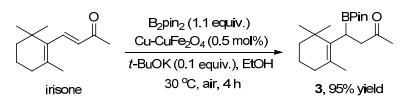
Scheme 1.
1, 4-Reduction of α, β, γ, δ-unsaturated alkenes

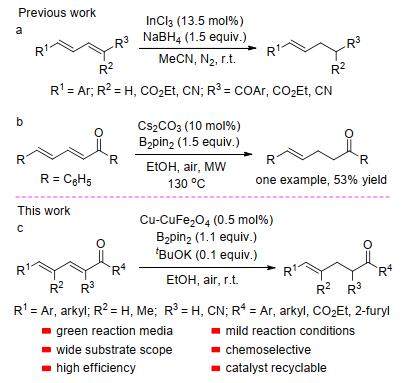
The selective reduction of a functional group in the presence of a number of other reducible functional groups has long been a problem that organic chemists have struggled to resolve.[1] The chemoselective 1, 4-reduction of α, β-unsaturated carbonyl compounds is well-established.[2] In contrast, very limited knowledge is available regarding the selective reduction of highly activated α, β, γ, δ-unsatu- rated carbonyl compounds.[3] This is due to the presence of three reducible positions (e.g., 1, 2-, 1, 4-, and 1, 6-reduc- tion), often resulting in mixtures of multi-reduced products under reductive conditions. To the best of our knowledge, there are only two general reports concerning regarding the 1, 4-reduction of α, β, γ, δ-unsaturated alkenes, previously divulged by Ranu et al.[4] (Scheme 1a) and Song et al.[5] (Scheme 1b). However, these methods suffer from a number of disadvantages, such as low yields, the need for high temperatures, low efficiency and the use of toxic solvents. The γ, δ-unsaturated alkenes are important intermediates and building blocks for the construction of other valuable fine chemicals, [6] and therefore, the development of a highly efficient catalyst for the environmentally benign reduction of α, β, γ, δ-unsaturated alkenes into γ, δ-unsaturated alkenes is thus highly desirable.
In recent, there is an explosive growth in the use of nanoparticles as highly reactive catalysts, [7] with the size and shape of various metal nanoparticles giving rise to their improved catalytic performance over traditionally employed catalysts.[8] Incorporation of metal nanoparticles in magnetic materials has recently gained attraction because a combination of magnetic materials and metal nanoparticles provides synergistically useful performance and stability.[9] In addition, they possess the added benefit of being separable by means of an external magnet after completion of the reaction. Au, [10] Cu, [11] Pt, [12] Pd, [13] Ru[14] and Rh[15] nanoparticles have been incorporated into a magnetic materials for use as catalysts in several transformations. In particular, iron-oxide nanoparticles (NPs) such as MFe2O4 (M=Mn, Fe, Co, Ni, Cu), Fe3O4 and Fe2O3, have been widely used as solid magnetic supports.[16] As a part of our continuous efforts in the field of nano-catalysis, the catalytic activity of Cu(0) incorporated into CuFe2O4 NPs for the hydroboration of alkynes and protodeboronation of ynones have been demonstrated.[17] Herein, we reveal the activity of the same Cu-CuFe2O4 NPs catalyst toward the 1, 4-reduction of α, β, γ, δ-unsaturated carbonyls with bis(pinacolato)diboron (B2pin2), employing an environmentally benign and inexpensive alcohol as a hydrogen donor and solvent (Scheme 1c).)
For our initial studies, reactions of commercially available B2pin2 and micro Cu powder with α, β, γ, δ-unsaturated ketone 1a in the presence of sodium methoxide, resulted in the formation of product 2a in 12% isolated yield (Table 1, Entry 1). Encouraged by this result, this reaction using our reported Cu-CuFe2O4 NPs in place of micro Cu powder was then tested. Gratifyingly, 2a could be obtained in 87% yield using this catalyst (Table 1, Entry 2). The use of CuFe2O4 NPs significantly reduced the yield of product 2a and no reaction occurred in the absence of catalyst (Table 1, Entries 3 and 4). From these results, it can be concluded that the Cu(0) NPs incorporated into CuFe2O4 provide improved catalytic performance. When t-BuOK was used in place of sodium methoxide, the desired product 2a was obtained in 94% yield (Table 1, Entry 5). Upon further optimization concerning the equivalents of base, catalyst loading, solvent and reaction time (Table 1, Entries 6~10), the final optimized reaction conditions were able to afford the desired product in 95% yield in only 4 h (Table 1, Entry 7).
 下载:
导出CSV
下载:
导出CSV
 |
|||||
| Entry | Catalyst | Solvent | Base | t/h | Yield b /% |
| 1 | Micro Cu powder | EtOH | MeONa | 6 | 12 |
| 2 | Nano Cu-CuFe2O4 | EtOH | MeONa | 6 | 87 |
| 3 | Nano CuFe2O4 | EtOH | MeONa | 6 | 28 |
| 4 | — | EtOH | MeONa | 6 | 0 |
| 5 | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 6 | 94 |
| 6 | Nano Cu-CuFe2O4 | MeOH | t-BuOK | 6 | 92 |
| 7 | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 4 | 95 |
| 8 | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 2 | 86 |
| 9 c | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 4 | 70 |
| 10 d | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 4 | 79 |
| a Unless otherwise noted, the reactions were carried out with 1a (0.2 mmol), B2pin2 (0.21 mmol), catalyst (0.5 mol%), base (0.1 equiv.), solvent (2 mL), 30 ℃, under air. b Isolated yield. c 5 mol% of base. d 0.25 mol% of catalyst. | |||||
Next, the substrate scope of α, β, γ, δ-unsaturated compounds was investigated. Firstly, the different aryl group-substituted α, β, γ, δ-unsaturated ketones were surveyed. As shown in Table 2, good to excellent E/Z selectivity and yield were maintained across a range of ketones with varying electronic demand at the carbonyl position (2a~2n). Particular attentions should be paid to compounds containing alkyl group substituted at the δ-position (2m and 2n), since in these cases, the corresponding products were obtained with either moderate E/Z selectivity (E/Z=10:1) or as the β-boration addition adduct. The amenability of sterically demanding α, β, γ, δ-unsaturated ketones to the catalytic conditions was also examined. Fortunately, the desired products bearing amethyl or methylene group substituted at the γ-position (2o~2r) and α-position (2s~2u) were obtained with good to excellent regioselectivity. The α, β, γ, δ-unsaturated carboxylic ester (2s) also reacted highly regioselectively. However, in this case, both 1, 4-reduction and 1, 6-reduction occured. Additionally, α, β, γ, δ-unsaturated cyano-ester (2t) was also examined and interestingly, only the 1, 4-reduced product was obtained in 75% yield and with a high E/Z selectivity of > 25:1.
下载:
导出CSV
 |
 |
| a Reaction conditions: 2 (0.2 mmol), B2(Pin)2 (0.22 mmol), EtOH (2 mL), KOtBu (0.02 mmol), and Cu-CuFe2O4 (0.6 mg, 0.5 mol%) at 30 ℃ under air for 4 h. The values in parentheses are the E/Z ratios detected by 1H NMR spectroscopy. b Isolated yield. c 60 ℃. |
Based on the β-boration addition of (E)-6-methylhepta- 3, 5-dien-2-one (1n), the utility of this methodology was further explored by applying our catalytic conditions to the reduction of natural products (Scheme 2). To our delight, irisone was successfully reduced to give their corresponding derivatives in 95% yield.
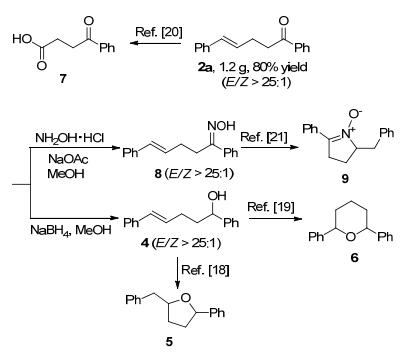
To further illustrate the synthetic utility of our reaction, we carried out the reduction of α, β, γ, δ-unsaturated carbonyl products 1a on a gram scale. The corresponding γ, δ-un-saturated ketone 2a was obtained in 80% isolated yield (Scheme 3). The γ, δ-unsaturated ketone was then successfully transformed into three structurally distinct synthons: (1) selective reduction of the ketone group under the reducing conditions gave the unsaturated alcohol 4 which was widely used for the synthesis of cyclic ethers 5[18] and 6[19] via intramolecular hydroalkoxylation; (2) selective oxidation of the carbon-carbon double bond under the oxidizing conditions gave the γ-keto acids 7[20] which could be further elaborated to other compounds by an amidation reaction; (3) condensation of the ketone with hydroxylamine hydrochloride afforded the oxime 8 which could be used to prepare cyclic nitrone 9.[21] It should be noted that nearly all of the cinnamaldehydes and ketones employed in the synthesis of α, β, γ, δ-unsaturated carbonyl compounds (Table 2) are commercially available, further demonstrating the advantages of directly employing α, β, γ, δ-unsaturated carbonyl compounds in this 1, 4-reduction reaction.
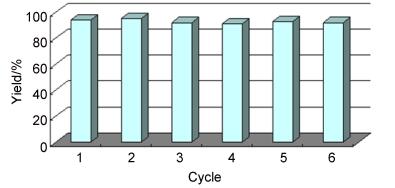
In our experiment, the magnetic CuFe2O4 composite with stabilized copper nanoparticles could be readily removed from the reaction mixture and recovered by simple filtration. The Cu-CuFe2O4 NPs may act as a heterogeneous catalyst in this reaction. To prove this hypothesis, the recyclability of Cu-CuFe2O4 NPs was tested in the reaction of 1a with B2Pin2. Remarkably, the catalyst maintained excellent activity, even after being used in six cycles, as shown in Figure 1. In each cycle, the recovered copper nanoparticles were washed with ethyl acetate and ethanol, then dried and reused for subsequent reactions without further activation. The transmission electron microscopy (TEM) and X-ray diffraction (XRD) images of the fresh and sixth recycled catalysts indicated that little morphological change had occurred. In addition, the color of the final reaction solution did not change, which indicated that the catalyst had not been oxidized.[22] Furthermore, inductively coupled plasma mass spectrometry (ICP-MS) analysis showed that the amount of Cu(0) lost to leaching was 0.021% after one catalytic circle. These results indicated that the Cu-CuFe2O4 NPs acted as a highly active heterogeneous catalyst for the 1, 4-reduction of α, β, γ, δ-unsaturated carbonyl compounds. The methodology possesses several advantages, such as easy recovery of the catalyst, being operational simple and the fact that the catalyst can be recycled multiple times without losing activity.
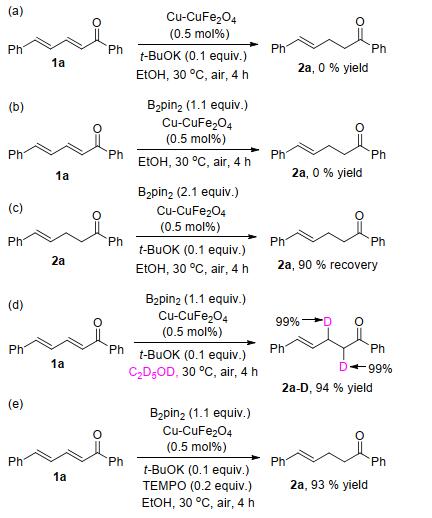
To investigate the key factors affecting the whole protodeboronation process, several control experiments were also conducted. No reaction occurred when B2pin2 and base were removed from the reaction system, respectively (Schemes 4a and 4b). Notably, the protodeboronation product 2a was still obtained in 90% yield when the amount of B2pin2 was increased to 2.1 equiv. (Scheme 4c). This proves that the system exhibits high chemical selectivity. A deuterium labeling experiment was carried out in ethanol-d6 solvent under the standard conditions (Scheme 4d), and the product was isolated and analyzed by NMR spectroscopy. The result revealed that deuterium was incorporated at the α-position and β-position of the reduced product with 99% ratio, clearly demonstrating that the solvent serves as the hydrogen source for this protodeboronation system. The reaction was not inhibited by adding 0.2 equiv. 2, 2, 6, 6- tetramethylpiperidine 1-oxyl (TEMPO) (4e).
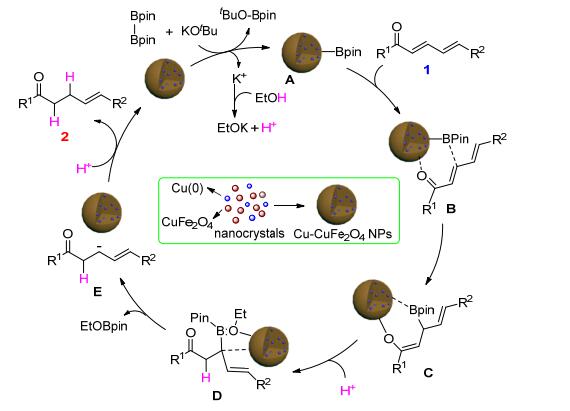
According to previous literature reports[23] and our experimental results, a plausible mechanism is described in Scheme 5. The catalytic cycle is initiated by reaction of Cu-CuFe2O4 with B2Pin2 in the presence of KOtBu, forming key intermediate A, K+ can react with EtOH to generate H+ and EtOK. Intermediate A, through adsorption of the α, β, γ, δ-unsaturated compounds, gives intermediate B. Subsequent boron addition occurs via a six-membered cyclic transition states to afford C, [23c] which is protonated by H+ to give β-boration addition species D.[5] Then the boron group of D is removed leading to the formation of the negatively charged species E.[24] Finally, E is further protonated by the remaining H+ to give the reduced product 2 together with the nano Cu-CuFe2O4 catalyst.
In summary, an efficient methodology for the Cu- CuFe2O4 NPs catalyzed the boron 1, 4-reduction of α, β, γ, δ- unsaturated carbonyl compounds in an alcohol medium under mild condition has been developed. This methodol-ogy tolerates a broad substrate scope and gives the desired products in good yields (up to 96% yield) and with high chemoselectivities (E/Z up to > 25:1). This new synthetic method is operationally simple and scalable, and the obtained γ, δ-unsaturated ketone products can be readily derivatized. Remarkably, the Cu-CuFe2O4 NPs could be easily recovered and reused at least six times with only a slight decrease in catalytic activity. Furthermore, the mechanistic studies concerning catalyst activation and intermediate formation have been discussed.
The chemicals used in this study were purchased from Energy Chemicals Co. Ltd. (Shanghai, P. R. China). The solvents used in this study were supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, P. R. China). All chemicals and solvents were used directly without any purification. Preparation of the catalyst was according to our previous report.[17]
CH3CH2OH (2 mL), α, β, γ, δ-unsaturated carbonyls (0.2 mmol), B2Pin2 (56 mg, 0.22 mmol), potassium t-butoxide (2.3 mg, 0.02 mmol) and Cu-CuFe2O4 nanoparticles (0.6 mg, 0.5 mol%) were added to a Schlenk tube under air atmosphere. The mixture was then stirred at 30 ℃ for 4 h. Next, H2O (2 mL) was added into the mixture. Then the mixture was extracted with CH2Cl2 (3 mL, three times). The combined organic phase was dried over anhydrous MgSO4 and concentrated, and the residue was purified by column chromatography using petroleum ether/ethyl acetate (V/V=30/1) as an eluent. Evaporation of the volatiles under vacuum resulted in the corresponding product.
(2E, 4E)-1, 5-Diphenylpenta-2, 4-dien-1-one (1a, 20 mmol), B2Pin2 (22 mmol), Cu-CuFe2O4 nanoparticles (12 mg, 0.5 mol%), t-BuOK (10 mol%), ethanol (30 mL), under air at room temperature for 12 h. In each run, after reaction, the catalyst was separated by filtration, washed thoroughly with ethyl acetate, water, ethanol and dried by vacuum. Then, the dried catalyst was used further, without any purification or reactivation. The filtrate was evaporated under vacuum, and the residue was purified by column chromatography.
(E)-1, 5-Diphenylpent-4-en-1-one (2a):[25] Light yellow oil (44.4 mg, 94% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.97~7.99 (m, 2H), 7.56~7.57 (m, 1H), 7.45~7.49 (m, 2H), 7.20~7.36 (m, 5H), 6.47 (d, J=16.0 Hz, 1H), 6.26~6.34 (m, 1H), 3.14~3.18 (m, 2H), 2.64~2.69 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 199.3, 137.4, 136.9, 133.1, 130.8, 129.1, 128.61, 128.5, 128.0, 127.1, 126.0, 38.3, 27.5.
(E)-5-Phenyl-1-(p-tolyl)pent-4-en-1-one (2b):[26] Light yellow oil (45.0 mg, 90% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.87~7.89 (m, 2H), 7.27~7.35 (m, 6H), 7.18~7.21 (m, 1H), 6.57 (d, J=15.6 Hz, 1H), 6.26~6.33 (m, 1H), 3.11~3.15 (m, 2H), 2.62~2.67 (m, 2H), 2.41 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 199.0, 143.8, 137.5, 134.4, 130.7, 129.3, 129.3, 128.5, 128.2, 127.0, 126.0, 38.1, 27.6, 21.7.
(E)-5-Phenyl-1-(m-tolyl)pent-4-en-1-one (2c): Light yellow oil (42.6 mg, 85% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.87~7.89 (m, 2H), 7.27~7.35 (m, 6H), 7.18~7.21 (m, 1H), 6.57 (d, J=15.6 Hz, 1H), 6.26~6.33 (m, 1H), 3.11~3.15 (m, 2H), 2.62~2.67 (m, 2H), 2.41 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 199.6, 138.4, 137.5, 136.9, 133.8, 130.7, 129.2, 128.6, 128.5, 127.0, 126.0, 125.3, 38.3, 27.5, 21.4. HRMS (ESI) calcd for C18H18ONa [M+Na]+: 273.1250, found 273.1247.
(E)-5-Phenyl-1-(o-tolyl)pent-4-en-1-one (2d): Light yellow oil (45.6 mg, 91% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.64~7.66 (m, 1H), 7.24~7.32 (m, 8H), 6.44 (d, J=16.0 Hz, 1H), 6.23~6.31 (m, 1H), 3.06~3.10 (m, 2H), 2.60~2.66 (m, 2H), 2.50 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 203.6, 138.0, 137.9, 137.4, 132.0, 131.3, 130.8, 129.0, 128.5, 128.4, 127.1, 126.0, 125.7, 41.1, 27.7, 21.3. HRMS (ESI) calcd for C18H18ONa [M+Na]+: 273.1250, found 273.1252.
(E)-1-(4-Ethylphenyl)-5-phenylpent-4-en-1-one (2e): Li- ght yellow oil (50.2 mg, 95% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.90~7.92 (m, 2H), 7.27~7.35 (m, 6H), 7.19~7.21 (m, 1H), 6.46 (d, J=16.0 Hz, 1H), 6.26~6.33 (m, 1H), 3.12~3.15 (m, 2H), 2.62~2.74 (m, 4H), 1.26 (t, J=7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 199.0, 150.0, 137.5, 134.6, 130.7, 129.3, 128.5, 128.3, 128.1, 127.0, 126.0, 38.2, 29.0, 27.6, 15.3. HRMS (ESI) calcd for C19H20ONa [M+Na]+: 287.1406, found 287.1405.
(E)-1-(4-Methoxyphenyl)-5-phenylpent-4-en-1-one (2f):[25] Light yellow oil (50.5 mg, 95% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.95~7.98 (m, 2H), 7.27~7.35 (m, 4H), 6.64 (d, J=9.2 Hz, 2H), 6.46 (d, J=16.0 Hz, 1H), 6.26~6.33 (m, 1H), 3.87 (s, 3H), 3.08~3.12 (m, 2H), 2.62~2.67 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 198.0, 163.4, 137.5, 130.6, 130.3, 130.0, 129.3, 128.5, 127.0, 126.0, 113.7, 55.5, 37.9, 27.7.
(E)-1-(4-Chlorophenyl)-5-phenylpent-4-en-1-one (2g):[25] Light yellow solid (49.7 mg, 92% yield, E:Z > 25:1). m.p. 51.0~52.1 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.91~7.93 (m, 2H), 7.43~7.47 (m, 2H), 7.18~7.35 (m, 4H), 7.02~7.17 (m, 1H), 6.46 (d, J=16.0 Hz, 1H), 6.24~6.31 (m, 1H), 3.11~3.14 (m, 2H), 2.62~2.67 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 198.1, 145.3, 142.4, 139.5, 137.3, 135.1, 130.9, 129.8, 129.5, 128.9, 128.9, 128.8, 128.5, 127.3, 127.1, 126.0, 38.3, 27.4.
(E)-1-(4-Fluorophenyl)-5-phenylpent-4-en-1-one (2h): Light yellow oil (46.7 mg, 92% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.99~8.02 (m, 2H), 7.27~7.35 (m, 4H), 7.11~7.16 (m, 3H), 6.46 (d, J=16.0 Hz, 1H), 6.24~6.32 (m, 1H), 3.13 (t, J=7.2 Hz, 2H), 2.65 (q, J=7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 197.7, 167.0, 164.4, 137.6, 133.3, 130.9, 130.7, 130.6, 128.9, 128.5, 127.1, 126.0, 115.8, 115.6, 38.2, 27.5. HRMS (ESI) calcd for C17H15OFNa [M+Na]+: 277.0999, found 277.0997.
(E)-1-(Naphthalen-2-yl)-5-phenylpent-4-en-1-one (2i):[25] White solid (53.8 mg, 94% yield, E:Z > 25:1). m.p. 62.0~63.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.50 (s, 1H), 8.05~8.05 (m, 1H), 7.96~7.98 (m, 2H), 7.87~7.92 (m, 2H), 7.56~7.61 (m, 2H), 7.25~7.37 (m, 5H), 6.50 (d, J=16.0 Hz, 1H), 6.31~6.38 (m, 1H), 3.28~3.32 (m, 2H), 2.70~2.75 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 199.3, 137.4, 135.6, 134.2, 132.5, 130.8, 129.7, 129.6, 129.2, 128.5, 128.5, 128.5, 127.8, 127.1, 126.8, 126.0, 123.9, 38.4, 27.7.
(E)-1-(4-Chlorophenyl)-4-methyl-5-phenylpent-4-en-1- one (2j): White solid (53.1 mg, 85% yield, E:Z > 25:1). m.p. 57.5~58.3 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.05~8.07 (m, 2H), 7.69~7.71 (m, 2H), 7.62~7.64 (m, 2H), 7.28~7.50 (m, 8H), 6.49 (d, J=15.6 Hz, 1H), 6.28~6.36 (m, 1H), 3.17~3.21 (m, 2H), 2.66~2.72 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 198.9, 145.7, 139.9, 137.4, 135.6, 130.8, 129.1, 128.9, 128.6, 128.5, 128.2, 127.3, 127.1, 126.0, 38.3, 27.6. HRMS (ESI) calcd for C23H20ONa [M+Na]+: 335.1406, found 335.1405.
(E)-1-(Furan-2-yl)-5-phenylpent-4-en-1-one (2k): Light yellow oil (43.4 mg, 96% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.58~7.59 (m, 1H), 7.28~7.32 (m, 4H), 7.20~7.21 (m, 2H), 6.53~6.54 (m, 1H), 6.45 (d, J=15.6 Hz, 1H), 6.22~6.30 (m, 1H), 3.01 (t, J=7.6 Hz, 2H), 2.61~2.66 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 188.6, 152.7, 146.3, 137.4, 130.9, 128.8, 128.5, 127.1, 126.0, 117.1, 112.2, 38.1, 27.4. HRMS (ESI) calcd for C15H14O2Na [M+Na]+: 249.0886, found 249.0884.
(E)-6-Phenylhex-5-en-2-one (2l): Light yellow oil (33.5 mg, 96% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.26~7.34 (m, 4H), 7.18~7.22 (m, 1H), 6.40 (d, J=15.6 Hz, 1H), 6.15~6.23 (m, 1H), 2.60~2.63 (m, 2H), 2.45~2.51 (m, 2H), 2.17 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.1, 137.3, 130.7, 128.8, 128.5, 127.1, 126.0, 43.2, 30.1, 27.1. HRMS (ESI) calcd for C12H14ONa [M+Na]+: 197.0937, found 197.0935.
(E)-1-Phenylhept-4-en-1-one (2m): Light yellow oil (34.6 mg, 92% yield, E:Z=10:1). 1H NMR (400 MHz, CDCl3) δ: 7.95~7.97 (m, 2H), 7.53~7.57 (m, 1H), 7.45~7.47 (m, 2H), 5.37~5.55 (m, 2H), 3.01~3.05 (m, 2H), 2.40~2.48 (m, 2H), 1.98~2.09 (m, 2H), 0.95 (t, J=7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 199.9, 137.0, 133.1, 132.9, 128.5, 128.0, 127.5, 38.6, 27.2, 25.5, 13.8. HRMS (ESI) calcd for C13H16ONa [M+Na]+: 211.1093, found 211.1089.
6-Methyl-4-(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolan-2-yl)hept-5-en-2-one (2n): Colorless oil (37.9 mg, 95% yield). 1H NMR (400 MHz, CDCl3) δ: 4.98 (d, J=13.6 Hz, 2H), 2.60~2.63 (m, 2H), 2.17~2.19 (m, 1H), 2.10 (s, 3H), 1.67 (s, 3H), 1.62 (s, 3H), 1.22 (s, 6H), 1.19 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 208.9, 132.0, 123.0, 83.0, 46.2, 29.5, 25.8, 24.6, 24.5, 18.0. HRMS (ESI) calcd for C14H25O3BNa [M+Na]+: 275.1789, found 275.1786.
(E)-4-Methyl-1, 5-diphenylpent-4-en-1-one (2o): Light yellow oil (46.0 mg, 91% yield, E:Z=10:1). 1H NMR (400 MHz, CDCl3) δ: 7.99~8.01 (m, 2H), 7.56~7.58 (m, 1H), 7.46~7.50 (m, 2H), 7.30~7.34 (m, 2H), 7.19~7.24 (m, 3H), 6.33 (s, 1H), 3.19~3.23 (m, 2H), 2.60~2.64 (m, 2H), 1.92 (d, J=0.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 199.8, 138.2, 137.6, 137.0, 133.1, 128.8, 128.6, 128.1, 128.0, 126.0, 125.5, 37.3, 34.9, 18.0. HRMS (ESI) calcd for C18H18ONa [M+Na]+: 273.1250, found 273.1254.
(E)-4-Methyl-5-phenyl-1-(p-tolyl)pent-4-en-1-one (2p): Light yellow oil (48.6 mg, 92% yield, E:Z=10:1). 1H NMR (400 MHz, CDCl3) δ: 7.89~7.91 (m, 2H), 7.19~7.33 (m, 7H), 6.33 (s, 1H), 3.15~3.19 (m, 2H), 2.55~2.63 (m, 2H), 2.42 (s, 3H), 1.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 199.5, 143.8, 138.2, 137.8, 134.4, 129.3, 128.81, 128.2, 128.0, 126.0, 125.4, 37.2, 35.0, 21.7, 18.0. HRMS (ESI) calcd for C19H20ONa [M+Na]+: 287.1406, found 287.1405.
(E)-1-(4-Chlorophenyl)-4-methyl-5-phenylpent-4-en-1- one (2q): Light yellow oil (51.2 mg, 90% yield, E:Z=7:1). 1H NMR (400 MHz, CDCl3) δ: 7.92~7.94 (m, 2H), 7.44~7.46 (m, 2H), 7.30~7.34 (m, 2H), 7.20~7.23 (m, 2H), 6.32 (s, 1H), 3.15~3.19 (m, 2H), 2.58~2.62 (m, 2H), 1.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 198.5, 139.5, 138.1, 137.4, 135.2, 129.5, 128.9, 128.9, 128.8, 128.5, 128.3, 128.1, 126.1, 125.6, 37.3, 34.8, 18.0. HRMS (ESI) calcd for C18H17OClNa [M+Na]+: 307.0860, found 307.0859.
(E)-1-(4-Fluorophenyl)-4-methyl-5-phenylpent-4-en-1- one (2r): Light yellow oil (49.3 mg, 92% yield, E:Z=9:1). 1H NMR (400 MHz, CDCl3) δ: 8.00~8.04 (m, 2H), 7.30~7.33 (m, 2H), 7.12~7.23 (m, 5H), 6.32 (s, 1H), 3.15~3.19 (m, 2H), 2.59~2.62 (m, 2H), 1.91 (d, J=0.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 198.2, 167.0, 164.4, 138.1, 137.5, 133.3, 130.7, 130.6, 128.8, 128.1, 126.1, 125.6, 115.8, 115.6, 37.3, 34.9, 18.0. HRMS (ESI) calcd for C18H17OFNa [M+Na]+: 291.1156, found 291.1151.
2-Cinnamyl-2, 3-dihydro-1H-inden-1-one (2s): Light yellow oil (45.6 mg, 91% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.76~7.78 (m, 1H), 7.57~7.60 (m, 1H), 7.44~7.46 (m, 1H), 7.20~7.39 (m, 6H), 6.48 (d, J=15.6 Hz, 1H), 6.17~6.24 (m, 1H), 3.28~3.35 (m, 1H), 2.72~2.92 (m, 3H), 2.39~2.47 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 208.1, 153.7, 137.2, 136.6, 134.8, 132.2, 128.5, 127.4, 127.2, 126.6, 126.1, 123.9, 47.1, 34.8, 32.1. HRMS (ESI) calcd for C18H16ONa [M+Na]+: 271.1093, found 271.1089.
(E)-2-(2-Methyl-3-phenylallyl)-2, 3-dihydro-1H-inden-1-one (2t): Light yellow oil (45.1 mg, 86% yield, E:Z=10:1). 1H NMR (400 MHz, CDCl3) δ: 7.77~7.79 (m, 1H), 7.58~7.60 (m, 1H), 7.45~7.47 (m, 1H), 7.31~7.40 (m, 3H), 7.21~7.26 (m, 3H), 6.34 (s, 1H), 3.26~3.30 (m, 1H), 2.91~2.96 (m, 3H), 2.19~2.23 (m, 1H), 1.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.4, 153.7, 138.0, 136.6, 134.8, 128.9, 128.1, 127.4, 126.9, 126.3, 126.2, 124.0, 45.8, 42.5, 32.2, 17.6. HRMS (ESI) calcd for C19H18ONa [M+Na]+: 285.1250, found 285.1247.
(13S)-16-Cinnamyl-3-methoxy-13-methyl-7, 8, 9, 11, 12, 13, 15, 16-octahydro-6H-cyclopenta[a]phenanthren-17(14 H)-one (2u): Light yellow solid (65.6 mg, 82% yield, E: Z=10:1). 1H NMR (400 MHz, CDCl3) δ: 7.49~7.51 (m, 1H), 7.17~7.39 (m, 6H), 6.95 (s, 1H), 6.71~6.73 (m, 1H), 6.64~6.66 (m, 1H), 3.78~3.79 (m, 3H), 2.87~2.93 (m, 2H), 2.76~2.80 (m, 1H), 2.39~2.51 (m, 5H), 1.19~2.05 (m, 3H), 1.39~1.59 (m, 5H), 0.95 (s, 1H), 0.85~0.87 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 208.8, 157.5, 141.1, 140.9, 137.8, 137.7, 135.7, 132.1, 128.8, 128.4, 128.4, 127.1, 126.3, 126.1, 124.6, 113.9, 111.5, 55.2, 48.3, 47.8, 44.0, 37.8, 34.6, 31.7, 31.6, 29.6, 26.7, 26.1, 25.9, 14.3. HRMS (ESI) calcd for C28H32O2Na [M+Na]+: 423.2295, found 423.2293.
(E)-Ethyl 5-phenylpent-4-enoate (2v): Colorless oil (16.3 mg, 40% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.27~7.35 (m, 3H), 7.17~7.22 (m, 2H), 6.43 (d, J=16.0 Hz, 1H), 6.19~6.24 (m, 1H), 4.12~4.41 (m, 2H), 3.38~3.43 (m, 1H), 2.76~2.78 (m, 1H), 2.48~2.55 (m, 2H), 1.24~1.30 (m, 3H); 13C NMR (100 MHz, CDCl3) δ: 172.9, 140.8, 130.9, 128.5, 127.1, 126.1, 126.0, 121.8, 60.2, 34.3, 28.3, 14.3. HRMS (ESI) calcd for C13H16O2Na [M+Na]+: 227.1043, found 227.1041.
(E)-Ethyl 2-cyano-5-phenylpent-4-enoate (2w):[26] Colorless oil (34.4 mg, 75% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.26~7.37 (m, 5H), 6.58 (d, J=16.0 Hz, 1H), 6.14~6.21 (m, 1H), 6.27 (q, J=7.2 Hz, 2H), 3.61~3.64 (m, 1H), 2.83~2.87 (m, 2H), 1.31 (t, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 165.5, 136.2, 135.0, 128.6, 128.0, 126.4, 122.4, 116.1, 45.8, 63.0, 37.9, 33.3, 14.0.
(E)-Ethyl 2-cyano-5-phenylpent-4-enoate (3): Colorless oil (60.8 mg, 95% yield). 1H NMR (400 MHz, CDCl3) δ: 3.02~3.09 (m, 1H), 2.25~2.29 (m, 1H), 2.16~2.22 (m, 1H), 2.10 (s, 3H), 1.83~1.86 (m, 2H), 1.56 (s, 1H), 1.37~1.41 (m, 4H), 1.17 (s, 6H), 1.13 (s, 6H), 1.02 (s, 3H), 0.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.8, 138.0, 127.4, 82.9, 45.9, 39.9, 36.1, 33.4, 30.0, 28.5, 27.6, 24.6, 24.5, 21.4, 19.5. HRMS (ESI) calcd for C19H33BO3Na [M+Na]+: 343.2415, found 343.2417.
(E)-1, 5-Diphenylpent-4-en-1-ol (4):[6] Colorless oil (41.0 mg, 85% yield, E:Z > 25:1). 1H NMR (400 MHz, CDCl3) δ: 7.28~7.38 (m, 8H), 7.13~7.22 (m, 2H), 6.41 (d, J=16.0 Hz, 1H), 6.22~6.27 (m, 1H), 4.73~4.76 (m, 1H), 3.35~3.39 (m, 1H), 2.29~2.31 (m, 2H), 1.90~1.99 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 144.6, 137.6, 133.2, 130.4, 130.0, 128.5, 128.49, 128.5, 127.6, 126.9, 125.9, 125.9, 74.0, 38.5, 29.3.
4-Oxo-4-phenylbutanoic acid (7):[20] White solid (27.0 mg, 75% yield). m.p. 117.0~118.2 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.96~7.98 (m, 2H), 7.55~7.58 (m, 1H), 7.44~7.48 (m, 2H), 3.29~3.32 (m, 2H), 2.79~2.82 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 197.8, 179.0, 136.3, 133.3, 128.6, 128.0, 33.1, 28.1.
(4E)-1, 5-Diphenylpent-4-en-1-one oxime (8):[21] White solid (34.0 mg, 67% yield, E:Z > 25:1). m.p. 115.3~116.6 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.68 (s, 1H), 7.62~7.64 (m, 2H), 7.40~7.40 (m, 3H), 7.19~7.39 (m, 5H), 6.42 (d, J=15.6 Hz, 1H), 6.21~6.28 (m, 1H), 2.98~3.02 (m, 2H), 2.48~2.53 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 159.2, 137.5, 130.5, 129.3, 129.3, 128.6, 128.5, 127.0, 126.4, 126.0, 29.7, 26.1.
Supporting Information Full experimental details, TEM images, XRD spectrums, photographs of before reaction and after reaction of B2Pin2 and 1a, 1H NMR spectrum of deuterium-labeling experiments, and 1H NMR and 13C NMR spectra for all compounds. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Smith, M. B.; March, J. March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Hoboken, NJ, 2007.
(b) Andersson, P. G.; Munslow, I. J. Modern Reduction Methods, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008.
For selected examples, see: (a) Wang, X.; Han, Z.; Wang, Z.; Ding, K. Angew. Chem., Int. Ed. 2012, 51, 936.
(b) Sasson, Y.; Blum, J. J. Org. Chem. 1975, 40, 1887.
(c) Li, W.; Wu, X-F. Eur. J. Org. Chem. 2015, 331.
(d) Ding, B.; Zhang, Z.; Liu, Y.; Sugiya, M.; Imamoto, T.; Zhang, W. Org. Lett. 2013, 15, 3690.
(e) Zhang, B.-H.; Shi, L.-X.; Guo, R.-X. Helv. Chim. Acta 2013, 96, 2152.
(f) Zhou, X.; Li, X.; Zhang, W.; Chen, J. Tetrahedron Lett. 2014, 55, 5137.
Nussim, M.; Mazur, Y.; Sondheimer, F. J. Org. Chem. 1964, 29, 1120. doi: 10.1021/jo01028a032
Ranu, B. C.; Samanta, S. J. Org. Chem. 2003, 68, 7130. doi: 10.1021/jo0347821
Huang, X.; Hu, J.; Wu, M.; Wang, J.; Peng, Y.; Song, G. Green Chem. 2018, 20, 255. doi: 10.1039/C7GC02863F
Zhou, Y.; Rao, C.; Song, Q. Org. Lett., 2016, 18, 4000. doi: 10.1021/acs.orglett.6b01816
(a) An, K.; Somorjai, G. A. Catal. Lett. 2015, 145, 233.
(b) Singh, A. K.; Xu, Q. ChemCatChem 2013, 5, 652.
(c) Sankar, M.; Dimitratos, N.; Miedziak, P. J.; Wells, P. P.; Kiely, C. J.; Hutchings, G. J. Chem. Soc. Rev. 2012, 41, 8099.
Valden, M.; Lai, X.; Goodman, D. W. Science 1998, 281, 1647. doi: 10.1126/science.281.5383.1647
Jin, C.; Wang, Y.; Wei, H.; Tang, H.; Liu, X.; Lu, T.; Wang, J. J. Mater. Chem. A 2014, 2, 11202. doi: 10.1039/c4ta00258j
Yang, W.; Wei, L.; Yi, F.; Cai, M. Catal. Sci. Technol. 2016, 6, 4554. doi: 10.1039/C5CY02159F
Wang, D.; Etienne, L.; Echeverria, M.; Moya, S.; Astruc, D. Chem.-Eur. J. 2014, 20, 4047. doi: 10.1002/chem.201304536
Jang, J.; Byun, S.; Kim, B. M.; Lee, S. Chem. Commun. 2018, 54, 3492. doi: 10.1039/C7CC09926F
Zhang, L.; Li, P.; Liu, C.; Yang, J.; Wang M.; Zhang, L. Catal. Sci. Technol. 2014, 4, 1979. doi: 10.1039/C4CY00040D
Polshettiwar, V.; Varma, R. S. Chem.-Eur. J. 2009, 15, 1582. doi: 10.1002/chem.200802264
Abu-Reziq, R.; Alper, H.; Wang, D.; Post, M. L. J. Am. Chem. Soc. 2006, 128, 5279. doi: 10.1021/ja060140u
Mohan, B.; Park, J. C.; Park, K. H. ChemCatChem 2016, 8, 2345. doi: 10.1002/cctc.201600280
Zeng, X.; Gong, C.; Guo, H.; Xu, H.; Zhang, J.; Xie, J. New J. Chem. 2018, 42, 17346. doi: 10.1039/C8NJ03708F
Perez-Mayoral, E.; Matos, I.; Fonseca, I.; Cejka, J. Chem.-Eur. J. 2010, 16, 12079. doi: 10.1002/chem.201001593
Francesco, I. N.; Cacciuttolo, B.; Pucheault, M.; Antoniotti, S. Green Chem. 2015, 17, 837. doi: 10.1039/C4GC01990C
Ma, Z.; Xie, F.; Yu, H.; Zhang, Y.; Wu, X.; Zhang, W. Chem. Commun. 2013, 49, 5292. doi: 10.1039/c3cc42088d
Peng, X.; Tong, B.; Hirao, H.; Chiba, S. Angew. Chem., Int. Ed. 2014, 53, 1959. doi: 10.1002/anie.201308617
Ding, W.; Song, Q. Org. Chem. Front. 2016, 3, 14. doi: 10.1039/C5QO00289C
Kobayashi, S.; Xu, P.; Endo, T.; Ueno, M.; Kitanosono, T. Angew. Chem., Int. Ed. 2012, 51, 12763. doi: 10.1002/anie.201207343
Zhou, X.-F.; Sun, Y.-Y.; Wu, Y.-D.; Dai, J.-J.; Xu, J.; Huang, Y.; Xu, H.-J. Tetrahedron 2016, 72, 5691. doi: 10.1016/j.tet.2016.07.079
Belhomme, M., Wang, D., Szabó, K. J. Org. Lett. 2016, 18, 2503. doi: 10.1021/acs.orglett.6b01132
Ranu, B. C., Samanta, S. J. Org. Chem. 2003, 68, 7130. doi: 10.1021/jo0347821
Table 1. Screening of reaction conditionsa
|
|||||
| Entry | Catalyst | Solvent | Base | t/h | Yield b /% |
| 1 | Micro Cu powder | EtOH | MeONa | 6 | 12 |
| 2 | Nano Cu-CuFe2O4 | EtOH | MeONa | 6 | 87 |
| 3 | Nano CuFe2O4 | EtOH | MeONa | 6 | 28 |
| 4 | — | EtOH | MeONa | 6 | 0 |
| 5 | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 6 | 94 |
| 6 | Nano Cu-CuFe2O4 | MeOH | t-BuOK | 6 | 92 |
| 7 | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 4 | 95 |
| 8 | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 2 | 86 |
| 9 c | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 4 | 70 |
| 10 d | Nano Cu-CuFe2O4 | EtOH | t-BuOK | 4 | 79 |
| a Unless otherwise noted, the reactions were carried out with 1a (0.2 mmol), B2pin2 (0.21 mmol), catalyst (0.5 mol%), base (0.1 equiv.), solvent (2 mL), 30 ℃, under air. b Isolated yield. c 5 mol% of base. d 0.25 mol% of catalyst. | |||||
 下载: 导出CSV
下载: 导出CSV
Table 2. α, β, γ, δ-Unsaturated carbonyl scopea, b
|
|
|
|
| a Reaction conditions: 2 (0.2 mmol), B2(Pin)2 (0.22 mmol), EtOH (2 mL), KOtBu (0.02 mmol), and Cu-CuFe2O4 (0.6 mg, 0.5 mol%) at 30 ℃ under air for 4 h. The values in parentheses are the E/Z ratios detected by 1H NMR spectroscopy. b Isolated yield. c 60 ℃. |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们