
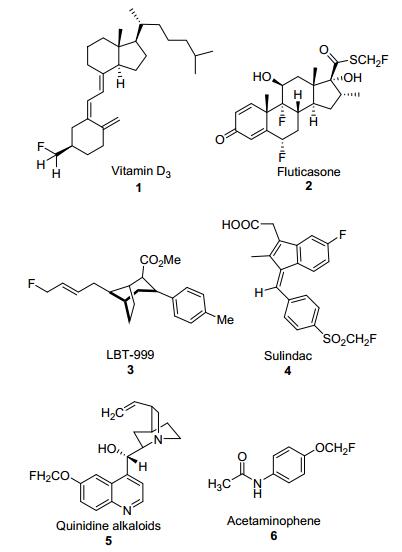
图 1.
含有单氟甲基结构的生物活性分子和药物
Figure 1.
Bioactive molecules and drugs containing monofluoromethyl structure

含氟化合物因为具有独特的性质已经成为制药和先进材料的潜在候选者, 尽管它是极其罕见的天然产物[1], 但由于含氟化合物具有较好的药理活性和生理活性, 例如在抗溃疡、抗菌和抗炎等[2]方面具有较好的活性, 市场上近30%的药品或农用化学品的结构中至少含有一个氟原子[3].因此, 含氟化合物高效合成方法的开发越来越引起合成化学家的关注.一个基团中氟原子的存在会影响其理化性质, 使合成的产物成为化学科学中独特且极具价值的支架[4], 近年来, 对有机氟化合物的需求迅速增长, 有机分子的亲脂性、代谢稳定性和生物利用度等性质可以通过引入氟原子而显著改变[5], 在药物开发的不同阶段, 选择性地掺入一个氟原子或氟烷基(如CF3, CF2H和CH2F)已经成为药物发现的常规做法[6].在这种情况下, 由于CH2F官能团可以模拟生物活性分子中经常遇到的CH3和CH2OH基团[7], 单氟甲基化合物特别有价值, 许多药物分子和生物活性分子都含有单氟甲基结构骨架(图 1), 但与大量三氟甲基化[8]和二氟甲基化反应[9]的成功例子相比, 单氟甲基化在研究中要少得多, 因此, 开发一种新的合成含CH2F基团有机化合物的策略仍然备受关注.
本文根据氟试剂的不同反应机理, 对近年来单氟甲基化反应中所涉氟甲基试剂分为5类, 较为详细地介绍了曾报道过的亲核单氟甲基化试剂(PhSO2CH2F, FBSM, TBTSO2CH2F和LiCH2F等)、自由基单氟甲基化试剂(CH2FSO2Na和CH2SO2Cl等)、亲电单氟甲基化试剂(单氟甲基卤化物、单氟硫盐试剂、单氟甲基亚砜亚胺试剂和单氟甲基硫叶立德试剂)、保护的单氟甲基化试剂[(PhSO2)2CFX和CHXFCO2R]和其它氟甲基试剂(PhSO2SCH2F和CH2FSSO3Na)等作为试剂的单氟甲基化反应研究.
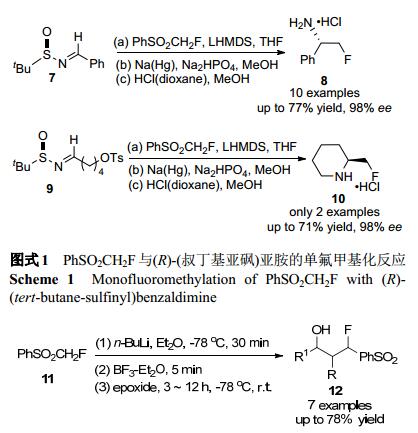
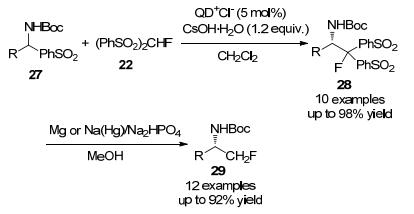
亲核氟烷基化是将氟烷基引入目标分子的一种直接方法, 然而, 亲核的单氟甲基化(“CH2F”基团向碳亲电体的转移)一直未见报道. 2006年, 胡金波课题组[10]首次报道了以PhSO2CH2F作为单氟甲基化试剂, 与(R)-N- (叔丁基亚砜)亚胺发生亲核单氟甲基化反应, 合成了相应的α-单氟甲胺, 具有高度立体选择性和简便性的特点, 该方法同样可采用对甲苯磺酸盐(OTs)-(R)-(叔丁基亚砜)亚胺为原料合成均手性α-单氟甲基化环仲胺(Scheme 1).
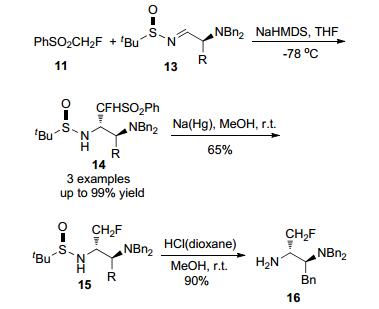
同年, 该课题组[11]以PhSO2CH2F作为试剂, 通过CHF-引入PhSO2, 与简单环氧化物进行了亲核单氟甲基化反应, 一步合成了产物β-单氟烷醇(Scheme 2).
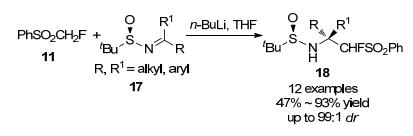
2007年, 刘俊及其同事[12]通过直接亲核单氟甲基化反应, 用PhSO2CH2F作为试剂与N-叔丁基磺酰亚胺合成对应的化合物14, 经还原脱磺酰化和酸催化醇解后, 很容易转化为手性α-单氟甲基化乙二胺(Scheme 3), 该反应具有产率高, 与非对映选择性好等优点, 这些优点可使此方法应用于有机合成中一些重要的应用.
2008年, 该实验室[13]深入研究, 以PhSO2CH2F为试剂, 通过(R)-N-叔丁基亚砜基酮亚胺的亲核单氟甲基化反应, 首次实现了α, α-单氟甲基胺的合成(Scheme 4), 同样具有高效、高非对映选择性特点, PhSO2CH2F与碱预生成PhSO2CHF-是反应的关键, PhSO2CHF-的热稳定性、良好的亲核性和相对较弱的碱性在当前反应中起着重要作用.
2009年, 胡金波等[14]发展了一种高效实用的亲核氟烷基化方法, 制备了α-官能化的单氟(苯磺酰基)甲烷, 以PhSO2CH2F为试剂对多种酯与亚砜进行亲核单氟甲基化反应, 高产率地合成了α-单氟酮砜和α-氟二砜(Scheme 5).
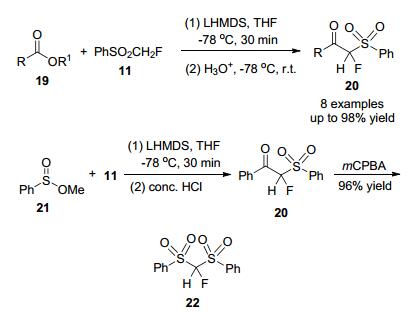
环氧化物和含氟碳离子之间的开环反应异常困难, 可以争议地归因于含氟碳离子的固有性质, 即它的热稳定性较低. 2006年, 李亚及其同事[11]报道了一种未知的化合物氟双(苯磺酰基)氟甲烷[(PhSO2)2CHF], 分别与环氧化物和氮杂环丙烷进行单氟甲基化反应(Scheme 6), 而这些环氧化物通常是惰性的.
大约在同一时间, Shibata等[15]报道的氟双(苯磺酰基)甲烷(FBSM)试剂也可以用于对映体选择性的单氟甲基化反应. 2007年, 该课题组[16]首次报道在FBSM作为试剂与Mannich反应结合条件下, α-氨基砜原位生成亚胺中间体, 由相转移催化剂(PTC)催化对映选择性氟二苯磺酰单氟甲基化反应, 然后通过还原脱磺酰化反应, 生成相应的不对称α-单氟甲基胺产物(Scheme 7).
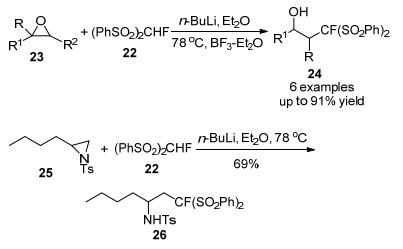
2007年, Olah课题组[17]报道了一种新的、高效的Mitsu-nobu反应, 该反应利用(PhSO2)2CHF作为亲核试剂与醇进行单氟甲基化反应, 以PPh3和偶氮二羧酸二异丙酯(DIAD)为氧化还原偶联剂, 在室温下进行苯氧化还原反应, 在中性条件下得到产率较高的产物(Scheme 8).
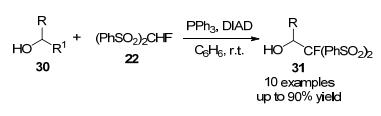
2009年, 该课题组[18]报道了一种以(PhSO2)2CHF为高通用性试剂与烷基、芳基卤化物的亲核单氟甲基化反应, 使用芳基卤化物, 可以实现立体一锅法合成α-单氟苯乙烯基砜, 产率适中, 该方法通过适当选择还原性脱磺酰化试剂体系, 进行逐步的脱磺酰化反应, 分别得到α-氟代-α-(苯磺酰基)烷烃和α-氟代烷烃(Scheme 9).
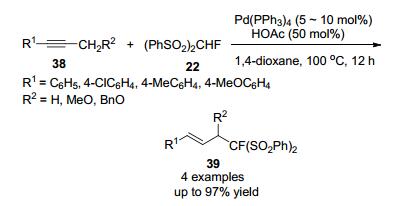
2009年, 胡金波[19]课题组报导了在乙酸条件下, 由Pd催化的炔烃与(PhSO2)2CFH试剂烯丙基化反应, 得到各种烯丙基化的单氟甲基化合物, 产率较高(Scheme 10), 检查底物的范围和限制条件, 发现该反应适用于1-芳基取代的丙炔和3-芳基取代的丙炔醚, 该反应具有高区域选择性和高立体选择性的特点.
2011年, Shibata课题组[20]首次用FBSM作为试剂, 实现了Morita-Baylis-Hillman碳酸盐的有机催化对映体选择性单氟甲基化反应, 获得较高产率与ee值(Scheme 11), 金鸡纳生物碱和FeCl2或者Ti(OiPr)4的协同催化可提高产物产率高达99%, 对映体选择性也相对提高.
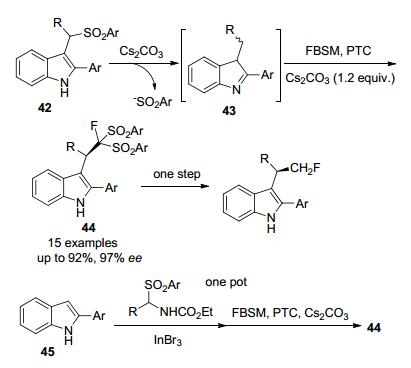
2013年, 该课题组[21]通过原位生成的乙烯亚胺中间体, 首次实现了相转移催化剂(PTC)催化的吲哚衍生物与FBSM的不对称单氟甲基化反应, 反应产率高, 对映体选择性高, 该反应有效地利用了芳基磺酰基激活底物和试剂FBSM, 在同种反应条件下, 2-芳基吲哚与FBSM的一锅转化也是可以实现的(Scheme 12).
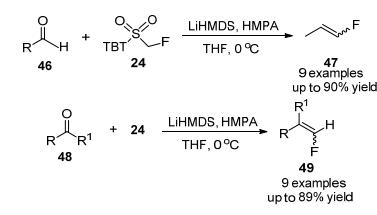
2010年, 朱林桂等[22]报道1-叔丁基-1H-四唑-5-氟甲基砜(TBTSO2CH2F)是一种新型高效的氟甲基试剂, 通过Julia-Kocienski烯化反应合成末端和内部单氟烯烃, TBTSO2CH2F与羰基化合物(醛和酮)之间发生碱-介导反应, 在碱为LiHMDS, 溶液为四氢呋喃(THF)时, 以较高产率和中等E/Z选择性合成末端单氟烯烃(Scheme 13), 该试剂被证明是制备结构多样的单氟烯烃的一种通用的关键起始原料, 具有许多潜在的应用价值.
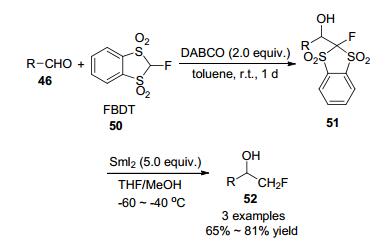
同年, Shibata等[23]开发了一种新的亲核单氟甲基化试剂2-氟-1, 3-苯并二硫代-1, 1, 3, 3-四氧化物(FBDT), 它适用于基于α-氟碳离子生成的醛的第一次亲核单氟甲基化反应, 可通过选择有机碱来控制FBDT对共轭醛的1, 2-加成和1, 4-加成反应的选择性, 用此方法仅需两步即可从醛中获得单氟甲基醇, 产率较好到较高(Scheme 14).
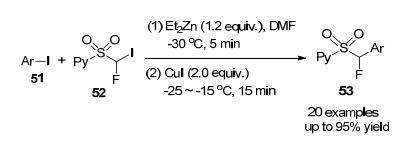
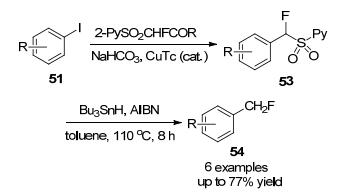
2012年, 胡金波课题组[24]报道了一种由铜催化, 以2-PySO2CFHI为试剂, 芳基碘脱苯甲酰的单氟甲基化反应, 产率高达95% (Scheme 15).利用这种方法, 无论是化学计量铜还是催化量的铜, 都能有效地将具有不同官能团的芳基碘化物转化为所需的单氟烷基化产物.该方法还适用于生物活性分子的晚期单氟甲基化, (2-吡啶基)磺酰基的存在不仅在铜介导的交叉偶联反应中起着关键作用, 而且还促进了产物的进一步转化.
2013年, 该课题组[25]进一步报道了一种新的芳基碘化物的单氟甲基化反应, 芳基碘化物可以通过铜催化的Hurtley型脱苄基氟烷基化反应以及随后的还原脱磺酰反应有效地转化为相应的单氟甲基化产物(Scheme 16), (2-吡啶基)磺酰基部分可以很容易地通过Bu3SnH介导的脱磺酰反应去除.
2017年,邹大鹏等[26]报道了由含路易斯酸的金属CuI催化的芳烃与二乙胺基三氟化硫(DAST)发生单氟甲基化反应, 其中底物为2-((甲亚砜)甲氧基)萘时, 目标产物产率高达99% (Scheme 17), 该反应在引入CuI (10 mol%)作为催化剂的条件下, 副产物消失, 改进了反应过程, 为单氟甲基芳醚的合成提供了一条有效的途径.该方法简单易行, 具有官能团选择性好, 底物范围广等特点.

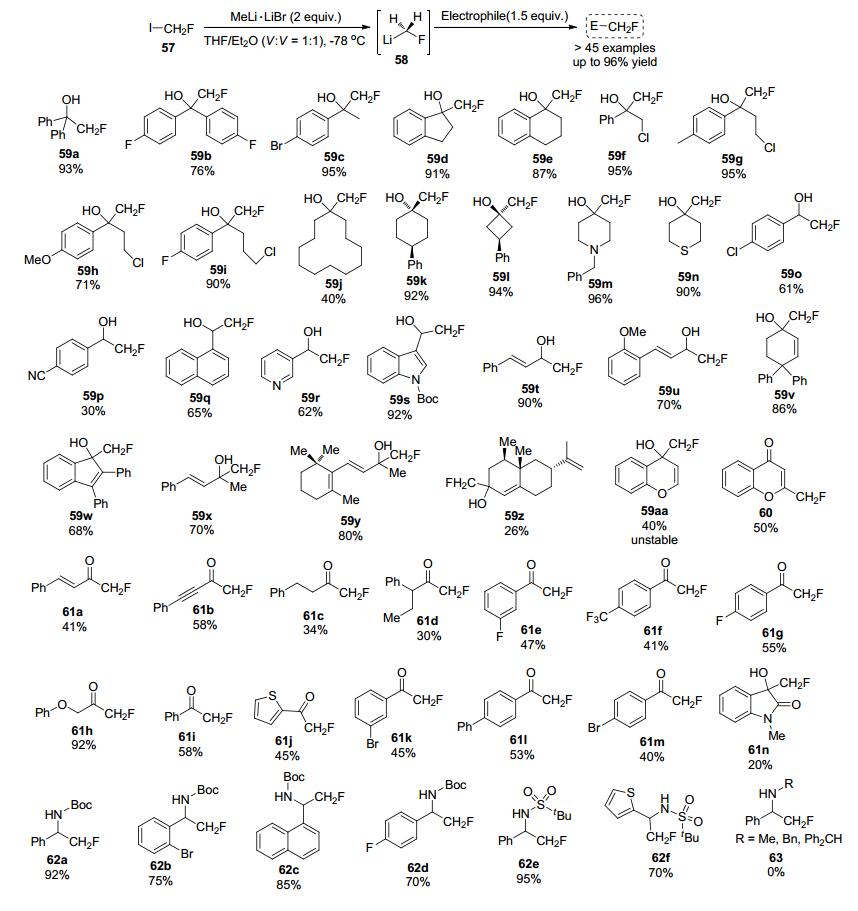
2017年, Luisi等[27]开发了一种直接亲核氟甲基化的新型一锅法, 该策略使用从市售的氟碘甲烷在-78 ℃下使用MeLi•LiBr作为锂化剂生成的氟甲基锂(LiCH2F)作为氟试剂, 使用各种亲电体进行单氟甲基化反应, 可以通过一次合成操作以非常高的收率获得一系列有价值的化学产物(Scheme 18), 这项工作为氟甲基化策略和氟化有机金属的进一步发展铺平了道路.
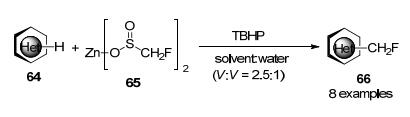
杂环化合物的固有特性使得它们在水溶性和作为配体的能力等生物应用中非常理想, 这使得它们在直接化学功能化方面具有挑战性, 2012年, Baran课题组[28]报道了在过氧化叔丁醇(TBHP)存在下, 杂芳烃与Zn(O2SCH2F)2直接单氟甲基化反应(Scheme 19), 该反应条件温和, 可以容忍高度敏感的官能团.
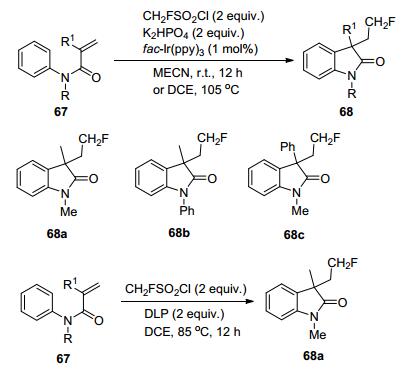
2014年, Dolbier等[29]使用氟化磺酰氯作为CH2F自由基的来源, 在可见光温和条件下, MeCH作为溶剂, K2HPO4作为碱, 底物N-芳基丙烯酰胺与CH2FSO2Cl单氟甲基化反应, 产物68a仅可获得16%的收率, 当反应在105 ℃下进行时, 以二氯乙烷作为溶剂, 环化产物(68a, 68b, 68c)的收率提高至45%~48%, 使用明确的热自由基引发剂过氧化二月桂酰过氧化物(DLP)会生成相应的环化产物, 收率达70% (Scheme 20).
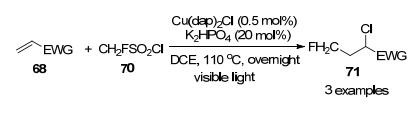
在这项工作的基础上, 该课题组[30]认为单氟甲基磺酰氯与缺电子烯烃的光诱导原子转移自由基加成(ATRA)反应是可能的, 随后他们报导了CH2FSO2Cl与缺电子烯烃在110 ℃下使用Cu介导的光化学条件可通过ATRA反应合成目标产物, 产率达到68% (Scheme 21), 该反应也可应用于各种氟烷基磺酰氯与各种缺电子烯烃反应, 但需要苛刻的条件或化学计量的过氧化物.
2015年, 胡金波等[31]通过NaBH4催化还原化合物合成了单氟甲基亚磺酸钠(CH2FSO2Na), 在Ag的催化下与共轭N-芳基磺酰胺单氟甲基化反应合成产物75 (Scheme 22), 此方法为快速大规模制备氟甲基亚磺酸钠提供了一条简单有效的途径.
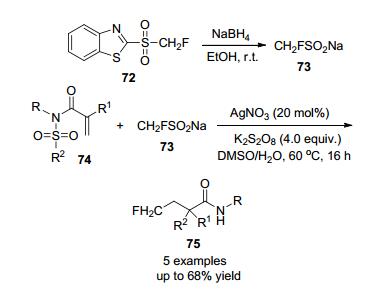
2019年, 付维军[32]课题组研究了银催化下CH2FSO2Na与炔烃化合物76的级联自由基单氟甲基化, 合成了多种单氟甲基香豆素(Scheme 23), 产率适中, 银催化剂和K2S2O8能有效地促进反应, 此外, 该方法操作简单, 反应条件温和, 官能团相容性好.
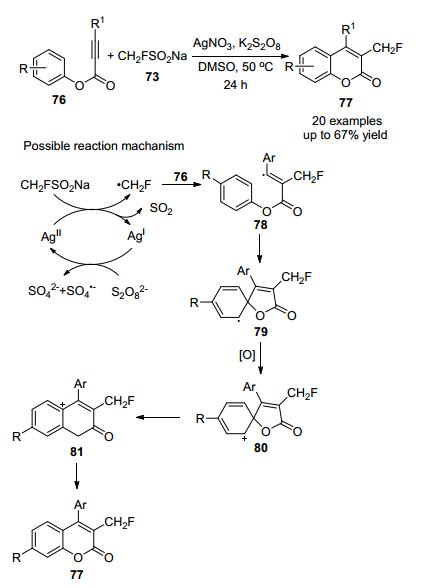
根据上述实验结果和文献报道[33-34], 提出一种合理的反应机理, 首先单氟甲基亚磺酸钠与AgNO3反应生成单氟甲基自由基(•CH2F), 然后与化合物76的叁键反应生成中间体78, 然后, 自由基中间体78在分子内5-外环化得到自由基中间体79, 自由基中间体79被氧化成环己二烯基80, 然后1, 2-酯迁移形成阳离子81, 最后, 中间体81通过消除H+得到产物.
[18F]氟甲基与[11C]甲基具有相似的电子和空间性质, [18F]氟甲基化配体[35-36]可能与从相同底物制备的[11C]甲基配体具有相似的示踪性质, 由于[18F]氟甲基化试剂的制备不便、反应活性的不确定型以及[18F]氟甲基化产物的不稳定性, [18F]氟甲基化的应用受到了很大的限制. 2004年, Suzuki等[37]制备了[18F]FCH2I, 该试剂在各种有机溶剂中稳定, 在碱性溶液中不稳定, 可与苯酚、噻酚、酰胺和胺官能团单氟甲基化反应(Scheme 24), 此结果可能也适用于非放射性FCH2I的反应.
2007年, 张伟等[38]发现CH2ClF可以作为一种有用的亲电单氟甲基化试剂, 用于各种含O-、S-和N-的亲核反应物, 合成相应的单氟甲基化产物(Scheme 25), 尽管氯氟甲烷与多种亲核物质发生反应, 但它是一种消耗臭氧的气体, 因此它的未来用途是值得怀疑的.
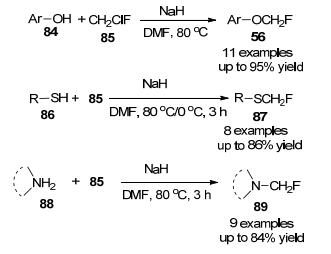
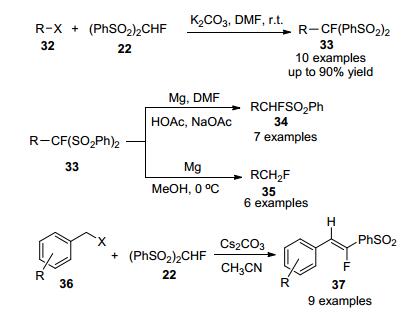
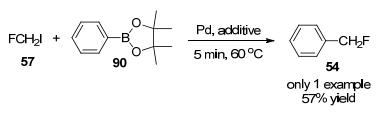
2009年, Suzuki课题组[39]报道了第二个芳烃直接单氟甲基化的例子, 首次用CH2FI作为限制反应物, 在N, N-二甲基甲酰胺(DMF)中用钯(1 equiv.)处理过量的萘苯硼酸酯(40 equiv.), 进行单氟甲基化反应, 产量为57% (Scheme 26), 但此反应操作不便.
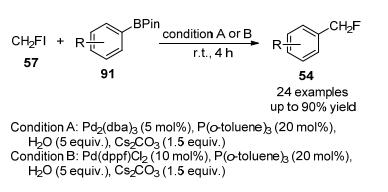
2015年, 胡金波等[40]实现了一种新的氟甲基碘化物(CH2FI)的制备, 并首次报道了在水体系中使用Pd2(dba)3/ Pd(dppf)Cl2为催化剂, 在碱为Cs2CO3, CH2Cl2和DMF为助溶剂, 三(邻甲苯基)膦作为支撑配体条件下, 芳基硼酸酯与该试剂进行直接单氟甲基化反应, 合成单氟甲基芳烃(Scheme 27), 目标产物获得较高收率, 该反应在室温下进行, 具有良好的官能团耐受性.
同年, 张新刚等[41]以工业原料氟甲基溴(CH2FBr)为偶联剂, 报道了一种制备单氟甲基化芳烃的有效而直接的方法, 该反应采用低成本的镍催化剂, 4-二甲氨基吡啶(DMAP)作为添加剂, 在温和的反应条件下多种芳基硼酸与CH2FBr单氟甲基化反应, 高效制备目标产物(Scheme 28), 该方案具有合成简单及官能团相容性良好等特点.
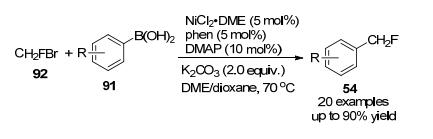
2019年, 王细胜课题组[42]开发了一种温和高效的镍催化还原交叉偶联直接单氟甲基化(杂)芳基溴化物的方法, 该方案以溴氟甲烷为试剂, 以溴代芳烃为底物, 在Ni和Mn催化下合成对应的产物(Scheme 29), 与已知的直接单氟甲基化方法相比[43], 该设计具备体系操作简便、底物经济易得以及对多种杂芳烃等具有优异的耐受性等特点, 该策略为药物开发提供了一条合成单氟甲基化分子的有效途径.
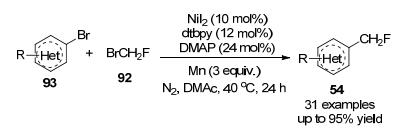
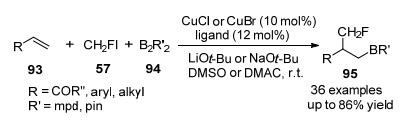
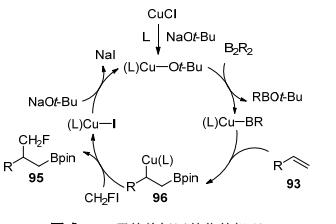
有机硼化合物是有机合成的通用中间体, 最近, 不饱和碳-碳键的硼化双官能团化已成为一种高度官能化烷基硼烷的有效方法[44], 2019年, 卿凤翎及其同事[45]开发出了一种以CH2FI为氟试剂的铜催化的烯烃与氟碘甲烷和二硼试剂的区域选择性单氟甲基化, 该方案以良好的收率和优异的区域选择性合成了以前未知且有用的硼烷基单氟甲基化烷烃(Scheme 30).
可能的机理如Scheme 31所示, 首先, 用NaOt-Bu处理CuCl并与配体配位生成(L)Cu-Ot-Bu.然后, (L)Cu-Ot-Bu与B2R2反应得到硼铜(L)Cu-BR, 随后, 烯烃的C=C键插入Cu—B键得到中间体96, 该中间体被CH2FI捕获以合成硼烷基氟甲基化的产物, 同时96形成(L)Cu-I并转化为(L)Cu-Ot-Bu, 用于下一个催化循环, 尚不清楚丙烯酰胺和苯乙烯需要两种不同反应体系的原因.
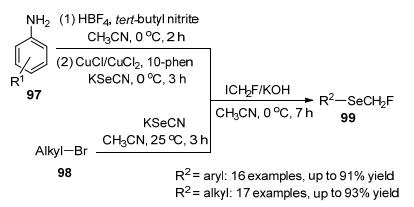
2019年, 易文斌等[46]成功地开发了第一个以有机硒氰酸酯为原料, 与ICFH2单氟甲基化反应, 通过一锅多步反应合成单氟甲基硒醚的例子, 反应从胺或烷基溴化物开始, 形成相应的C(sp2)—SeCFH2或C(sp3)—SeCFH2键, 合成相应的目标产物(Scheme 32), 该反应操作简单, 底物范围广, 官能团广, 可应用于类药物化合物的晚期单氟甲基硒化反应.
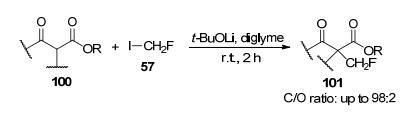
2019年, 该课题组[47]首次实现了高碳选择性单氟甲基化反应, 在室温下, 采用β-酮酯为原料, 氟碘甲烷为试剂, 二甘醇为溶剂, 并且在锂盐作为碱的条件下, β-酮酯被转化为约27种碳选择性CH2F-产物, 该反应底物范围广, 区域选择性高, 且产率高达91% (Scheme 33).
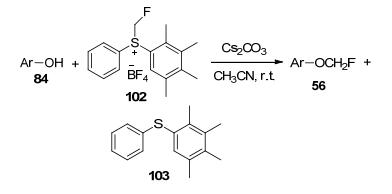
除了首次发表的氟甲醇直接亲电单氟甲基化反应外[48], 2008年, Olah及其同事[49]开发了一种新的亲电单氟甲基化试剂, S-(单氟甲基)二芳基硫四氟硼酸盐, 其可与芳基化合物进行单氟甲基化反应, 生成产率较好的氟甲醚(Scheme 34), 这种方法还可应用于各种生物和药用重要化合物的合成.
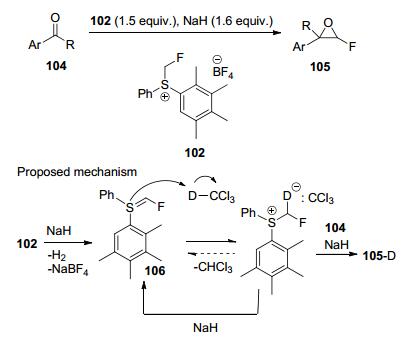
2019年, Veliks等[50]将二芳基氟甲基硫盐102作为氟甲基转移试剂, 在温和条件下与各种芳基取代酮和醛单氟甲基化反应, 合成了相应的单氟环氧化物(Scheme 35), 该反应具有易于操作和收率较高等优点, 而且该反应首次证明了氟甲基亚叶酸硫106作为反应中间体的可行性.
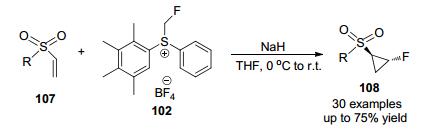
环丙基部分和氟原子通常存在于药物分子中, 从代谢稳定性、亲脂性和药代动力学方面来看, 这两种基团通常会改善潜在药物分子的物理化学性质[51], 然而, 这两个部分的结合形成氟环丙基部分相当罕见, 而且研究较少. Melngaile课题组[52]发现二芳基氟甲基硫盐是一种有效的氟代亚甲基转移试剂, 在室温下用THF溶剂和NaH处理乙烯基砜, 以较好产率合成官能化的单氟环丙烷(Scheme 36).
亚砜亚胺和亚砜亚胺盐具有重要的生理和化学性质, 在有机合成中得到了广泛的应用[53].最近, 氟化亚砜和亚砜亚胺盐作为烷基化试剂的应用引起了人们的极大关注[54], 但涉及单氟甲基化反应的研究甚少.
2011年, Shibata及其同事[55]开发出了新的亲电性单氟甲基化试剂单氟甲基亚砜盐109, 它们与β-酮酯进行了亲电的单氟甲基化反应, 得到β-酮酯的单氟甲基烯醇醚110, 产率高达96%, 在所有情况下均观察到选择性的O-单氟甲基化, 基于Pd/C催化的还原体系, 可以将未知的单氟甲基烯醇醚转化为单氟甲醚(Scheme 37).
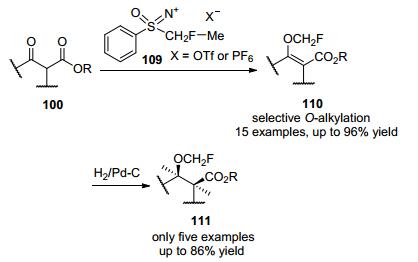
此方法不仅是烯醇酸盐选择性O-烷基化反应的少有实例之一, 而且在制药和农业化工业方面, 为含氟化合物的合成提供了一种新的途径, 但O-选择性单氟甲基化的机理仍尚不清楚, 有待于进一步深入了解.
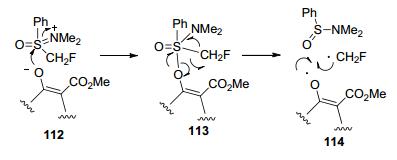
2012年, 该课题组[56]深入研究了单氟甲基亚砜盐109与β-酮酯的单氟甲基化反应, 并且揭示了β-酮酯单氟甲基化的反应机理.如Scheme 38所示, 单氟甲基化将通过烯醇氧对112的硫中心的攻击来进行, 以合成硫烷型中间体113, 该中间体与二甲氨基苯亚砜产生氧和CH2F自由基, 发现C/O偏好依赖于氟甲基中的氟原子数目, 在β-酮酯的单氟甲基化反应中, O-区域选择性主要归因于单氟甲基自由基的性质.
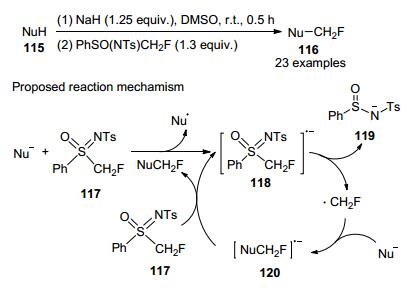
2014年, 胡金波等[57]以PhSO(NTS)CH2F作为一种新型的单氟甲基化试剂, 分别对O-、S-、N-和P-亲核物质进行了直接单氟甲基化反应, 合成了相应的目标产物(Scheme 39).
基于实验结果, 他们提出可能的机理, 从亲核物质到117的单电子转移得到自由基阴离子118, 然后协同或逐步消除119, 得到单氟甲基自由基, 它与另一亲核物质结合形成自由基阴离子中间体120, 在120到117的单电子转移与中间体118的形成后, 形成NuCH2F产物, 初步的机理研究表明, 这是一种涉及固定过程的机制.
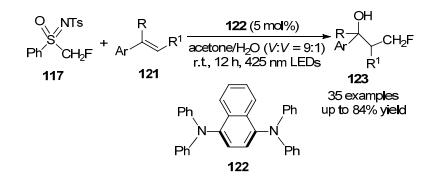
2019年, Koike课题组[58]开发了一种具有强还原性的光还原体系, 使用1, 4-双(二苯基氨基)萘(122)作为催化剂, 在温和的条件下使用PhSO(NTs)CH2F试剂促进烯烃的无金属羟基单氟甲基化, 轻松合成了γ-氟代醇(Scheme 40).本工作为开发具有高还原能力但激发态寿命短的可见光有机光催化剂提供了新的策略, 这对于常规Ir光催化剂难以实现的反应是有用的, 需要更高还原能力的反应有待于进一步开发.
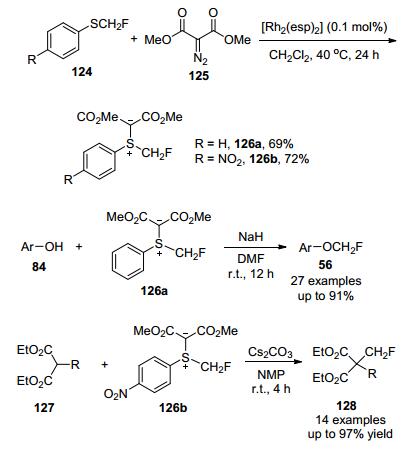
2017年, 沈其龙及其同事[59]成功发现两种亲电性单氟甲基硫叶立德试剂126, 它们可以直接从容易获得的原料中合成, 且亲电性较高, 可与多种亲核物质发生单氟甲基化反应, 当醇类化合物与试剂126a进行单氟甲基化反应时, 产率高达91%, β-酮酯作为底物与试剂126b在温和条件下进行反应时, 生成对应的C-选择性单氟甲基化产物, 产率高达97% (Scheme 41).
2008年, Olah课题组[60]以氟双(苯磺酰基)甲烷(22)为原料, 通过碘化反应定量制备了(PhSO2)2CFI, 在三乙基硼烷条件下, 与烯烃单氟甲基化反应, 在1, 5-二氮杂二环[5.4.0]十一-5-烯(DBU)存在的情况下, 加合物经过脱氢碘化一锅法立体选择性合成双(苯磺酰基)单氟甲基烯烃, 产物总收率良好到中等(Scheme 42).
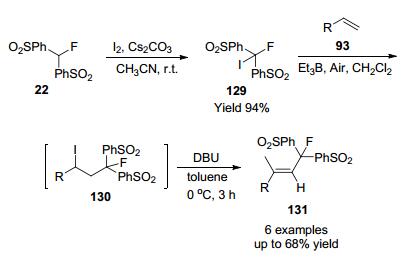
2014年,俞寿云及其同事[61]使用溴氟乙酸乙酯(EBFA)为试剂, 通过与联苯异氰化物在室温下通过可见光促进的烷基化和随后的脱羧反应制备了一系列单氟甲基化菲啶衍生物, 最终得到良好至理想的收率(Scheme 43), 已经开发的分步反应和一锅反应均运行良好, 此外, EBFA价格便宜且易于获得, 使此方案成为合成单氟甲基化杂芳烃的理想选择.
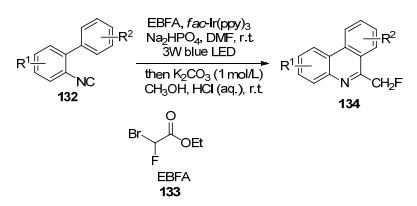
2015年, 王细胜等[62]开发了金属镍催化芳基硼酸新的单氟甲基化反应, 氟甲基试剂含有苯砜或乙氧基羰基, 在CH2Cl2中, PPh3为配体, K2CO3为碱时, 合成了相应的目标产物, Na(Hg)介导的磺酰基产物通过还原脱磺酰反应(Scheme 44), 以优异的产率得到单氟甲基芳烃, 但该反应步骤与反应条件相对苛刻.
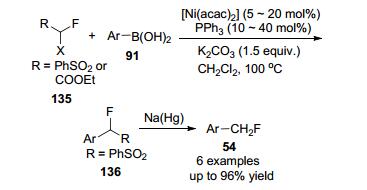
2017年, Ackermann课题组[63]报道了由膦/羧酸盐辅助钌(II)催化的间位C—H单氟甲基化, 在P(3- C6H4CF3)3作为添加剂条件下, 合成了6种目标产物, 其中在吡啶、嘧啶、吡唑基甚至嘌呤的辅助下完成了前所未有的远程碳氢单氟甲基化反应, 顺利地合成了所需的产品137a~137d (Scheme 45).
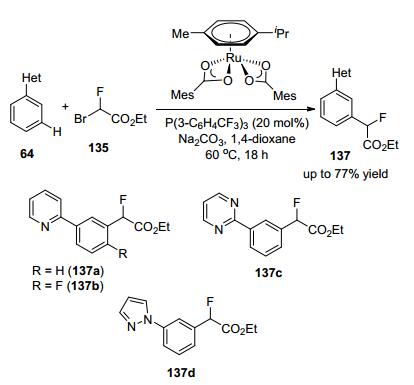
2017年, 许华建等[64]开发了在Eosin Y催化体系中肉桂酸新的单氟甲基化反应, 通过可见光介导的光氧化还原催化α, β-不饱和羧酸的有效烯烃反应合成C—CH2F键, 与试剂135发生单氟甲基化反应, 以良好的收率和高E/Z选择性合成了相应的单氟甲基化烯烃(Scheme 46), 该反应在常压和不含金属条件下进行, 具有广泛的官能团耐受性.
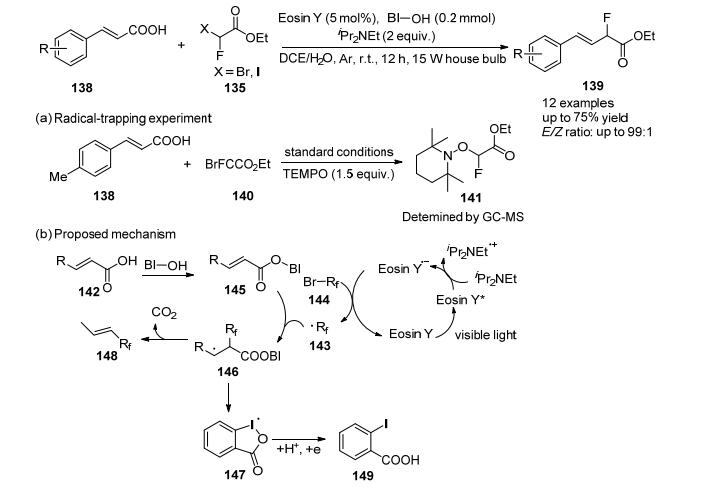
自由基捕获实验表明, 加入1.5 equiv.的2, 2, 6, 6-四甲基哌啶-1-氧基(TEMPO), 完全抑制了标准条件下的模型反应, 气相色谱-质谱联用(GC-MS)技术检测到了TEMPO-CFHCO2Et加合物, 表明该反应途径涉及自由基的参与, 根据上述结果和文献报告[65, 66], 提出了一种可能的机制.最初, 反应首先生成关键中间体α-氟乙酸乙酯(143), 它是通过将单电子从Eosin Y自由基阴离子转移到144形成的, 同时, 苯并碘氧基乙烯基羧酸络合物(BI-OOCCH=CHR, 145)是由BI-OH与乙烯基羧酸的结合生成的, 随后, α-氟乙酸乙酯自由基143加成到155的α碳原子上, 最终, 中间体146经过二氧化碳和苯并碘唑自由基147的消除以生成所需的产品148, 苯并二噁唑自由基147还原质子化生成碘代苯甲酸149.
2019年, 该实验室[67]开发一种可见光诱导的方法, 在室温下无需复杂的额外配体或添加剂, 使用(三苯基锡)钴肟作为催化剂来活化溴氟乙酸乙酯, 使一系列苯乙烯衍生物经过本反应条件, 以良好的收率和优异的E/Z选择性生成相应的单氟甲基化烯烃(Scheme 47), 该方法反应条件温和, 催化剂成本低, 底物范围广, 成为进一步修饰生物活性化合物的潜在有用方法.
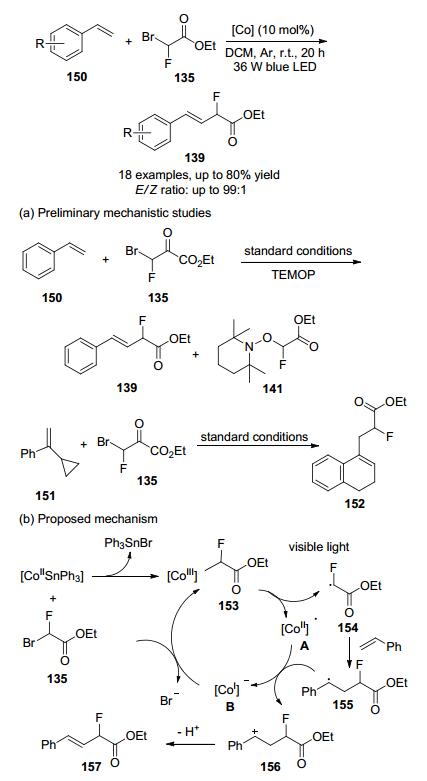
通过自由基捕获实验深入了解反应机理, 在标准条件下加入自由基清除剂TEMPO时, 反应被完全抑制, 在这种情况下, GC-MS也检测到TEMPO-CHFCO2Et加合物, 结果表明自由基参与了反应路径.另外, 将自由基捕捉剂151引入反应体系, 以42%的收率分离出开环产物152, 这些结果均证明在催化循环中确实存在游离的单氟烷基.
根据上述调查和文献报告[68-69], 提出可能的机理.最初, [CoII-SnPh3]与溴氟乙酸乙酯(135)反应产生中间体153.通过光辐照[CoIII]—CFHCO2Et键解离产生[CoII]•A和关键的中间体α-单氟乙酸乙酯基团154[69b, 70], 后者加到烯烃中得到自由基155[71], 随后, 155向[CoII]•A提供一个电子产生阳离子络合物156, 其失去质子而产生烯烃产物157.最后, [CoI]•B与溴氟乙酸乙酯(135)的反应将再生中间体153, 从而完成催化循环.
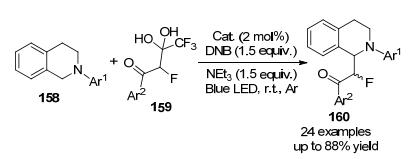
可见光诱导的光氧化还原催化引起了越来越多的关注, 并且通过可见光照射引起了一些基本的化学转变[72], 可见光氧化还原催化已应用于氟甲基化反应[73], 2015年, 朱成建课题组[74]报道了四氢异喹啉与三氟甲基β-氟代间二醇在可见光催化下的光氧化还原反应, 通过C(sp3)—H键活化生成单氟甲基化的四氢异喹啉(Scheme 48), 该方案提供了一种引入稳定的、易于制备的单氟间二醇作为CF源的新方法.
2017年, 沈其龙及其同事[75]成功合成了一种单氟甲基硫代试剂S-(氟甲基)苯磺酸盐, 该试剂可由易得的原料以较高产率合成, 在铜作为催化剂条件下, 试剂161能与多种芳基硼酸在温和的条件下发生单氟甲基化反应, 得到相应的单氟甲基硫代芳烃, 产率高达93%, 此外, 试剂161在银催化剂存在下与烷基烯烃反应得到高产率的烷基单氟甲基硫醚(Scheme 49).
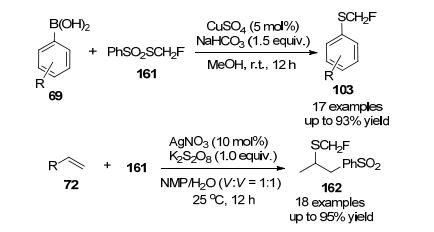
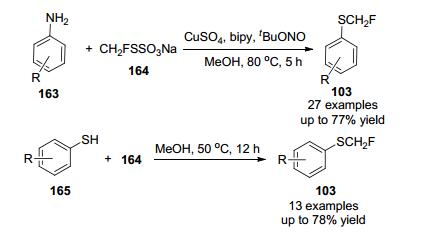
2018年,易文斌等[76]发现了一种典型的Bunte盐S-(氟甲基)硫代硫酸钠, 该试剂可用廉价易得的原料一步高效合成.其与苯胺单氟甲基化反应, 在CuSO4作为催化剂, Bipy作为配体, MeOH作为溶剂条件下, 以中等到较好的产率得到相应的单氟甲基硫醇产物.此外, 它还可以与芳基硫醇单氟甲基化反应(Scheme 50), 该方法具有操作方便、官能团相容性好、底物范围广适用于药物类化合物的后期研究等特点.
按照氟甲基试剂进行分类, 综述了近十多年单氟甲基化反应的研究进展, 目前常见的氟甲基试剂包括亲核单氟甲基化试剂、自由基单氟甲基化试剂、亲电单氟甲基化试剂、保护的单氟甲基化试剂以及其它氟甲基试剂等, 从以上的论述可以看出, 将单氟甲基引入到有机分子结构中已经引起合成化学家的广泛关注, 并且已经取得了很大的进步.
尽管单氟甲基化反应已取得重大进展, 但寻找成本低、反应条件温和、催化剂环保安全的单氟甲基化反应仍是最值得关注的发展方向.除此之外, 氟试剂种类较为丰富, 基本能完成各类单氟甲基化反应的需要, 但是存在着部分毒性大和环境有害等问题, 寻找和发展新型的单氟甲基试剂仍是将来研究的重点方向, 将其应用于工业化与药物生产, 将是值得关注和发展的另一个研究目标, 单氟甲基化反应机理的进一步研究值得探讨.
Harpere, D. B.; O'Hagan, D. Nat. Prod. Rep. 1994, 11, 123.
(a) Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765.
(b) Xu, X.-H.; Matsuzaki, K.; Shibata, N. Chem. Rev. 2015, 115, 731.
(c) Boiko, V. N. Beilstein J. Org. Chem. 2010, 6, 880.
(a) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320.
(b) Jeschke, P. ChemBioChem 2004, 5, 590.
(a) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320.
(b) Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432.
(c) Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315.
Some examples: (a) Wang, J.; Sánchez-Roselló, M.; Aceña, J.; del Pozo, C. L.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432.
(b) Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881.
(c) Hagmann, W. K. J. Med. Chem. 2008, 51, 4359.
(a) Wong, D. T.; Perry, K. W.; Bymaster, F. P. Nat. Rev. 2005, 4, 764.
(b) Kirk, K. L. Org. Process Res. Dev. 2008, 12, 305.
(c) Wang, J.; Sanchez-Rosello, M.; Acena, J.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432.
Some examples: (a) Kollonitsch, J.; Patchett, A. A.; Marburg, S.; Maycock, A. L.; Perkins, L. M.; Doldouras, G. A.; Duggan, D. E.; Aster, S. D. Nature 1978, 274, 906.
(b) Mckeage, K.; Keam, S. J. Drugs 2009, 69, 1799.
(c) Rojas, R. A.; Paluga, I.; Goldfrad, C. H.; Duggan, M. T.; Barnes, N. J. Asthma 2007, 44, 437.
For selected examples, see: (a) Liu, X.; Xu, C.; Wang, M.; Liu, Q. Chem. Rev. 2015, 115, 683.
(b) Charpentier, J.; Fruh, N.; Togni, A. Chem. Rev. 2015, 115, 650.
(c) Chu, L.; Qing, F.-L. Acc. Chem. Res. 2014, 47, 1513.
(d) Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214.
倪传法, 朱林桂, 胡金波, 化学学报, 2015, 73, 90.For selected examples, see: (a) Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765.
(b) Ni, C.; Zhu, L.; Hu, J. Acta Chim. Sinica 2015, 73, 90(in Chinese).
Li, Y.; Ni, C.; Liu, J.; Zhang, L.; Zheng, J.; Zhu, L.; Hu, J. Org. Lett. 2006, 8, 1693.
Ni, C.; Li, Y.; Hu, J. J. Org. Chem. 2006, 71, 6829.
Liu, J.; Li, Y.; Hu, J. J. Org. Chem. 2007, 72, 3119.
Liu, J.; Zhang, L.; Hu, J. Org. Lett. 2008, 10, 5377.
Ni, C.; Zhang, L.; Hu, J. J. Org. Chem. 2009, 74, 3767.
Fukuzumi, T.; Shibata, N.; Sugiura, M.; Yasui, H.; Nakamura, S.; Toru, T. Angew. Chem., Int. Ed. 2006, 45, 4973.
Mizuta, S.; Shibata, N.; Goto, Y.; Furukawa, T.; Nakamura, S.; Toru, T. J. Am. Chem. Soc. 2007, 129, 6394.
Prakash, G. K. S.; Chacko, S.; Alconcel, S.; Stewart, T.; Mathew, T.; Olah, G. A. Angew. Chem., Int. Ed. 2007, 46, 4933.
Prakash, G. K. S.; Chacko, S.; Vaghoo, H.; Shao, N.; Gurung, L.; Mathew, T.; Olah, G. A. Org. Lett. 2009, 11, 1127.
Ni, C.; Hu, J.; Tetrahedron Lett. 2009, 50, 7252.
Furukawa, T.; Kawazoe, J.; Zhang, W.; Nishimine, T.; Tokunaga, E.; Matsumoto, T.; Shiro, M.; Shibata, N. Angew. Chem., Int. Ed. 2011, 50, 9684.
Matsuzaki, K.; Furukawa, T.; Tokunaga, E.; Matsumoto, T.; Shiro, M.; Shibata, N. Org. Lett. 2013, 15, 3282.
Zhu, L.; Ni, C.; Zhao, Y.; Hu, J. Tetrahedron 2010, 66, 5089.
Furukawa, T.; Goto, Y.; Kawazoe, J.; Tokunaga, E.; Nakamura, S.; Yang, Y.; Du, H.; Kakehi, A.; Shiro, M.; Shibata, N. Angew. Chem., Int. Ed. 2010, 49, 1642.
Zhao, Y.; Gao, B.; Ni, C.; Hu, J. Org. Lett. 2012, 14, 6080.
Zhao, Y.; Ni, C.; Jiang, F.; Gao, B.; Shen, X.; Hu, J. ACS Catal. 2013, 3, 631.
Geng, Y.; Liang, A. P.; Gao, X. Y.; Niu, C. S.; Li, J. Y.; Zou, D. P.; Wu, Y. S.; Wu, Y. J. J. Org. Chem. 2017, 82, 8604.
Parisi, G.; Colella, M.; Monticelli, S.; Romanazzi, G.; Holzer, W.; Langer, T.; Degennaro, L.; Pace, V.; Luisi, R. J. Am. Chem. Soc. 2017, 139, 13648.
Fujiwara, Y.; Dixon, J. A.; O'Hara, F.; Funder, E. D.; Dixon, D. D.; Rodriguez, R. A.; Baxter, R. D.; Herle, B.; Sach, N.; Collins, M. R.; Ishihara, Y; Baran, P. Nature 2012, 492, 95.
Tang, X.; Thomoson, C. S.; Dolbier Jr., W. R. Org. Lett. 2014, 16, 4594.
Tang, X.; Thomoson, C. S.; Dolbier Jr., W. R. Angew. Chem., Int. Ed. 2015, 54, 4246.
He, Z.; Tan, P.; Ni, C.; Hu, J. Org. Lett. 2015, 17, 1838.
Fu, W.-J.; Sun, Y.-N.; Li, X.-Y. Synth. Commun. 2019, 169, 7452.
(a) Li, Y.; Lu, Y.; Qiu, G.; Ding, Q. Org. Lett. 2014, 16, 4240.
(b) Moskvina, V. S.; Khily, V. P. Chem. Heterocycl. Comp. 2019, 55, 300.
(c) Zhang, X.; Li, Y.; Hao, X.; Jin, K.; Zhang, R.; Duan, C. Tetrahedron 2018, 74, 7358.
(d) Chen, L.; Wu, L.; Duan, W.; Wang, T.; Li, L.; Zhang, K.; Zhu, J.; Peng, Z.; Xiong, F. J. Org. Chem. 2018, 83, 8607.
(e) Bu, M.-J.; Lu, G.-P.; Cai, C. Catal. Commun. 2018, 114, 70.
(f) Ni, S.-Y.; Zhou, J.; Mei, H.-B.; Han, J.-L. Tetrahedron Lett. 2018, 59, 1309.
(a) Deng, X.; Chao, H.; Chen, C.; Zhou, H.; Yu, L. Sci. Bull. 2019, 64, 1280.
(b) Yu, L.; Huang, X. Synlett 2006, 2136.
(c) Yu, L.; Huang, X. Synlett 2007, 1371.
(d) Liu, M.; Li, Y.; Yu, L.; Xu, Q.; Jiang, X. Sci. China: Chem. 2018, 61, 294.
Zessin, J.; Eskola, O.; Brust, P.; Bergman, J.; Steinbach, J.; Lehikoinen, P.; Solin, O.; Johannsen, B. Nucl. Med. Biol. 2001, 28, 857.
Zhang, M. R.; Maeda, J.; Furutsuka, K.; Yoshida, Y.; Ogawa, M.; Suhara, T.; Suzuki, K. Bioorg. Med. Chem. Lett. 2003, 13, 201.
Zhang, M. R.; Ogawa, M.; Furutsuka, K.; Yoshida, Y.; Suzuki, K. J. Fluorine Chem. 2004, 125, 1879.
Zhang, W.; Zhu, L.; Hu, J. Tetrahedron 2007, 63, 10569.
Doi, H.; Ban, I.; Nonoyama, A.; Sumi, K.; Kuang, C.; Hosoya, T.; Tsukada, H.; Suzuki, M. Chem.-Eur. J. 2009, 15, 4165.
Hu, B.; Gao, L.; Li, C.; Hu, J. Org. Lett. 2015, 17, 3086.
An, L.; Xiao, Y.-L.; Min, Q.-Q.; Zhang, X. Angew. Chem., Int. Ed. 2015, 54, 9079.
Yin, H.; Sheng, J.; Zhang, K.-F.; Zhang, Z.-Q.; Bian, K.-J.; Wang, X.-S. Chem. Commun. 2019, 55, 7635.
(a) Doi, H.; Ban, I.; Nonoyama, A; Sumi, K.; Kuang, C.; Hosoya, T.; Tsukada, H.; Suzuki, M. J. Chem.-Eur. 2009, 15, 4165.
(b) Hu, J.; Gao, B.; Li, L.; Ni, C. Org. Lett. 2015, 17, 3086.
(c) An, L.; Xiao, Y.-L.; Zhang, X. Angew. Chem., Int. Ed. 2015, 54, 9079.
(a) Collins, B. S. L.; Wilson, C. M.; Myers, E. L.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2017, 56, 11700.
(b) Cuenca, A. B.; Shishido, R.; Ito, H.; Fernández, E. Chem. Soc. Rev. 2017, 46, 415.
(c) Fyfe, J. W. B.; Watson, A. J. B. New Tricks. Chem. 2017, 3, 31.
(d) Hemming, D.; Fritzemeier, R.; Westcott, S. A.; Santos, W. L.; Steel, P. G. Chem. Soc. Rev. 2018, 47, 7477.
Wu, N.-Y.; Xu, X.-H.; Qing, F. L. ACS Catal. 2019, 9, 5726.
Cao, Y.; Jiang, L.; Yi, W. Adv. Synth. Catal. 2019, 361, 4360.
Ding, T.; Jiang, L.; Yang, J.; Xu, Y.; Wang, G.; Yi, W. Org. Lett. 2019, 21, 6025.
Olah, G. A.; Pavlath, A. Acta Chim. Acad. Sci. Hung. 1953, 3, 425.
Prakash, G. K. S.; Ledneczki, I.; Chacko, S.; Olah, G. A. Org. Lett. 2008, 10, 557.
Veliks, J.; Kazia, A. Chem.-Eur. J. 2019, 25, 3786.
Meanwell, N. A. J. Med. Chem. 2018, 61, 5822.
Melngaile, R.; Sperga, A.; Baldridge, K. K.; Veliks, J. Org. Lett. 2019, 21, 7174.
For reviews, see: (a) Okamura, H.; Bolm, C. Chem. Lett. 2004, 33, 482.
(b) Johnson, C. R. Acc. Chem. Res. 1973, 6, 341.
For selected examples, see: (a) Shen, X.; Zhang, W.; Ni, C.; Gu, Y.; Hu, J. J. Am. Chem. Soc. 2012, 134, 16999.
(b) Shen, X.; Zhang, W.; Zhang, L.; Luo, T.; Wan, X.; Gu, Y.; Hu, J. Angew. Chem., Int. Ed. 2012, 51, 6966.
(c) Zhang, W.; Huang, W.; Hu, J. Angew. Chem., Int. Ed. 2009, 48, 9858.
(d) Zhang, W.; Wang, F.; Hu, J. Org. Lett. 2009, 11, 2109.
Nomura, Y.; Tokunaga, E.; Shibata, N. Angew. Chem., Int. Ed. 2011, 50, 1885.
Yang, Y.-D.; Lu, X.; Liu, G.; Tokunaga, E.; Tsuzuki, S.; Shibata, N. ChemistryOpen 2012, 1, 221.
Shen, X.; Zhou, M.; Ni, C.; Zhang, W.; Hu, J. Chem. Sci. 2014, 5, 117.
Noto, N.; Koike, T.; Akita, M. ACS Catal. 2019, 9, 4382.
Liu, Y.; Lu, L.; Shen, Q. Angew. Chem., Int. Ed. 2017, 56, 9930.
Prakash, G. K. S.; Zhao, X.; Chacko, S.; Wang, F.; Vaghoo, H.; Olah, G. A. J. Fluorine Chem. 2008, 129, 1036.
Sun, X.; Yu, S. Org. Lett. 2014, 16, 2938.
Su, Y.-M.; Feng, G.-S.; Wang, Z.-Y.; L, Q.; Wang, X.-S. Angew. Chem., Int. Ed. 2015, 54, 6003.
Ruan, Z.; Zhang, S.-K.; Zhu, C.; Ruth, P. N.; Stalke, D.; Ackermann, L. Angew. Chem., Int. Ed. 2017, 56, 2045.
Tang, W.-K.; Feng, Y.-S.; Xu, Z.-W.; Cheng, Z.-F.; Xu, J.; Dai, J.-J.; Xu, H.-J. Org. Lett. 2017, 19, 5501.
Huang, H.; Jia, K.; Chen, Y. Angew. Chem., Int. Ed. 2015, 54, 1881.
(a) Jadhav, S. D.; Bakshi, D.; Singh, A. J. Org. Chem. 2015, 80, 10187.
(b) Majek, M.; Jacobi von Wangelin, A. Acc. Chem. Res. 2016, 49, 2316.
(c) Meyer, A. U.; Slanina, T.; Yao, C.-J.; König, B. ACS Catal. 2016, 6, 369.
(d) Neumann, M.; Fuldner, S.; Konig, B.; Zeitler, K. Angew. Chem., Int. Ed. 2011, 50, 951.
(e) Yu, C.; Iqbal, N.; Park, S.; Cho, E. J. Chem. Commun. 2014, 50, 12884.
Tang, W.-K.; Xu, Z.-W.; Xu, J.; Tang, F.; Li, X.-X.; Dai, J.-J.; Xu, H.-J.; Feng, Y.-S. Org. Lett. 2019, 21, 196.
(a) Weiss, M. E.; Kreis, L. M.; Lauber, A.; Carreira, E. M. Angew. Chem. 2011, 123, 11321.
(b) Kreis, L. M.; Krautwald, S.; Pfeiffer, N.; Martin, R. E.; Carreira, E. M. Org. Lett. 2013, 15, 1634.
(a) Koide, T.; Aritome, I.; Saeki, T.; Morita, Y.; Shiota, Y.; Yoshizawa, K.; Shimakoshi, H.; Hisaeda, Y. ACS Omega 2018, 3, 4027.
(b) Tada, M.; Kaneko, K. J. Org. Chem. 1995, 60, 6635.
(a) Gupta, B. D.; Roy, S. Inorg. Chim. Acta 1988, 146, 209.
(b) Giese, B.; Hartung, J.; He, J.; Huter, O.; Koch, A. Angew. Chem. 1989, 101, 334.
Okabe, M.; Abe, M.; Tada, M. J. Org. Chem. 1982, 47, 1775.
(a) Xia, X.-D.; Ren, Y.-L.; Chen, J.-R.; Yu, X.-L.; Lu, L.-Q.; Zou, Y.-Q.; Wan, J.; Xiao, W.-J. Chem.-Asian. J. 2015, 10, 124.
(b) Hu, X.-Q.; Chen, J.-R.; Wei, Q.; Liu, F.-L.; Deng, Q.-H.; Beauche-min, A. M.; Xiao, W.-L. Angew. Chem., Int. Ed. 2014, 53, 12163.
(c)Ding, W.; Zhou, Q.-Q.; Xuan, J.; Li, T.-R.; Lu, L.-Q.; Xiao, W.-J. Tetrahedron Lett. 2014, 55, 4648.
Some examples: (a) Wang, L.; Wei, X.; Jia, W.; Zhong, J.; Wu, L.-Z.; Liu, Q. Org. Lett. 2014, 16, 5842.
(b) Jung, J.; Kim, E.; You, Y.; Cho, E. J. Adv. Synth. Catal. 2014, 356, 2741.
(c) Su, Y. M.; Hou, Y.; Yin, F.; Xu, Y. M.; Li, Y.; Zheng, X.; Wang, X. S. Org. Lett. 2014, 16, 2958.
Li, W.; Zhu, Y.; Duan, Y.; Zhang, M.; Zhu, C. Adv. Synth. Catal. 2015, 357, 1277.
Zhao, Q.; Lu, L.; Shen, Q. Angew. Chem., Int. Ed. 2017, 56, 11575.
Liu, F.; Jiang, L.; Qiu, H.; Yi, W. Org. Lett. 2018, 20, 6270.

图 1 含有单氟甲基结构的生物活性分子和药物
Figure 1 Bioactive molecules and drugs containing monofluoromethyl structure

图式 1 PhSO2CH2F与(R)-(叔丁基亚砜)亚胺的单氟甲基化反应
Scheme 1 Monofluoromethylation of PhSO2CH2F with (R)- (tert-butane-sulfinyl)benzaldimine

图式 3 PhSO2CH2F与N-叔丁基磺酰亚胺的单氟甲基化反应
Scheme 3 Monofluoromethylation of PhSO2CH2F with N-tert- butyl sulfonamide

图式 4 PhSO2CH2F与(R)-N-叔丁基亚砜基酮亚胺的单氟甲基化反应
Scheme 4 Monofluoromethylation of PhSO2CH2F with (R)-N- tert-butyl sulfoxide ketamine

图式 5 PhSO2CH2F与酯或亚砜的单氟甲基化反应
Scheme 5 Monofluoromethylation of PhSO2CH2F with ester or sulfoxide

图式 6 (PhSO2)2CHF与环氧化物或氮杂环丙烷的单氟甲基化反应
Scheme 6 Monofluoromethylation of (PhSO2)2CHF with epoxide or azacyclopropane

图式 9 (PhSO2)2CHF与烷基、芳基卤化物的单氟甲基化反应
Scheme 9 Monofluoromethylation of (PhSO2)2CHF with alkyl and aryl halides

图式 10 (PhSO2)2CFH与炔烃的单氟甲基化反应
Scheme 10 Monofluoromethylation of (PhSO2)2CFH with alkynes

图式 12 吲哚衍生物与FBSM的单氟甲基化反应
Scheme 12 Monofluoromethylation of FBSM with indole derivatives

图式 13 TBTSO2CH2F与醛和酮的单氟甲基化反应
Scheme 13 Monofluoromethylation of TBTSO2CH2F with aldehydes and ketones

图式 15 芳基碘化物与氟碘甲基2-吡啶砜的单氟甲基化反应
Scheme 15 Monofluoromethylation of aryl iodides with fluoro- iodomethyl 2-pyridyl sulfone

图式 16 芳基碘化物与2-PySO2CHFCOR的单氟甲基化反应
Scheme 16 Monofluoromethylation of aryliodide with 2-Py- SO2CHFCOR

图式 17 芳烃与二乙胺基三氟化硫单氟甲基化反应
Scheme 17 Monofluoromethylation of aromatics with diethylamine sulfur trifluoride

图式 19 杂芳烃与Zn(O2SCH2F)2的单氟甲基化反应
Scheme 19 Monofluoromethylation of heteroaromatics with Zn(O2SCH2F)2

图式 20 CH2FSO2Cl与N-芳基丙烯酰胺的单氟甲基化反应
Scheme 20 Monofluoromethylation of N-arylacrylamides with CH2FSO2Cl

图式 21 CH2FSO2Cl与缺电子烯烃的单氟甲基化反应
Scheme 21 Monofluoromethylation of CH2FSO2Cl with electron deficient olefin

图式 22 CH2FSO2Na与共轭N-芳基磺酰胺的单氟甲基化反应
Scheme 22 Monofluoromethylation of CH2FSO2Na with conjugated N-arylsulfonylated amides

图式 23 CH2FSO2Na与炔烃化合物的单氟甲基化反应
Scheme 23 Monofluoromethylation of CH2FSO2Na with alkyne compound

图式 25 氟氯甲烷与亲核物质的亲电单氟甲基化反应
Scheme 25 Electrophilic monofluoromethylation of nucleophiles with chlorofluoromethane

图式 26 氟碘甲烷与萘苯硼酸酯的单氟甲基化反应
Scheme 26 Monofluoromethylation of fluoromethyl iodide with naphthalene phenylborate

图式 27 氟碘甲烷与芳基硼酸酯的单氟甲基化反应
Scheme 27 Monofluoromethylation of fluoroiodomethane and arylborate

图式 28 溴氟甲烷与芳基硼酸的单氟甲基化反应
Scheme 28 Monofluoromethylation of fluoromethyl bromide with arylboric acid

图式 29 溴氟甲烷与溴代芳烃的单氟甲基化反应
Scheme 29 Monofluoromethylation of bromofluoromethane with brominated aromatics

图式 30 氟甲基碘与烯烃的二硼试剂单氟甲基化反应
Scheme 30 Monofluoromethylation reaction of olefins with fluoromethyl iodine and diboron reagent

图式 32 胺或烷基溴化物与ICH2F的单氟甲基化反应
Scheme 32 Monofluoromethylation of amines or alkyl bromides with ICH2F

图式 34 芳基化合物和S-(单氟甲基)二芳基硫四氟硼酸盐的单氟甲基化反应
Scheme 34 Monofluoromethylation of aryl compounds with S- (monofluoromethyl) diaryl thiotetrafluoroborates

图式 35 二芳基氟甲基硫盐与芳基化合物的单氟甲基化反应
Scheme 35 Monofluoromethylation of diarylfomethylsul- fonates with aryl compounds

图式 36 二芳基氟甲基硫盐与乙烯基砜的单氟甲基化反应
Scheme 36 Monofluoromethylation of diarylfluoromethylsul- fonate with vinyl sulfone

图式 37 单氟甲基亚砜盐109与β-酮酯的单氟甲基化反应
Scheme 37 Monofluoromethylation of sulfoxide 109 with β- ketoesters

图式 38 β-酮酯单氟甲基化可能的反应机理
Scheme 38 Proposed reaction mechanism of monofluoromethylation of β-ketoesters

图式 40 PhSO(NTs)CH2F与烯烃的单氟甲基化反应
Scheme 40 Monofluoromethylation of PhSO(NTs)CH2F with alkene

图式 41 单氟甲基硫叶立德试剂126的单氟甲基化
Scheme 41 Monofluoromethylation of monofluoromethyl thiolylide reagent 126

图式 42 氟碘双(苯磺酰基)甲烷与烯烃的单氟甲基化反应
Scheme 42 Monofluoromethylation of fluoroiodobis(phenyl- sulfonyl)methane with alkenes

图式 43 联苯异氰酸酯与溴氟乙酸乙酯的单氟甲基化反应
Scheme 43 Monofluoromethylation of benzidine isocyanate with ethyl bromofluoroacetate

图式 44 芳基硼酸与氟甲基试剂135的单氟甲基化反应
Scheme 44 Monofluoromethylation of arylboric acid with fluoromethyl reagent 135

图式 45 芳基化合物与溴单氟酯的单氟甲基化反应
Scheme 45 Monofluoromethylation of aryl compounds with bromo-monofluoroesters

图式 46 α, β-不饱和羧酸与溴氟乙酸乙酯(碘氟乙酸乙酯)的单氟甲基化反应
Scheme 46 Monofluoromethylation of α, β-unsaturated carboxylic acids with ethyl bromofluoroacetate (ethyl iodofluor-oacetate)

图式 47 溴氟乙酸乙酯与烯烃的单氟甲基化反应
Scheme 47 Monofluoromethylation of ethyl bromofluoroacetate with alkene

图式 48 四氢异喹啉与三氟甲基β-氟代间二醇的单氟甲基化反应
Scheme 48 Monofluoromethylation of tetrahydroisoquinoline with trifluoromethyl β-fluoro-m-diol

图式 49 S-(氟甲基)苯磺酸盐与芳基硼酸或烷基烯烃的单氟甲基化反应
Scheme 49 Monofluoromethylation of S-(fluoromethyl) benzene sulfonate with arylboric acid or alkylalkene

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
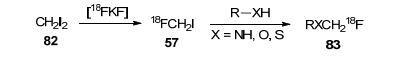
 下载:
下载:










