thermally stable in nitrogen up to 300 ℃. Its BET surface area was determined to be 830 m2·g-1. P-14 exhibited high photocatalytic activity for the Aza-Henry reactions of tertiary amines 45 and nitroalkanes 46, including Me, Et and n-Pr derivatives, to afford the C—C coupling products 47 (Eq. 18). All the reactions gave rise to the C—C coupling products 43 in 80%~97% yields. The Ar group of 45 might be the benzene ring bearing the electron-donating or withdrawing group(s) or the naphthalene ring. The liquid nitroalkanes also acted as the solvents. After ten recycling times, the conversion of the tertiary amine substrate still exceeded 90%.
|
|
(18) |
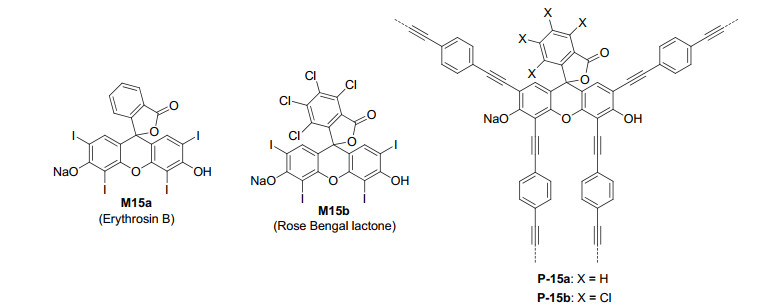
Cai et al.[39] further prepared Erythrosin B- and Rose Bengal lactone-based polymers P-15a and P-15b from the Sonogashira-Hagihara cross-coupling polycondensations of Erythrosin B (M15a) or Rose Bengal lactone (M15b) with M14b under the similar reaction conditions (Figure 11). These two polymers had a BET surface area of 330 or 398 m2·g-1. Both polymers exhibited efficient photocatalysis for the reversible addition-fragmentation chain transfer (RAFT) polymerization of N, N-dimethylacrylamide in dimethyl sulfoxide (DMSO). In comparison with P-15a, the electron-withdrawing chlorine-substituted phenyl groups in P-15b gave rise to higher triplet quantum yield and therefore provided better oxygen tolerance during the polymerization.
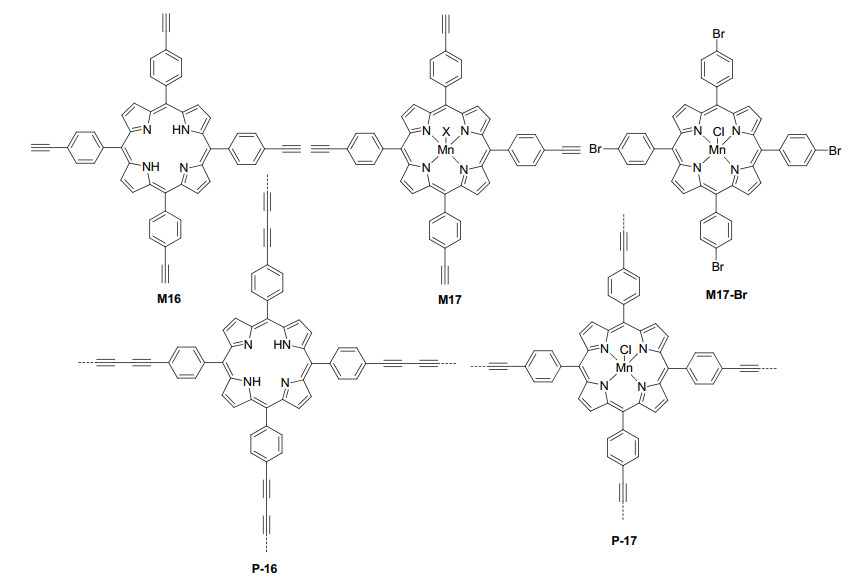
Porphyrin polymerization provides one valuable way for improving the stability and recyclability of porphyrin catalysts.[40] In this category, Liu et al.[41] first synthesized polymer P-16 from the alkyne-alkyne homocoupling polycondensation of porphyrin monomer M16 (Figure 12). The polymer had a BET surface area of 662 m2·g-1 and was found to act as an organocatalyst for the Knoevenagel condensation reaction of malononitrile 48 with aromatic and heterocyclic aldehydes 49 to afford 2-methylene- malononitrile products 50 in 77%~98% yields (Eq. 19). The reaction of 48 with cyclohexanone also proceeded to afford the coupling product in 91% yield. The solid catalyst could be recovered easily by filtration and reused for at least ten times for the reaction of benzaldehyde. Yang et al.[42] also conducted the coupling polymerization of compounds M17 and M17-Br to prepare porphyrin polymer P-17 (Figure 12), which had a BET surface area of 380 m2·g-1. P-17 was developed as a heterogeneous catalyst for the activation of molecular oxygen to oxidize toluene under mild conditions. Benzaldehyde was obtained selectively in 70% with a conversion of toluene being up to 10.2%. The catalysis did not need the initiation of visible light, but no control experiments were available for the reactions conducted by shielding the room light. The turnover number was as high as 13653, and the catalyst remained structurally stable and the toluene conversion rate hardly decreased after 5 h of reaction and five cycles of reuse.
|
|
(19) |
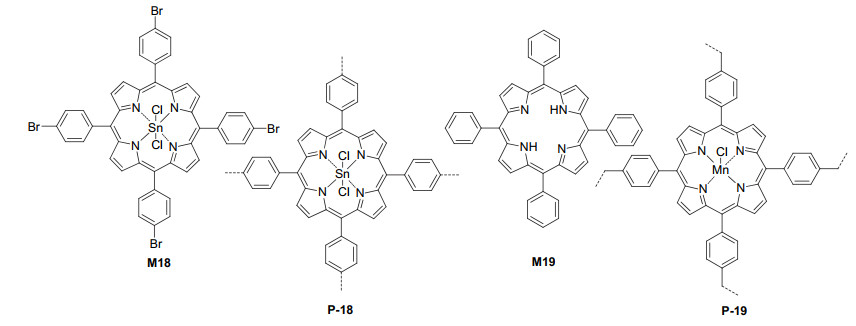
Ji et al.[43] synthesized polymer P-18 via the Yamamoto homo-coupling reaction of porphyrin tetrabromide M18 (Figure 13). The polymer had a BET surface area of 987 m2·g-1. P-18 efficiently catalyzed the visible light-induced molecular oxygen oxidation of alkyl aryl sulfides, dialkyl sulfides and diphenyl sulfide. The resulting sulfoxides were formed in 71%~97% yields. The solid catalyst could be readily recycled and reutilized for at least four times without any apparent loss of catalytic activity. Moreover, P-18 was also able to catalyze visible light-induced oxidative coupling reactions of various primary amines into imines (Eq. 1).
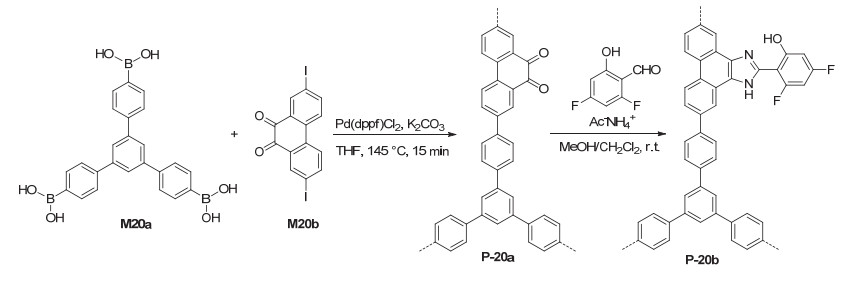
Postmodification or functional group-attachment is a widely used strategy for the preparation of functional porous systems such as covalent organic frameworks and metal organic frameworks.[5, 44] POPs as highly stable conjugated polymers are intrinsically suitable for further introduction of catalytic sites. Sánchez et al.[45] prepared polymer P-20b from the coupling polymerization of M20a and M20b followed by the introduction of the 3, 5-difluoro- phenol unit to the resulting polymer P-20a (Scheme 2). P-20b had a BET surface area of 285 m2·g-1. The attached 3, 5-difluorophenol units in P-20b were efficient heterogeneous catalysts for visible light-promoted aza-Henry reaction of compounds 39 and 41a to afford the nitro derivative 42a (Eq. 16). After 72 h, 42a could be formed in 99% yield. Ru complexes were also attached to the polymer by treating P-20b with Ru(bpy)3Cl2. The resulting attached Ru complexes also catalyzed the aza-Henry reaction to afford 42a in a comparable yield.
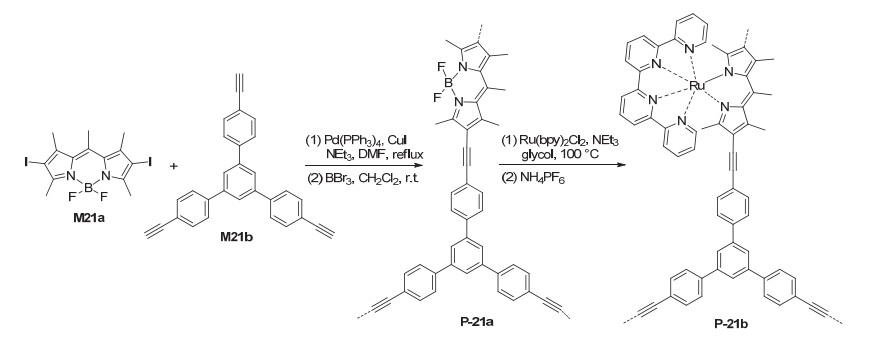
Liras et al.[46] prepared polymer P-21a from the coupling polymerization of compounds M21a and M21b (Scheme 3). The polymer was further treated with Ru(bpy)2Cl2 to attach ruthenium complexes to the 4, 4- difluoro-4-bora-3a, 4a-diaza-s-indacene (BODIPY) units of the backbone to afford new polymer P-21b, which had a BET surface area of ca. 306 m2·g-1. P-21b also efficiently catalyzed the visible light-induced aza-Henry reaction of compound 39 with different nitro derivatives (Eq. 16), including MeNO2, EtNO2, n-PrNO2 and EtOCOCH2NO2. The resulting C—C coupling products 42 were obtained in 77%~96% yields, together with the oxidative amide as minor product (4%~18%). P-21b was also highly stable and could be recycled for seven times with no significant decrease of the activity. Su and Wang et al.[47] reported that by simple ion exchange, [Ru(bpy)3]2+ complexes could be loaded to the carboxylate anchors of 3D porous organic frameworks prepared through Click coupling polymerization. The resulting Ru-containing porous polymers exhibited excellent catalytic activity for visible light-induced oxidative hydroxylation of 4-methoxyphenylboronic acid to 4-methoxyphenol (Eq. 5) and could be recycled for at least six times.
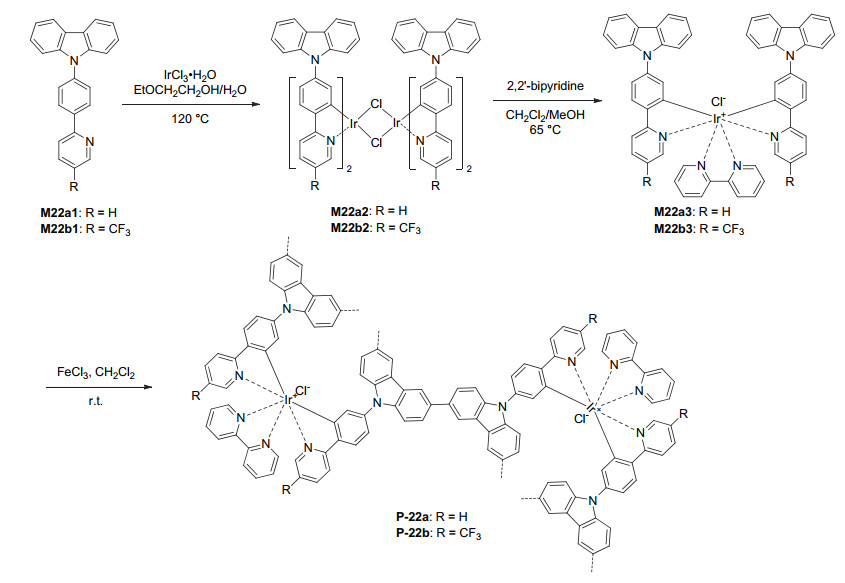
Ruthenium and iridium complexes are two kinds of most widely exploited complexes for visible light photoredox catalysis of organic reactions.[8, 48] Both kinds of complexes are highly stable and structurally rigid and thus suitable to be incorporated into porous organic frameworks. Lin et al.[49] pioneered the incorporation of both kinds of complexes into 3D porous polymers and revealed their efficient photocatalysis for aza-Henry reaction and α-alkylation of bromomalonate. Han et al.[50] utilized the above-mentioned FeCl3-mediated oxidative hyper-crosslinked coupling of the carbazole unit to synthesize polymers P-22a and P-22b from M22a3 or M22b3 (Scheme 4). For these synthesis, iridium complex intermediates M22a2 and M22b2 were first prepared from the reactions of M22a1 and M22a2 with iridium chloride and then treated with 2, 2'-bipyridine to produce M22a3 and M22b3, respectively. The BET surface areas of P-22a and P-22b were determined to be 97 and 76 m2·g-1, respectively. Both polymers catalyzed the visible light-induced air photooxidation of methyl phenyl sulfides (Eq. 3), which selectively afforded the corresponding sulfoxide products (93%~99%) and sulfones of low yields (1%~4%). For the photocatalytic hydroxylation of arylboronic acids, the resulting phenol products were also formed in high yields (67%~99%) (Eq. 5). Generally, P-22b exhibited higher catalytic activity, which was elucidated in terms of photophysical and electrochemical properties. The two polymers could catalyze the visible light-induced cross-dehydrogenative coupling reactions of tertiary amines 45 with nitromethane or nitroethane to afford the corresponding C—C coupling nitro derivatives (47) (Eq. 18). In particular, the reactions of N-aryltetrahydroisoquinolines 39, 39a and 39b with phosphonates 41g and 41i afforded the C—P coupling products 42g, 42i and 42j in excellent yields (Eq. 20).
|
|
(20) |
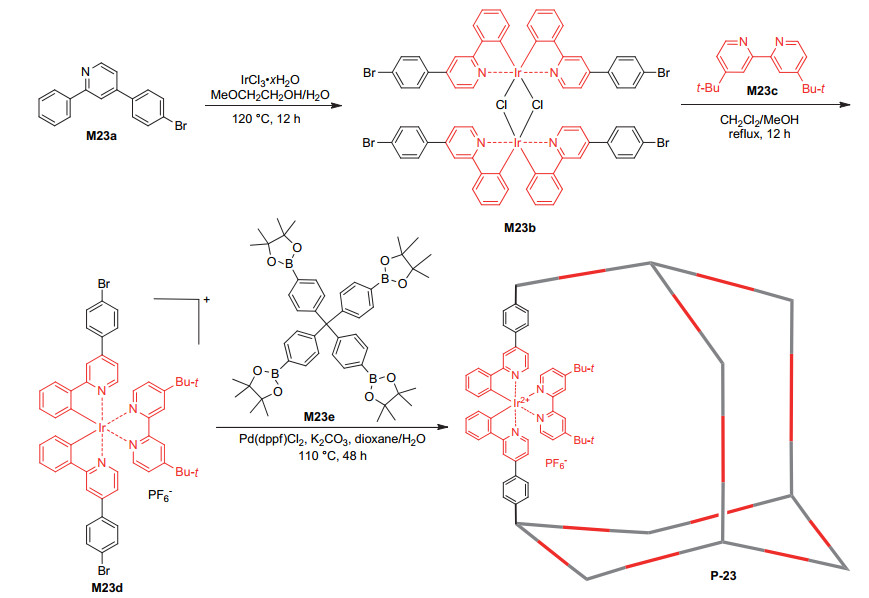
Zhang et al.[51] developed another approach for incorporating iridium complexes into porous polymers such as P-23, the synthesis of which is shown in Scheme 5. Compound M23a was first reacted with iridium chloride to afford complex M23b, which further reacted with M23c to produce complex intermediate M23d. The coupling polymerization of M23d with M23e gave rise to polymer P-23, which had a BET surface area of 124 m2·g-1. This polymer was found to be an efficient heterogeneous catalyst for the visible light-induced molecular oxygen oxidation of sulfides and arylboronic acids (Eqs. 3 and 5). Moreover, it was able to catalyze the Smiles-Truce rearrangement of alkyliodides 51a~51m to afford the corresponding rearrangement products 52a~52m (Eq. 21).[52] Notably, the activity of the iridium complex in P-23 was comparable with that of a homogeneous prototype complex. P-23 was also revealed to catalyze the recently reported desulfurative conjugate addition of substrates 53a and 53b to Michael acceptors 54a~54f, which afforded compounds 55a~55l (Eq. 22). The recyclability of the catalyst for all the four reactions was studied, which revealed the high stability of the catalyst. For the addition of 53a to 54a, after recycling for 19 times, the catalyst still exhibited important activity.
|
|
(21) |
|
|
(22) |
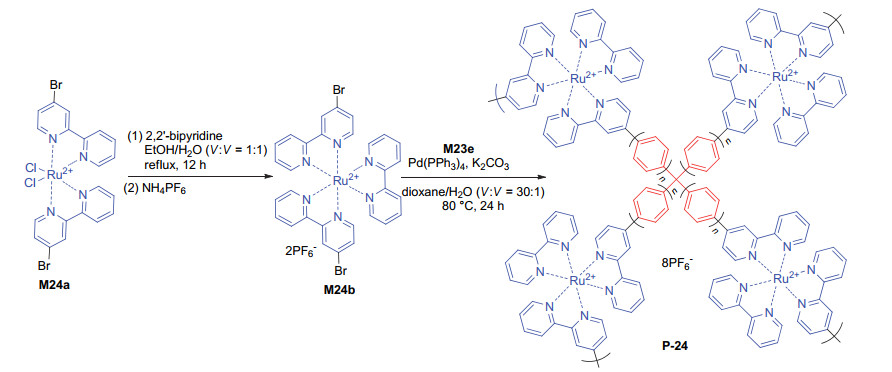
Zhang et al.[53] also introduced the [Ru(bpy)3]2+ units to the backbone of polymer P-24. For the synthesis of P-24 (Scheme 6), M24a was first reacted with 2, 2'-bipyridine to afford ruthenium complex M24b. This intermediate was further coupled with M23e to give rise to P-24 as a porous, insuluble polymer, which had a BET surface area of 15 m2·g-1. The existence of optical isomers of the complexes, inclusion of counterions, and the stoichiometric amount of the heavy Ru atoms in the polymeric backbones were proposed to account for the low surface area. P-24 was revealed to exhibit high heterogeneous photocatalytic activities for the enantioselective α-alkylation of aldehydes 56a and 56b with 57a~57d in the presence of chiral catalyst C1, which led to the formation of chiral products 58 (Eq. 23). The C—C coupling products were obtained in 77%~92% yields, while the ee values reached 90%~95%, which were comparable with the corresponding value obtained through homogeneous reactions mediated by an soluble control catalyst.[54] The reaction of 56a and 57a was used to exploit the recyclability of P-24. It was found that after ten times of recycling, the yield of the product was slightly reduced from 92% to 73%. However, the ee values were nearly maintained unchanged, which suggested that the polymer catalyst might suffer loss during the processes, but it was stable enough to sustain its porous structure.
|
|
(23) |
Visible light-induced heterogeneous catalysis has received increasing attention for organic transformations. Porous organic polymers are ideal platforms for exploiting the green and sustainable catalysis of organic transformations due to their straightforward synthesis and characterization. From the point of fundamental research, chemists pursue structural ordering or periodicity for porous materials, which is crucially important for some specific applications. As porous heterogeneous catalysts, ready synthesis and low cost, high activity and good sustainability through easy recovery and recycling are more important factors for achieving practical applications, which are all the intrinsic features of porous organic frameworks. Currently, studies for the applications of porous organic polymers are highly limited to the reactions that have been well-established homogeneously. Although the feature of the reuse and recyclability of porous organic frameworks has been well-demonstrated, their practicability as heterogeneous catalysts is still to be addressed. In the future, we may expect the report of studies for practical organic transformations or the synthesis of useful and highly valuable organic molecules through this strategy. In principle, the catalytic sites of heterogeneous porous organic frameworks can realize the photoredox activity of homogeneous catalysts. For this target, the pores should be large enough to allow the free entering and exiting of substrates and products as well as high transparency for light penetrating. Therefore, the development of porous organic frameworks with large pores should be important.
Cole-Hamilton, D. J.; Tooze, R. P. In Catalyst Separation, Recovery and Recycling, Springer, Dordrecht, 2006, pp. 1~8.
Heterogeneous Catalysis: Fundamentals and Applications, Ed.: Ross, J. R. H., Elsevier, Amsterdam, 2012, p. 222.
(a) Wang, Z.; Chen, G.; Ding, K. Chem. Rev. 2009, 109, 322.
(b) Fechete, I.; Wang, Y.; Vedrine, J. C. Catal. Today 2012, 189, 2.
(c) Thomas, J. M. Top. Catal. 2014, 57, 1115.
(d) Sun, Z.; Liu, Y.; Chen, J.; Huang, C.; Tu, T. ACS Catal. 2015, 5, 6573.
(e) Kaneda, K.; Mizugaki, T. Green Chem. 2019, 21, 1361.
(a) Lin, W. J. Solid State Chem. 2005, 178, 2486.
(b) Zeng, L.; Guo, X.; He, C.; Duan, C. ACS Catal. 2016, 6, 7935.
(c) Ma, D.; Li, B.; Shi, Z. Chin. Chem. Lett. 2018, 29, 827.
(d) Wang, P.; Deng, L. Chin. J. Chem. 2018, 36, 1222.
(e) Hou, J.; Hao, J.; Wang, Y.; Liu, J. Chem. Res. Chin. Univ. 2019, 35, 860.
(f) Kang, X.-M.; Shi, Y.; Cao, C.-S.; Zhao, B. Sci. China: Chem. 2019, 62, 622.
(g) Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.-Y. Chem. Soc. Rev. 2014, 43, 6011.
(h) Wu, Z.; Shi, Y.; Li, C.; Niu, D.; Chu, Q.; Xiong, W.; Li, X. Acta Chim. Sinica 2019, 77, 758 (in Chinese).
(武卓敏, 石勇, 李春艳, 牛丹阳, 楚奇, 熊巍, 李新勇, 化学学报, 2019, 77, 758.)
(i) Liu, Y.; Xuan, W.; Cui, Y. Adv. Mater. 2010, 22, 4112.
(a) Ding, S.-Y.; Wang, W. Chem. Soc. Rev. 2013, 42, 548.
(b) Bisbey, R. P.; Dichtel, W. R. ACS Cent. Sci. 2017, 3, 533.
(c) Peng, Z.; Ding, H.; Chen, R.; Gao, C.; Wang, C. Acta Chim. Sinica 2019, 77, 681 (in Chinese).
(彭正康, 丁慧敏, 陈如凡, 高超, 汪成, 化学学报, 2019, 77, 681.)
(d) Liang, R.-R.; Zhao, X. Org. Chem. Front. 2018, 5, 3341.
(e) Yuan, F.; Tan, J.; Guo, J. Sci. China: Chem. 2018, 61, 143.
(f) Wang, T.; Xue, R.; Wei, Y.; Wang, M.; Guo, H.; Yang, W. Prog. Chem. 2018, 30, 753.
(g) Kandambeth, S.; Dey, K.; Banerjee, R. J. Am. Chem. Soc. 2019, 141, 1807.
(a) Zhang, Y.; Riduan, S. N. Chem. Soc. Rev. 2012, 41, 2083.
(b) Sun, Q.; Dai, Z.; Meng, X.; Xiao, F.-S. Chem. Soc. Rev. 2015, 44, 6018.
(c) Kramer, S.; Bennedsen, N. R.; Kegnaes, S. ACS Catal. 2018, 8, 6961.
(d) Wei, Z.; Li, Y.; Wang, J.; Li, H.; Wang, Y. Chin. Chem. Lett. 2018, 29, 815.
(e) Li, C.-Y.; Yan, L.; Wang, W.-L.; Wang, Y.-Q.; Jiang, M.; Ding, Y.-J. Chin. Polym. Bull. 2018, 6, 32 (in Chinese).
(李存耀, 严丽, 汪文龙, 王玉清, 姜淼, 丁云杰, 高分子通报, 2018, 6, 32.)
(f) Pang, C.; Luo, S.; Hao, Z.; Gao, J.; Huang, Z.; Yu, J.; Yu, S.; Wang, Z. Chin. J. Org. Chem. 2018, 38, 2606 (in Chinese).
(庞楚明, 罗时荷, 郝志峰, 高健, 黄召昊, 余家海, 余思敏, 汪朝阳, 有机化学, 2018, 38, 2606.)
(g) Zhang, T.; Xing, G.; Chen, W.; Chen, L. Mater. Chem. Front. 2020, 4, 332.
(h) Liang, J.; Huang, Y.-B.; Cao, R. Coord. Chem. Rev. 2019, 378, 32.
(i) Zhao, Y.; Ma, W.; Xu, Y.; Zhang, C.; Wang, Q.; Yang, T.; Gao, X.; Wang, F.; Yan, C.; Jiang, J.-X. Macromolecules 2018, 51, 9502.
(j) Yuan, Y.; Huang, H.; Chen, L.; Chen, Y. Macromolecules 2017, 50, 4993.
(k) Kong, S.; Malik, A. U.; Qian, X.; Shu, M.; Xiao, W. Chin. J. Org. Chem. 2018, 38, 656 (in Chinese).
(孔胜男, Abaid Ullah Malik, 钱雪峰, 舒谋海, 肖文德, 有机化学, 2018, 38, 656.)
(l) Liu, M.; Zhou, B.; Zhou, L.; Xie, Z.; Li, S.; Chen, L. J. Mater. Chem. A 2018, 6, 9860.
(m) Kong, S.; Qian, X.; Shu, M.; Xiao, W. Chin. J. Org. Chem. 2018, 38, 2754 (in Chinese).
(孔胜男, 钱雪峰, 舒谋海, 肖文德, 有机化学, 2018, 38, 2754.)
(n) Gao, X.; Shu, C.; Zhang, C.; Ma, W.; Ren, S.-B.; Wang, F.; Chen, Y.; Zeng, J. H.; Jiang, J.-X. J. Mater. Chem. A 2020, 8, 2404.
(a) Hennig, H.; Rehorek, D.; Stich, R.; Weber, L. Pure Appl. Chem. 1990, 62, 1489.
(b) Chatterjee, D.; Dasgupta, S. J. Photochem. Photobiol. C 2005, 6, 186.
(c) Zhao, J.; Chen, C.; Ma, W. Top. Catal. 2005, 35, 269.
(d) Tung, C.-H.; Wu, L.-Z.; Zhang, L.-P.; Chen, B. Acc. Chem. Res. 2003, 36, 39.
(a) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322.
(b) Narayanam, J. M. R.; Stephenson, C. R. J. Chem. Soc. Rev. 2011, 40, 102.
(c) Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828.
(d) Xi, Y.; Yi, H.; Lei, A. Org. Biomol. Chem. 2013, 11, 2387.
(e) Xie, J.; Jin, H.; Xu, P.; Zhu, C. Tetrahedron Lett. 2014, 55, 36.
(f) Lang, X.; Ma, W.; Chen, C.; Ji, H.; Zhao, J. Acc. Chem. Res. 2014, 47, 355.
(g) Cao, M.-Y.; Ren, X.; Lu, Z. Tetrahedron Lett. 2015, 56, 3732.
(h) Zhang, X.; Rakesh, K. P.; Ravindar, L.; Qin, H.-L. Green Chem. 2018, 20, 4790.
(i) Jia, K.; Chen, Y. Chem. Commun. 2018, 54, 6105.
(j) Zhang, H.; Yu, S. Chin. J. Org. Chem. 2019, 39, 95 (in Chinese).
(张昊, 俞寿云, 有机化学, 2019, 39, 95.)
(k) Chen, B.; Wu, L.-Z.; Tung, C.-H. Acc. Chem. Res. 2018, 51, 2512.
(l) Dai, J.; Lei, W.; Liu, Q. Acta Chim. Sinica 2019, 77, 911 (in Chinese).
(戴建玲, 雷文龙, 刘强, 化学学报, 2019, 77, 911.)
(m) Liu, Y.-C.; Zheng, X.; Huang, P.-Q. Acta Chim. Sinica 2019, 77, 850 (in Chinese).
(刘玉成, 郑啸, 黄培强, 化学学报, 2019, 77, 850.)
(n) Chen, D.; Liu, J.; Zhang, X.; Jiang, H.; Li, J. Chin. J. Org. Chem. 2019, 39, 3353 (in Chinese).
(陈丹, 刘剑沉, 张馨元, 蒋合众, 李加洪, 有机化学, 2019, 39, 3353.)
(o) Yi, X.; Huang, F.; Baell, J. B.; Huang, H.; Yu, Y. Prog. Chem. 2019, 31, 505.
(p) Wang, T.-X.; Liang, H.-P.; Anito, D. S.; Ding, X.; Han, B.-H. J. Mater. Chem. A 2020, 8, 7003.
Ma, T.; Kapustin, E. A.; Yin, S. X.; Liang, L.; Zhou, Z.; Niu, J.; Li, L.-H.; Wang, Y.; Su, J.; Li, J.; Wang, X.; Wang, W. D.; Wang, W.; Sun, J.; Yaghi, O. M. Science 2018, 361, 48. doi: 10.1126/science.aat7679
(a) Fang, Q.; Wang, J.; Gu, S.; Kaspar, R. B.; Zhuang, Z.; Zheng, J.; Guo, H.; Qiu, S.; Yan, Y. J. Am. Chem. Soc. 2015, 137, 8352.
(b) Gao, Z.-Z.; Wang, Z.-K.; Wei, L.; Yin, G.; Tian, J.; Liu, C.-Z.; Wang, H.; Zhang, D.-W.; Zhang, Y.-B.; Li, X.; Liu, Y.; Li, Z.-T. ACS Appl. Mater. Interfaces 2020, 12, 1404.
(a) McKeown, N. B.; Budd, P. M.; Msayib, K. J.; Ghanem, B. S.; Kingston, H. J.; Tattershall, C. E.; Makhseed, S.; Reynolds, K. J.; Fritsch, D. Chem.-Eur. J. 2005, 11, 2610.
(b) McKeown, N. B. Sci. China: Chem. 2017, 60, 1023.
(a) Zhou, Y.-B.; Zhan, Z.-P. Chem.-Asian J. 2018, 13, 9.
(b) He, Q.; Zhang, C.; Li, X.; Wang, X.; Mu, P.; Jiang, J. Acta Chim. Sinica 2018, 76, 202 (in Chinese).
(贺倩, 张崇, 李晓, 王雪, 牟攀, 蒋加兴, 化学学报, 2018, 76, 202.)
(c) Lee, J.-S. M.; Cooper, A. I. Chem. Rev. 2020, 120, 2171.
(a) Yuan, Y.; Zhu, G. ACS Cent. Sci. 2019, 5, 409.
(b) Yan, T.; Xing, G.; Ben, T.; Qiu, S. Chem. J. Chin. Univ. 2018, 39, 1072 (in Chinese).
(闫婷婷, 邢国龙, 贲腾, 裘式纶, 高等学校化学学报, 2018, 39, 1072.)
(c) Xia, L.; Zhang, H.; Feng, B.; Yang, D.; Bu, N.; Zhao, Y.; Yan, Z.; Li, Z.; Yuan, Y.; Zhao, X. Chem. J. Chin. Univ. 2019, 40, 2456 (in Chinese).
(夏立新, 张红翠, 冯彬, 杨东奇, 布乃顺, 赵云波, 闫卓君, 李樟楠, 元野, 赵晓君, 高等学校化学学报, 2019, 40, 2456.)
(d) Ben, T.; Qiu, S. CrystEngComm 2013, 15, 17.
(a) Hou, S.; Tan, B. Macromolecules 2018, 51, 2923.
(b) Gao, H.; Ding, L.; Bai, H.; Li, L. ChemSusChem 2017, 10, 618.
(c) Tan, L.; Tan, B. Chem. Soc. Rev. 2017, 46, 3322.
(a) Chen, Q.; Luo, M.; Hammershøj, P.; Zhou, D.; Han, Y.; Laursen, B. W.; Yan, C.-G.; Han, B.-H. J. Am. Chem. Soc. 2012, 134, 6084.
(b) Chen, Q.; Han, B.-H. Macromol. Rapid Commun. 2018, 39, 1800040.
Su, C.; Tandiana, R.; Tian, B.; Sengupta, A.; Tang, W.; Su, J.; Loh, K. P. ACS Catal. 2016, 6, 3594. doi: 10.1021/acscatal.6b00443
(a) Shang, T.-Y.; Lu, L.-H.; Cao, Z.; Liu, Y.; He, W.-M.; Yu, B. Chem. Commun. 2019, 55, 5408.
(b) Yuan, W.; Yang, H.; Zhang, M.; Hu, D.; Wan, S.; Li, Z.; Shi, C.; Sun, N.; Tao, Y.; Huang, W. Chin. Chem. Lett. 2019, 30, 1955.
(c) Bi, K.; Tan, R.; Hao, R.; Miao, L.; He, Y.; Wu, X.; Zhang, J.; Rui, X. Chin. Chem. Lett. 2019, 30, 545.
(d) Li, J.; Grimsdale, A. C. Chem. Soc. Rev. 2010, 39, 2399.
Luo, J.; Zhang, X.; Zhang, J. ACS Catal. 2015, 5, 2250. doi: 10.1021/acscatal.5b00025
(a) Wang, P.-Z.; Chen, J.-R.; Xiao, W.-J. Org. Biomol. Chem. 2019, 17, 6936.
(b) Chen, X.; Chen, J.; Bao, Z.; Yang, Q.; Yang, Y.; Ren, Q.; Zhang, Z. Chin. J. Org. Chem. 2019, 39, 1681 (in Chinese).
(陈晓玲, 陈静雯, 鲍宗必, 杨启炜, 杨亦文, 任其龙, 张治国, 有机化学, 2019, 39, 1681.)
(c) Huang, W.; Cheng, X. Synlett 2017, 28, 148.
Zhi, Y.; Yao, Z.; Jiang, W.; Xia, H.; Shi, Z.; Mu, Y.; Liu, X. ACS Appl. Mater. Interfaces 2019, 11, 37578. doi: 10.1021/acsami.9b10958
Zhi, Y.; Li, K.; Xia, H.; Xue, M.; Mu, Y.; Liu, X. J. Mater. Chem. A 2017, 5, 8697. doi: 10.1039/C7TA02205K
Luo, J.; Lu, J.; Zhang, J. J. Mater. Chem. A 2018, 6, 15154. doi: 10.1039/C8TA05329D
(a) Rueping, M.; Vila, C.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C. Chem. Commun. 2011, 47, 2360.
(b) Wang, H.; Zheng, C.; Zhao, G. Chin. J. Chem. 2019, 37, 1111.
(c) Ye, J.; Huang, P. Chin. J. Org. Chem. 2018, 38, 2215 (in Chinese).
(叶剑良, 黄培强, 有机化学, 2018, 38, 2215.)
(d) Cao, W.; Liu, X.; Feng, X. Chin. Chem. Lett. 2018, 29, 1201.
Luo, J.; Zhang, X.; Lu, J.; Zhang, J. ACS Catal. 2017, 7, 5062. doi: 10.1021/acscatal.7b01010
Chen, Y.; Lu, L.-Q.; Yu, D.-G.; Zhu, C.-J.; Xiao, W.-J. Sci. China: Chem. 2019, 62, 24. doi: 10.1007/s11426-018-9399-2
Feng, L.-J.; Chen, Q.; Zhu, J.-H.; Liu, D.-P.; Zhao, Y.-C.; Han, B.-H. Polym. Chem. 2014, 5, 3081. doi: 10.1039/c3py01430d
Zhang, J.; Chen, W.; Rojas, A. J.; Jucov, E. V.; Timofeeva, T. V.; Parker, T. C.; Barlow, S.; Marder, S. R. J. Am. Chem. Soc. 2013, 135, 16376. doi: 10.1021/ja4095878
Li, R.; Byun, J.; Huang, W.; Ayed, C.; Wang, L.; Zhang, K. A. I. ACS Catal. 2018, 8, 4735. doi: 10.1021/acscatal.8b00407
Wang, Z. J.; Ghasimi, S.; Landfester, K.; Zhang, K. A. I. J. Mater. Chem. A 2014, 2, 18720. doi: 10.1039/C4TA03887H
Wang, Z. J.; Li, R.; Landfester, K.; Zhang, K. A. I. Polymer 2017, 126, 291. doi: 10.1016/j.polymer.2017.04.052
Li, R.; Wang, Z.-J.; Wang, L.; Ma, B. C.; Ghasimi, S.; Lu, H.; Landfester, K.; Zhang, K. A. I. ACS Catal. 2016, 6, 1113. doi: 10.1021/acscatal.5b02490
Zhang, K.; Kopetzki, D.; Seeberger, P. H.; Antonietti, M.; Vilela, F. Angew. Chem., Int. Ed. 2013, 52, 1432. doi: 10.1002/anie.201207163
Li, R.; Ma, B. C.; Huang, W.; Wang, L.; Wang, D.; Lu, H.; Landfester, K.; Zhang, K. A. I. ACS Catal. 2017, 7, 3097. doi: 10.1021/acscatal.7b00490
Lu, J.; Khetrapal, N. S.; Johnson, J. A.; Zeng, X. C.; Zhang, J. J. Am. Chem. Soc. 2016, 138, 15805. doi: 10.1021/jacs.6b08620
Zhi, Y.; Ma, S.; Xia, H.; Zhang, Y.; Shi, Z.; Mu, Y.; Liu, X. Appl. Catal. B 2019, 244, 36. doi: 10.1016/j.apcatb.2018.11.032
Wu, W.; Xu, S.; Qi, G.; Zhu, H.; Hu, F.; Liu, Z.; Zhang, D.; Liu, B. Angew. Chem., Int. Ed. 2019, 58, 3062. doi: 10.1002/anie.201811067
徐雯秀, 戴小强, 徐涵靖, 翁建全, 有机化学, 2018, 38, 2807. doi: 10.6023/cjoc201805031Xu, W.; Dai, X.; Xu, H.; Weng, J. Chin. J. Org. Chem. 2018, 38, 2807 (in Chinese). doi: 10.6023/cjoc201805031
Jiang, J.-X.; Li, Y.; Wu, X.; Xiao, J.; Adams, D. J.; Cooper, A. I. Macromolecules 2013, 46, 8779. doi: 10.1021/ma402104h
Li, X.; Zhang, Y. C.; Zhao, Y.; Zhao, H. P.; Zhang, B.; Cai, T. Macromolecules 2020, 53, 1550. doi: 10.1021/acs.macromol.0c00106
(a) Wang, S.; Song, K.; Zhang, C.; Shu, Y.; Li, T.; Tan, B. J. Mater. Chem. A 2017, 5, 1509.
(b) Dou, Z.; Xu, L.; Zhi, Y.; Zhang, Y.; Xia, H.; Mu, Y.; Liu, X. Chem.-Eur. J. 2016, 22, 9919.
Liu, X.; A, S.; Zhang, Y.; Luo, X.; Xia, H.; Li, H.; Mu, Y. RSC Adv. 2014, 4, 6447. doi: 10.1039/c3ra46988c
Li, Y.; Sun, B.; Zhou, Y.; Yang, W. Appl. Organomet. Chem. 2017, 31, 3578. doi: 10.1002/aoc.3578
Jiang, J.; Luo, R.; Zhou, X.; Chen, Y.; Ji, H. Adv. Synth. Catal. 2018, 360, 4402. doi: 10.1002/adsc.201800730
(a) Cohen, S. M. Chem. Rev. 2012, 112, 970.
(b) Cohen, S. M. J. Am. Chem. Soc. 2017, 139, 2855.
Monterde, C.; Navarro, R.; Iglesias, M.; Sánchez, F. ACS Appl. Mater. Interfaces 2019, 11, 3459. doi: 10.1021/acsami.8b18053
Liras, M.; Pintado-Sierra, M.; Iglesias, M.; Sánchez, F. J. Mater. Chem. A 2016, 4, 17274. doi: 10.1039/C6TA07696C
Li, L.; Cui, W.; Su, W.; Wang, Y.; Wang, R. Nano Res. 2016, 9, 779. doi: 10.1007/s12274-015-0957-x
(a) Angerani, S.; Winssinger, N. Chem.-Eur. J. 2019, 25, 6661.
(b) Marzo, L.; Pagire, S. K.; Reiser, O.; König, B. Angew. Chem., Int. Ed. 2018, 57, 10034.
(c) Wu, Y.-P.; Yan, M.; Gao, Z.-Z.; Hou, J.-L.; Wang, H.; Zhang, D.-W.; Zhang, J.; Li, Z.-T. Chin. Chem. Lett. 2019, 30, 1383.
(d) Naumann, R.; Goez, M. Green Chem. 2019, 21, 4470.
Xie, Z.; Wang, C.; deKraffit, K. E.; Lin, W. J. Am. Chem. Soc. 2011, 133, 2056. doi: 10.1021/ja109166b
Liang, H.-P.; Chen, Q.; Han, B.-H. ACS Catal. 2018, 8, 5313. doi: 10.1021/acscatal.7b04494
Xu, Z.-Y.; Luo, Y.; Zhang, D.-W.; Wang, H.; Sun, X.-W.; Li, Z.-T. Green Chem. 2020, 22, 136. doi: 10.1039/C9GC03688A
Xue, F.; Wang, F.-L.; Liu, J.-Z.; Di, J.-M.; Liao, Q.; Lu, H.-F.; Zhu, M.; He, L.-P.; He, H.; Zhang, D.; Song, H.; Liu, X.-Y.; Qin, Y. Angew. Chem., Int. Ed. 2018, 57, 6667. doi: 10.1002/anie.201802710
Luo, Y.; Xu, Z.-Y.; Wang, H.; Sun, X.-W.; Li, Z.-T.; Zhang, D.-W. ACS Macro Lett. 2020, 9, 90. doi: 10.1021/acsmacrolett.9b00872
Nicewicz, D. A.; MacMillan, D. W. C. Science 2008, 322, 77. doi: 10.1126/science.1161976

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





















 下载:
下载:

















