图 1.
富电子炔烃及代表性例子
Figure 1.
Electron-rich alkynes and representative examples

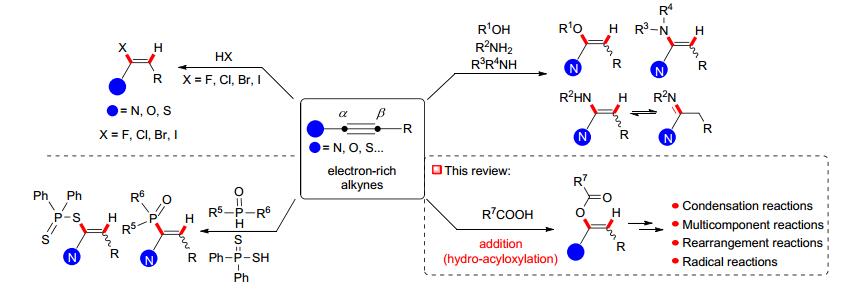
富电子炔烃(electron-rich alkynes)是一类碳碳叁键与一个供电子原子或供电子基团直接连接的炔烃, 这些炔烃的结构以及共振如图 1A所示, 这种共振使富电子炔烃呈现出亲电和亲核的双重反应活性.常见的炔酰胺(ynamides)、炔醚(ynol ethers)、炔硫醚(alkynyl thioethers)、炔胺(ynamines)和炔基三氮烯(1-alkynyl- triazenes)等都属于富电子炔烃(图 1B).这些富电子炔烃易于制备[1], 在杂环化合物合成[2]、自由基化学[3]、卡宾卡拜化学[4]、有机环加成和重排反应[5]以及天然产物全合成[6]等领域广泛应用.
氢-官能团化加成反应(hydro-functionalization)是富电子炔烃化学中的一类重要反应, 常见的炔酰胺、炔醚及炔硫醚等富电子炔烃和氢卤酸[7]、膦酸[8]、酚或醇[9]、氨基化合物[10]等质子性试剂都可以发生该类加成反应实现氢-官能团化(Scheme 1).其中, 羧酸与非活性炔烃的加成反应是制备烯醚的重要方法[11].近年来, 羧酸和富电子炔烃的加成反应也见诸报道, 其氢-酰氧化(hydro-acyloxylation)加成产物可以继续作为反应中间体, 参与缩合反应、多组分反应、重排反应及自由基反应.本文对富电子炔烃和有机羧酸的加成反应及其合成应用做一个介绍.
炔酰胺因为易于制备、稳定性好, 是富电子炔烃化学中最受关注的研究对象. 2012年, Lam课题组[12]首次报道了炔酰胺和羧酸的氢-酰氧化加成反应, 对各类不同结构的炔酰胺开展活性筛选, 发现在催化量的醋酸钯(2 mol%)存在及70 ℃的反应温度下, 炔酰胺1和羧酸2在甲苯中可以发生加成反应, 得到E型α-酰氧基烯酰胺(α-acyloxy enamides)类化合物3 (Scheme 2).该加成反应的效率较高(收率从中等到95%以上)并具有良好的区域选择性和立体选择性.在底物范围方面, 羧酸涵盖脂肪羧酸、芳香羧酸、烯酸和炔酸, 炔酰胺底物则适用于噁唑烷酮衍生的炔酰胺和炔磺酰胺等.不足的是, 该加成反应需要钯金属催化剂.
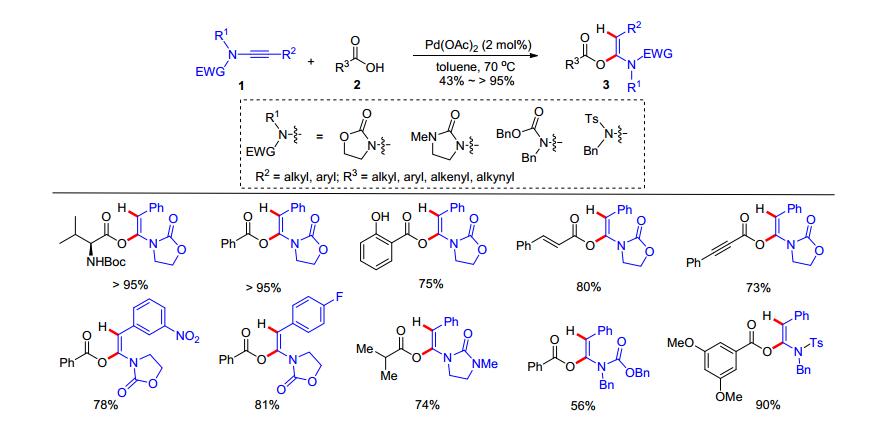
2015年, 毕锡和课题组[13]报道了无催化剂条件下的炔酰胺和羧酸的氢-酰氧化加成反应.以甲苯为溶剂于100 ℃的反应温度下, 炔酰胺4和羧酸5可以发生加成反应得到E型α-酰氧基烯酰胺6, 该反应具有良好的区域选择和立体选择性(Scheme 3, A).另外, 炔酰胺7和醋酸8在丙酮和水的混合溶剂中反应, 得到产物N-Ts酰胺9 (Scheme 3, B), 其反应历程包括了炔酰胺和醋酸加成反应形成α-乙酰氧基烯酰胺, 随后在高温下发生水解反应得到产物9.该反应的底物多样性良好, 原子利用率高且不需要任何催化剂催化, 是一个高效的绿色合成反应.
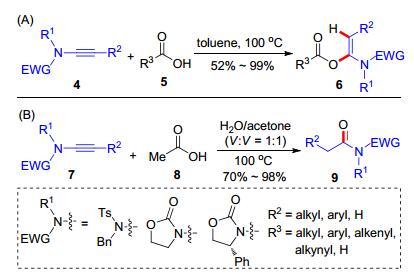
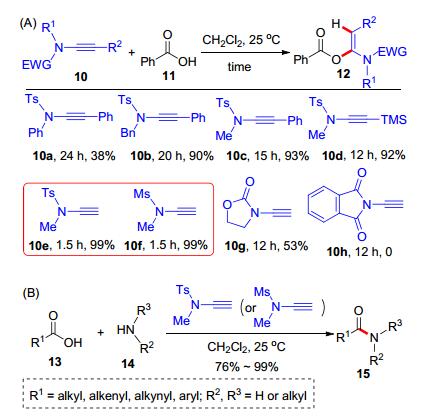
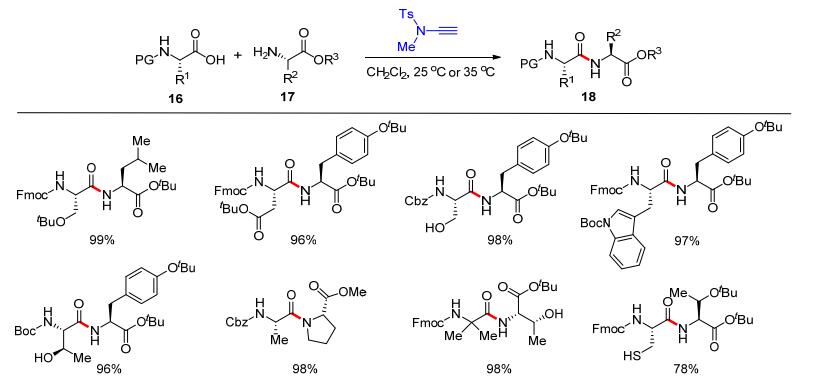
2016年, 赵军锋课题组[14]利用炔酰胺在多肽合成研究领域取得了重要进展.他们将炔酰胺作为羧酸和胺的缩合试剂构建酰胺键, 并将该方法应用于氨基酸的缩合反应中, 发展了一个中性的多肽缩合反应.首先, 测试了各类炔酰胺10和苯甲酸(11)的加成反应, 发现炔酰胺的结构与加成反应活性呈现相关性, 最高效的端炔酰胺10e和10f可以在室温下与苯甲酸发生加成反应生成12 (Scheme 4, A).因此, 炔酰胺也被选为缩合试剂开展了缩合反应的尝试; 结果表明, 羧酸13和炔酰胺10e或10f加成得到的α-酰氧基烯酰胺, 可以与一级或二级胺14高效生成酰胺15.底物范围方面, 各类脂肪酸、烯酸、炔酸和芳香酸以及各类一级脂肪胺、二级脂肪胺都适用于该缩合反应(Scheme 4, B).在此基础上, 该缩合反应被用于肽类化合物的合成.以炔酰胺10e为缩合剂, 各类氨基酸16和氨基酸酯17可以转化为多肽产物18 (Scheme 5).该反应具有效率高及多肽产物不消旋等优点, 具有重要的应用价值.
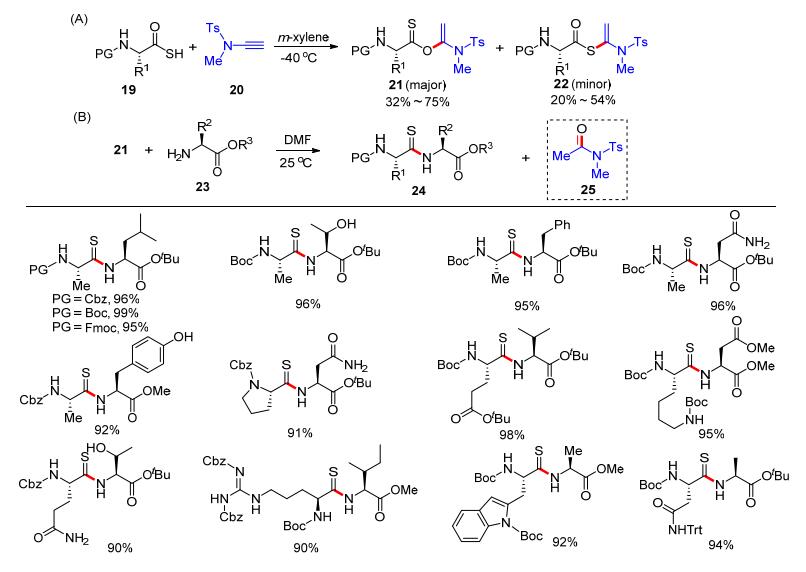
2019年, 赵军锋课题组[15]再次报道了将炔酰胺作为缩合试剂用于硫肽的合成.在使硫代氨基酸和炔酰胺进行加成反应时, 会同时生成α-硫酰氧基烯胺和α-酰硫基烯胺, 两种产物通过柱层析可以分离.经过条件优化, 硫代氨基酸19和炔酰胺20在-40 ℃下于间二甲苯中发生反应, 主要形成α-硫酰氧基烯胺21 (Scheme 6, A).将分离出来的21和氨基酸酯23反应, 在室温下以N, N-二甲基甲酰胺(DMF)作溶剂, 可以制备硫代酰胺化合物24以及副产物25 (Scheme 6, B).该工作提供了一条高效合成硫代酰胺键的路线, 具有良好的应用前景.
同时, 他们将该方法应用到了亮氨酸脑啡肽衍生物34等硫肽类生物活性分子的全合成(Scheme 7, A):首先, 芴甲氧羰基(Fmoc)保护的硫代苯丙氨酸与炔酰胺加成以65%的分离产率得到化合物26, 随后26与亮氨酸叔丁酯(27)进行缩合反应构建酰胺键, 并用二乙胺脱去Fmoc保护基得到二肽化合物28; 二肽28与Fmoc-甘氨酸和炔酰胺的加成产物29缩合, 用二乙胺脱去Fmoc保护基后得到了三肽化合物30; 甘氨酸叔丁酯(31)与α-硫酰氧基烯胺32缩合后脱去叔丁基得到二肽33; 最后, 将得到的33与30使用炔酰胺进行缩合反应即可得到目标化合物34.类似的, 细菌DNA促旋酶抑制剂Clos- thioamide (40)的全合成也按照该策略进行(Scheme 7, B): Fmoc保护的β-硫代丙氨酸与炔酰胺发生加成反应制备α-硫代酰氧基烯胺35; 随后, 两分子35与丙二胺(36)进行缩合得到化合物37; 脱去Fmoc保护基后与两分子35再进行一次缩合反应可以得到化合物38, 这两步的收率达到85%;脱去化合物38上的Fmoc保护基, 并与α-酰氧基烯胺39进行再次缩合, 然后用四丁基氟化铵(TBAF)进行羟基脱保护, 即得到目标产物40.在此过程中, 炔酰胺作为缩合剂构建酰胺键, 具有高效和无消旋化等优点, 是肽类和硫肽类化合物合成中新的重要策略.
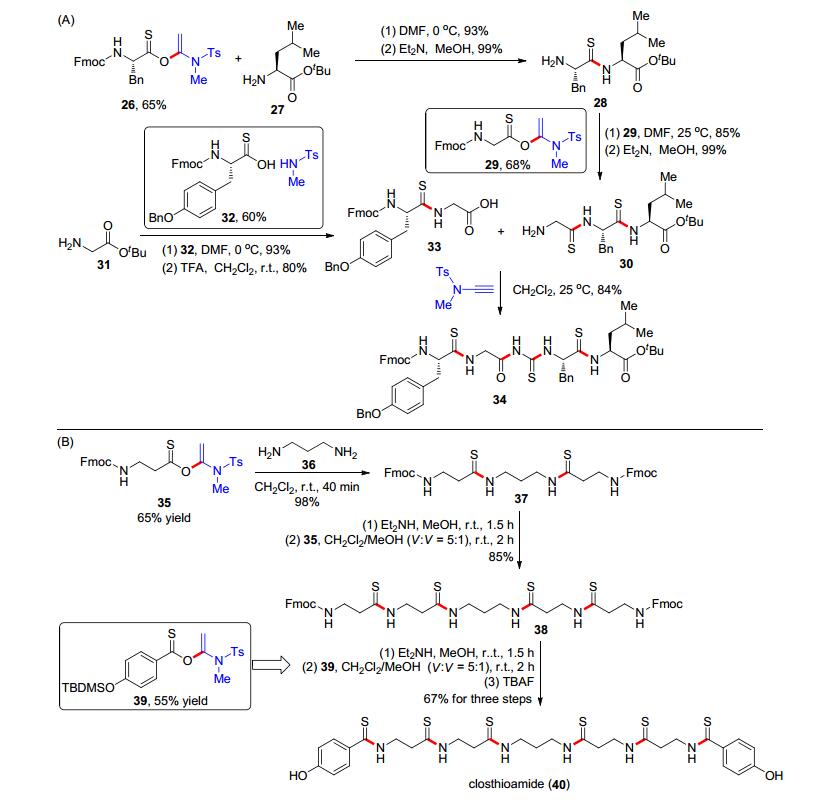
近期, 在炔酰胺介导的酰胺键构建的基础上, 赵军锋课题组[16]又成功发展了炔酰胺介导的酯键形成反应.羧酸41和炔酰胺42在二氯甲烷中完成加成反应形成α-烷氧基烯酰胺43后, 移去二氯甲烷并加入乙腈; 随后, 往体系中加入羟基化合物44和催化量的碱, 在室温下就可以高效地实现酯化反应得到产物45 (Scheme 8, A).该反应的羧酸底物可以涵盖脂肪羧酸、烯酸、炔酸和芳香酸; 羟基化合物方面, 酚类和醇类化合物均可以参与该酯化反应, 体现出良好的普适性.需要注意的是, 当使用酚类作为反应底物时, 三乙胺(TEA)或者N, N-二异丙基乙胺(DIPEA)可以有效催化该酯化反应; 而对于醇类底物, 碱则需要调整为N, N, N', N'-四甲基-1, 3-丙二胺(TMPDA).同时, 该研究组[17]成功开发了炔酰胺介导的大环内酯化反应.他们将羟基羧酸46与炔酰胺42在二氯甲烷中进行加成反应, 分离得到α-酰氧基烯酰胺47; 随后在对甲苯磺酸水合物(TsOH•H2O)的催化下, 以低反应浓度于二氯甲烷中成功实现了分子内的缩合反应, 得到了一系列的七元环到十七元环的大环内酯产物48 (Scheme 8, B).
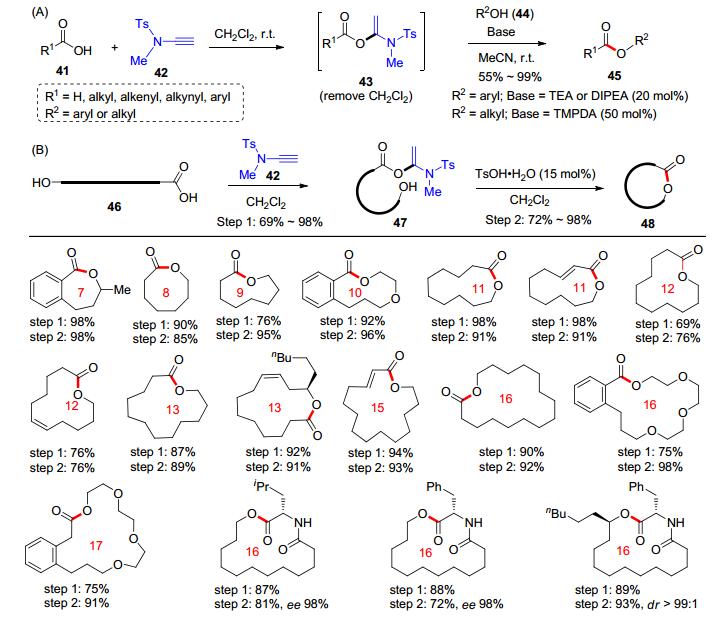
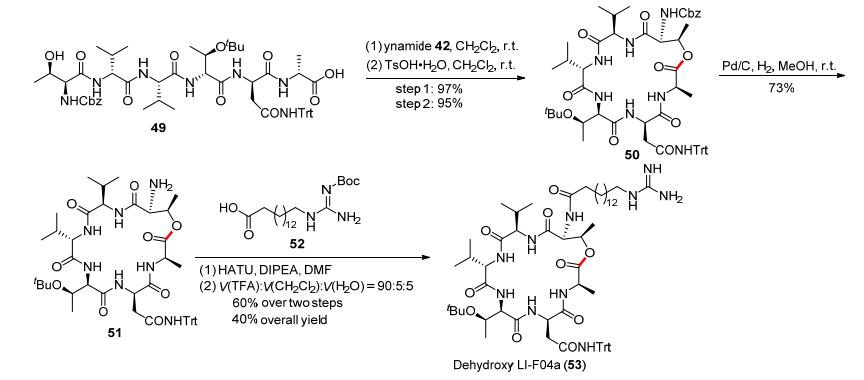
该酯键形成反应具有条件温和、产率高和产物无消旋化等特点, 是一种具有良好应用潜力的酯键合成方法.尤其是炔酰胺介导的大环内酯化反应的合成价值, 可以在活性大环内酯分子的全合成中得到展现.例如, 该反应可以用于抗真菌类环肽类似物Dehydroxy LIF04a (53)的快速全合成(Scheme 9):首先, 肽类化合物49利用炔酰胺介导的大环内酯化可以高效地制备十九元环产物50, 两步的反应收率分别可以达到97%和95%;随后, 利用氢化反应脱去50上的Cbz保护基, 以73%的收率得到化合物51; 51与羧酸52先后经过缩合反应和脱去Boc保护基即可得到目标化合物53.此路线仅需要五步反应就能以40%的总收率得到结构复杂的生物活性分子.该工作完善了炔酰胺作为缩合试剂在有机合成中的应用.在酰胺键和酯键的构建方面, 赵军锋课题组将炔酰胺推向了举足轻重的地位.
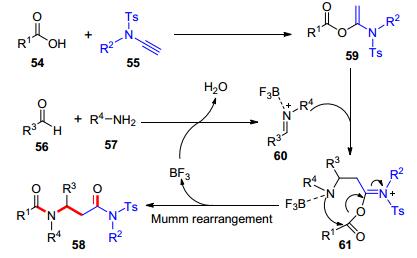
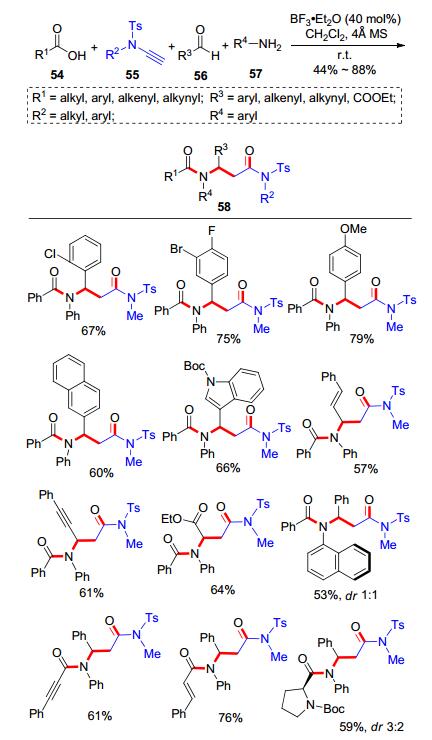
除了用于酰胺键和酯键的构建, 炔酰胺和羧酸的氢-酰氧化加成反应还可应用于多组分反应. 2017年, 我们课题组[18]通过“亲核/亲电双重活性设计”, 发现炔酰胺的互变异构呈现了“亲核/亲电双重活性”, 该性质与异腈具有相似性, 并基于该设计揭示了炔胺的C2合成模块性质, 开发了一个新型的炔胺、羧酸、醛、胺的类Ugi四组分反应, 合成了β-氨基酰胺类化合物(Scheme 10).反应历程包括了羧酸54和炔酰胺55的加成反应形成α-酰氧基烯胺59, α-酰氧基烯胺59对三氟化硼乙醚(BF3•Et2O)活化的亚胺鎓离子60进行加成得到61, 后发生Mumm重排得到产物58 (Scheme 11).反应产物属于Ugi反应的同系化产物.底物范围方面, 各类羧酸、芳香醛和芳香胺都可以很好地参与该多组分反应.该工作为多组分合成模块与反应类型的发展提供了新的视角.
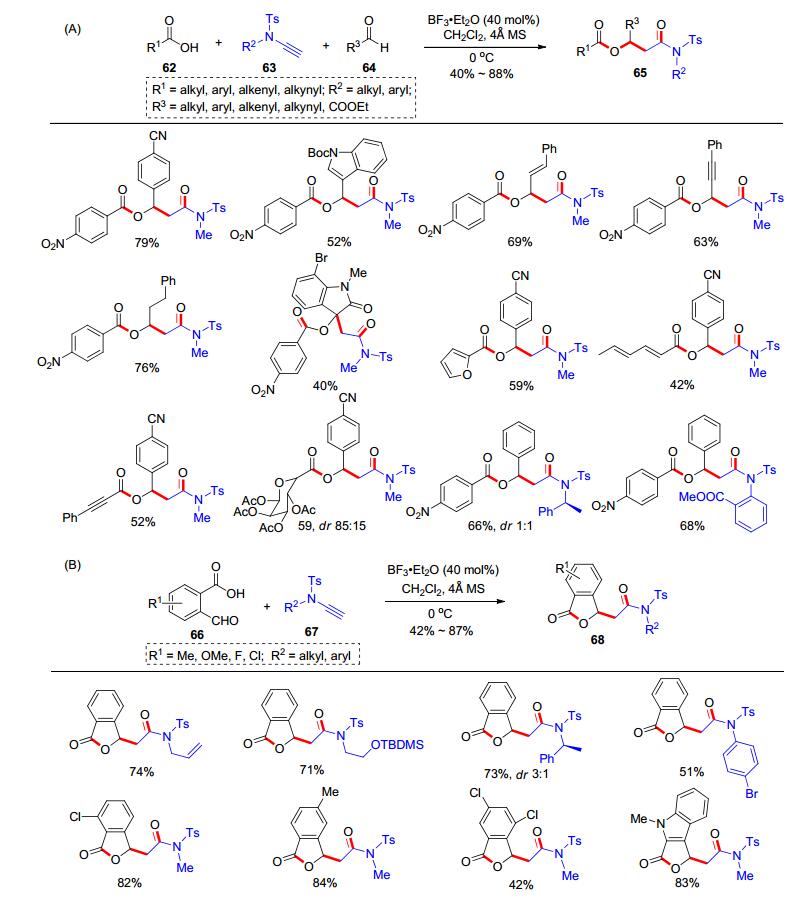
在此基础上, 我们课题组[19]继续利用炔酰胺作为合成模块, 发展了炔酰胺、羧酸、醛的类Passerini三组分反应合成β-酰氧基酰胺类化合物(β-acyloxyamides).类似地, 羧酸62和炔酰胺63发生加成反应原位生成α-酰氧基烯胺, 随后与醛64和三氟化硼乙醚(40 mol%), 反应可生成β-羟基酰胺65 (Scheme 12, A).底物范围方面, 各类羧酸、脂肪醛、芳香醛、烯醛和炔醛等都可以参与该三组分反应.此外, 我们还将羧酸和醛内置到同一分子中, 利用邻醛基苯甲酸(66)和炔酰胺67的反应, 实现了苯并呋喃酮类化合物68的合成(Scheme 12, B); 从而拓展了炔酰胺与羧酸的氢-酰氧化反应在多组分反应领域的应用.
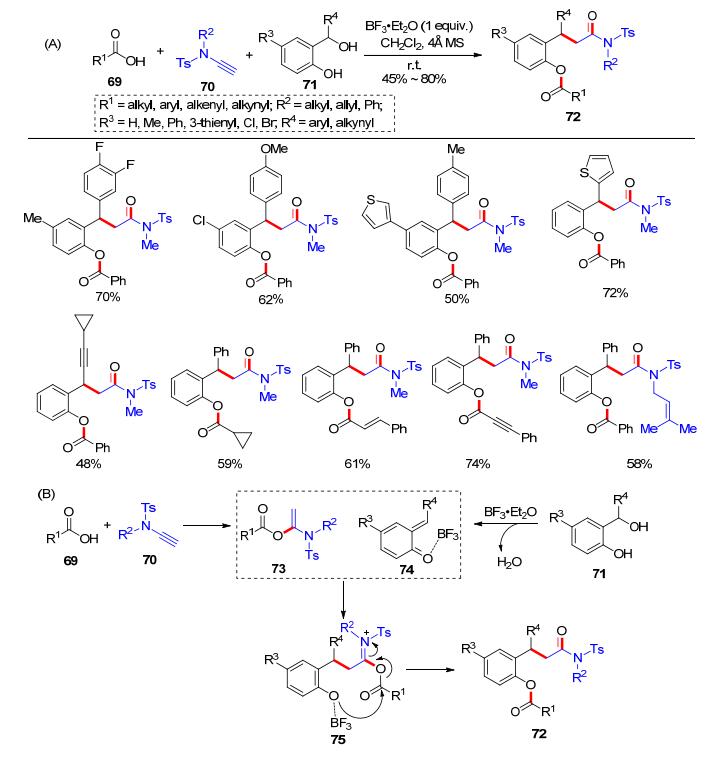
除了醛、亚胺等常见的亲电试剂, 我们还进一步引入各类亲电试剂, 开发了各类基于炔酰胺的新型多组分反应.比如, 邻亚甲基苯醌类化合物(ortho-quinone me- thides)是有机化学和化学生物中一类常见中间体[20], 并具有较高的化学反应活性.我们[21]认为邻亚甲基苯醌类化合物可以作为亲电试剂应用于多组分反应:利用羧酸和炔酰胺加成反应生成的α-酰氧基烯酰胺, 对原位生成的邻醌亚甲基化合物进行1, 4-加成/酯化反应, 实现多组分串联反应.通过条件优化发现, 加入化学计量的三氟化硼乙醚可以促进该反应, 获得2-酰氧基苯乙酰胺类化合物72 (Scheme 13, A).可能的反应机理是:羧酸69和炔酰胺70完成氢-酰氧化得到α-酰氧基烯酰胺73; 加入的邻羟基苄醇71在三氟化硼乙醚作用下原位生成邻亚甲基苯醌化合物74; 在三氟化硼乙醚作用下, 73和74发生加成反应后得到75, 然后发生酰基迁移可生成终即产物72 (Scheme 13, B).三氟化硼乙醚不仅活化了邻羟基苄醇释放出活性邻亚甲基苯醌物种, 也在后续的1, 4-加成反应中充当催化剂的角色.该反应的底物普适性好, 是一个理性设计的多组分串联反应.
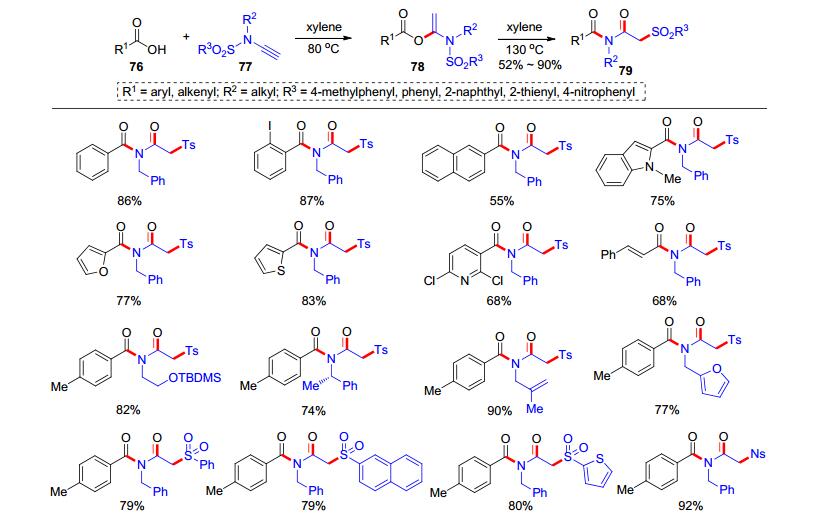
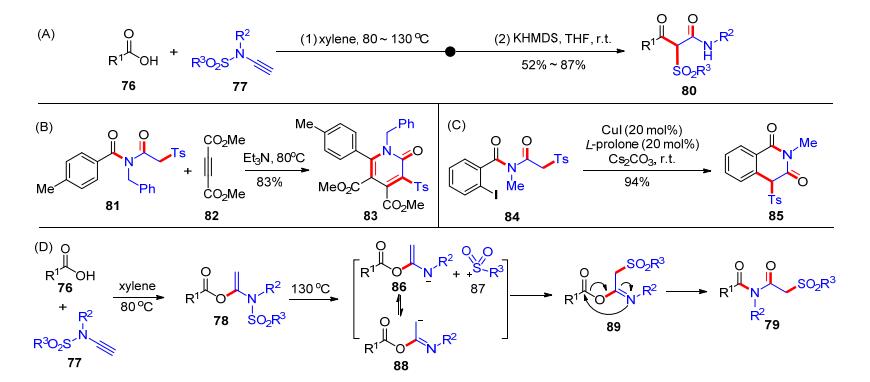
2018年, 我们课题组[22]发展了一个高温促进的氢-酰氧化加成串联磺酰基迁移反应.首先在二甲苯中于80 ℃的反应温度下, 羧酸76和炔酰胺77发生加成反应得到氢-酰氧化产物78; 升高温度至130 ℃, 78可以转化为化合物79 (Scheme 14).该反应具有较好的合成实用性, 可以放大到克级反应.比如, 当羧酸76和炔酰胺77在标准条件下完成反应后, 加入KHMDS可以引发分子内酰基迁移反应, 实现一锅法制备β-羰基酰胺类产物80 (Scheme 15, A); 而酰亚胺产物81和乙炔二甲酸二甲酯(DMAD, 82)可以在三乙胺作用下发生[4+2]环化, 高效地制备多取代的吡啶酮产物83 (Scheme 15, B); 另外, 产物84可以实现分子内的偶联反应得到环化产物85 (Scheme 15, C).该反应历程可能是: 76和77加成形成氢-酰氧化产物后, 在继续高温下发生碳氮键裂解得到86和87, 氮负离子86异构成碳负离子88后, 继续与87反应生成中间体89; 随后, 中间体89发生Mumm重排即可得到酰亚胺产物79 (Scheme 15, D).该反应基于炔酰胺和羧酸的加成反应发展了新型一锅法串联反应, 并开展了合成应用.
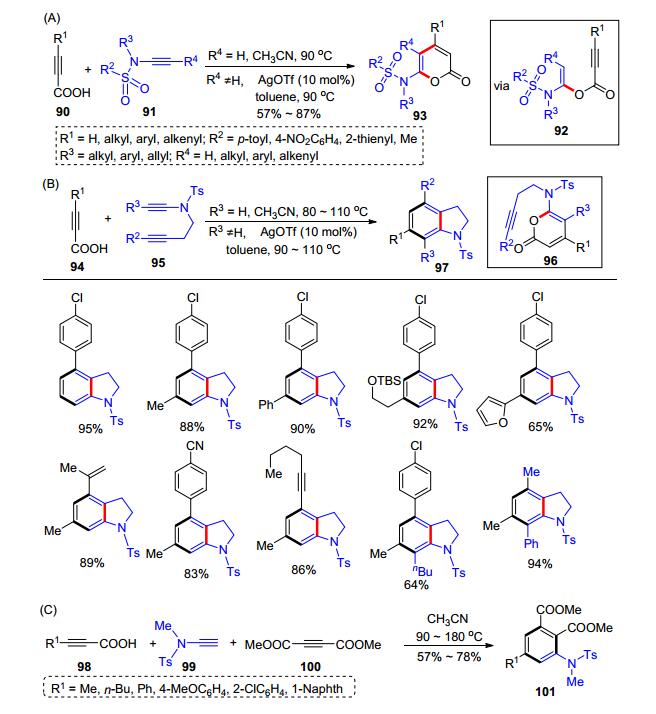
在以上的反应研究中, 羧酸显示了良好的普适性.进一步探索的时候[23], 我们还发现:炔酸90和炔酰胺91在室温下反应可高效合成α-酰氧基烯胺92; 但随着温度的升高, 有发生分子内环化得到α-吡喃酮93的倾向.在大量优化之后, 我们得到了最优条件:炔酸90和端炔酰胺在乙腈中于90 ℃下反应, 可以得到吡喃酮93; 当使用内炔酰胺作为底物时, 则需要加入催化量的三氟甲磺酸银(AgOTf, 10 mol%)实现该转化(Scheme 16, A).由于α-吡喃酮是一类优良的Diels-Alder反应底物, 我们继续发展了炔酰胺和炔酸参与的加成反应/串联Diels- Alder环化.我们设计了炔基支链取代的炔酰胺95, 与炔酸94在优化的反应条件下, 实现了炔酸和炔酰胺的加成反应/串联Diels-Alder环化(Scheme 16, B).该反应的历程包含了炔酸94与炔酰胺95形成α-吡喃酮96, 高温下引发了分子内的Diels-Alder反应得到了产物97.同时, 我们发现, 得到的α-吡喃酮参与分子间的Diels- Alder反应难度相对较高, 只有使用高活性的乙炔二甲酸二甲酯(100)作为底物, 炔酸98和端炔酰胺99才可以与之实现加成环合/串联Diels-Alder环化, 得到四取代苯类产物101 (Scheme 16, C).该反应以炔酰胺和炔基羧酸的加成反应为基础, 发展了三类新型有机合成方法, 为α-吡喃酮、二氢吲哚和多取代苯提供了一条新的合成路线.
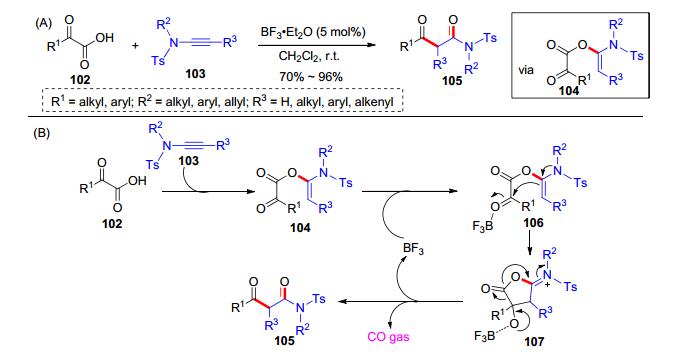
α-酮酸是一类特殊的高活性羧酸, 其在有机化学和生物化学中都有重要的作用, 常用作脱羧偶联反应和Minisci反应的自由基前体等, 并释放出CO2[24].在此过程中, 我们课题组[25]认为α-酮酸具有羧基和亲电性的酮羰基, 可以开展其与炔酰胺的反应活性探索.为此, 我们使用α-酮酸102和炔酰胺103进行尝试, 结果显示, 与羧酸相比, α-酮酸和炔酰胺的加成反应速度更快, 反应产物是β-羰基酰亚胺类化合物105 (Scheme 17, A), 反应过程也释放了大量气体.我们将释放出的气体通过气相色谱-热传导检测(GC-TCD), 确定气体为CO.于是, 我们提出了可能的反应历程:首先, α-酮酸102和炔酰胺103发生加成反应原位生成中间体104; 随后加入的三氟化硼乙醚活化了α-羰基并诱发了分子内重排, 释放出CO气体, 得到了产物105 (Scheme 17, B).值得一提的是, 在试图分离中间体104时, 我们发现该类化合物十分活泼并难以捕获, 因此这个反应只可适用一锅法操作.该方法提供了一种简捷高效的β-羰基酰亚胺合成方法.
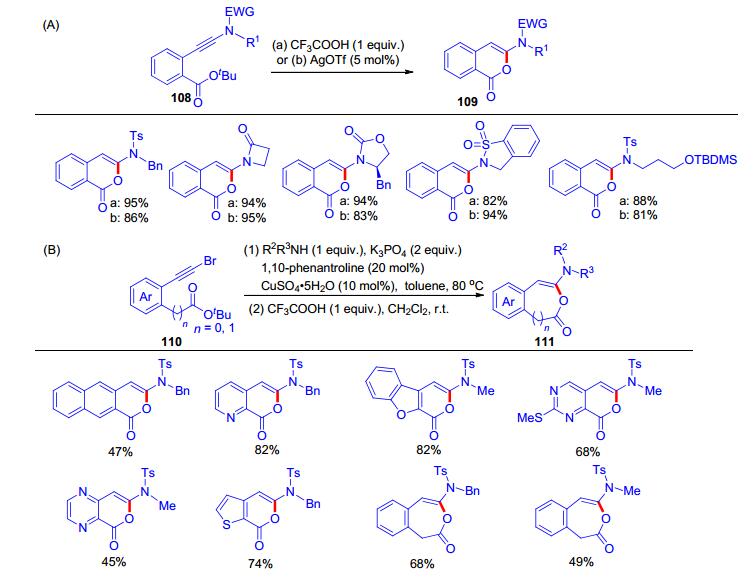
上述加成反应都是分子间的, 而分子内的加成反应却鲜有报道, 主要是由于该类原料制备困难. 2018年, Gillaizeau课题组[26]制备了炔酰胺108, 其结构含有一个叔丁酯保护的羧基.在化学计量的三氟乙酸(TFA)的作用下或者三氟甲磺酸银(5 mol%)的催化下, 该类炔酰胺可以发生分子内的环化反应得到氢-酰氧化产物109 (Scheme 18, A).此外, 他们还探索了从炔溴110出发, 一锅法偶联反应串联分子内环化制备了环状化合物111 (Scheme 18, B), 该反应适用于六元环和七元环的构建, 是炔酰胺和羧酸的分子内氢-酰氧化加成的唯一例证.
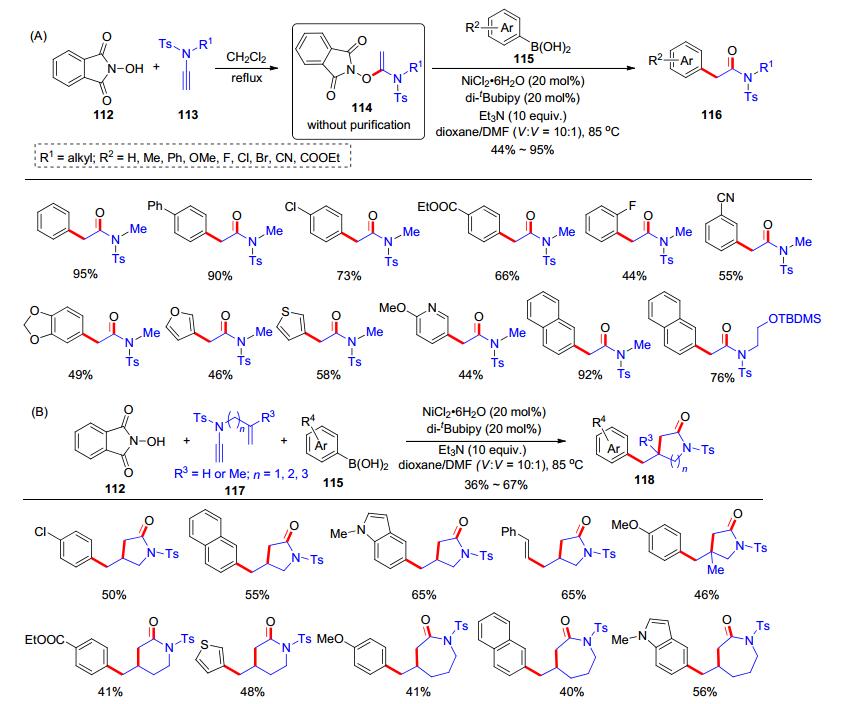
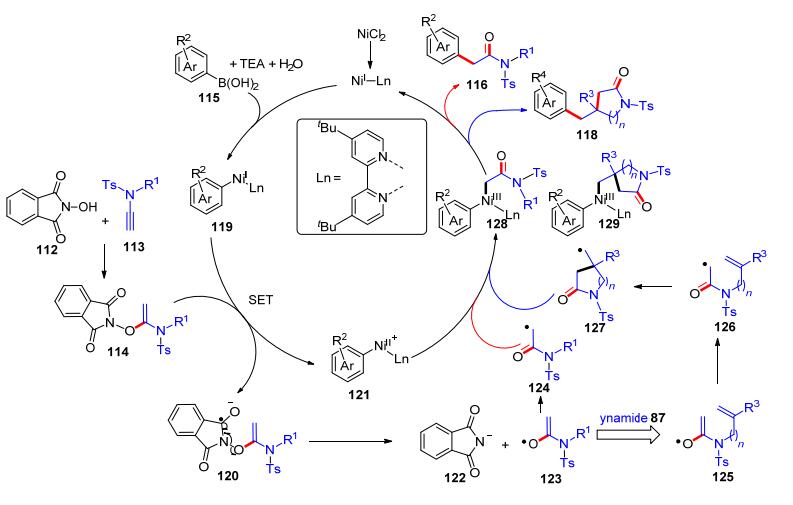
除了羧酸等经典的酸性底物外, N-羟基邻苯二甲酰亚胺(NHPI)也具有弱酸性, 其pKa约为6.1, 并呈现了单电子转移等特殊的化学反应活性[27].因此, 我们课题组[28]认为NHPI和炔酰胺也可能发生加成反应, 并可能适用于一系列的自由基反应.我们开展了大量的尝试与优化: NHPI (112)和端炔酰胺113在二氯甲烷中回流, 可以发生加成反应并以化学计量的收率得到产物114; 除去二氯甲烷溶剂后, 加入芳基硼酸115、NiCl2•6H2O (20 mol%)、二叔丁基联吡啶配体(di-tBubipy, 20 mol%)和三乙胺(10 equiv.), 在二氧六环和DMF (V:V=10:1)中于85 ℃下反应, 发生自由基偶联反应得到α-芳基酰胺类产物116 (Scheme 19, A).当使用N-侧链有烯烃取代的炔酰胺117时, 可以捕获自由基中间体得到环化的内酰胺产物118 (Scheme 19, B).我们也提出了可能的反应机理(Scheme 20):首先氯化镍和二叔丁基联吡啶配体及苯硼酸在三乙胺的作用下形成镍物种119, NHPI与炔酰胺113加成生成化合物114, 114在镍物种119作用下发生单电子转移(SET)得到自由基物种120和镍物种121; 物种120随后发生自由基裂解生成氧自由基物种123, 并马上重排形成碳自由基124; 124和镍物种121发生自由基加成形成中间体128, 随后还原消除释放出产物116, 并完成镍催化剂循环.对于炔酰胺117, 在形成氧自由基物种125后, 发生重排得到碳自由基126后继续与分子内烯烃发生自由基环化反应得到碳自由基127后, 发生自由基加成形成中间体129后还原消除生成产物118.在这个工作中, 我们发展的NHPI、炔酰胺和芳基硼酸的自由基串联反应, 实现了炔酰胺的自由基多组分反应.
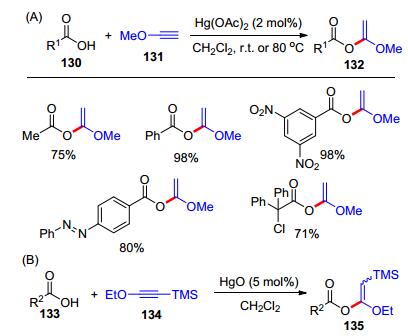
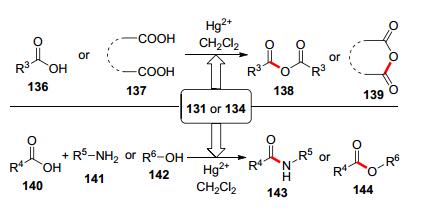
除了炔酰胺, 炔醚也是常见的富电子炔烃, 并呈现了差异化的反应活性.早在1958年, Wharton课题组[29]就制备了乙炔甲醚131, 并利用醋酸汞(2 mol%)作催化剂, 开发了乙炔甲醚与羧酸130的加成反应, 合成了α-烷氧基烯醇酯(α-alkoxy enol esters) 132 (Scheme 21, A).为了克服端炔醚稳定性差的问题, Kita研究组[30]制备了TMS取代的炔醚134; 发现在氧化汞(5 mol%)的催化下, 羧酸133和炔醚134可发生加成反应, 生成α-烷氧基烯醇酯135 (Scheme 21, B).同时, 炔醚131或134可以作为有机合成缩合剂(Scheme 22).例如, 在Hg试剂的作用下, 炔醚可以作为缩合试剂, 将羧酸136和二羧酸137分别转化成酸酐138和139[31].而且, 羧酸140和炔醚发生加成反应得到α-烷氧基烯醇酯产物后, 加入胺141和羟基化合物142可以分别制备酰胺143和酯144[32].然而, 上述工作都需要用到剧毒的Hg试剂, 这限制了炔醚和羧酸加成反应的应用.因此, 发展温和条件下的炔醚的氢-酰氧化反应非常迫切.
2014年, 朱钢国研究组[33]报道了银催化的炔醚和羧酸的加成反应实现氢-酰氧化.他们通过条件筛选发现:在氧化银(5 mol%)作用下, 将羧酸145和炔醚146在1, 4-二氧六环(dioxane)中于100 ℃下加热反应可以高效制备Z式α-烷氧基烯醚147 (Scheme 23, A); 该反应具有优秀的区域选择性和立体选择性.底物范围广, 各类脂肪酸、芳香酸、不饱和烯酸和炔酸, 以及各类端炔醚、烷基、芳基或烯丙基取代的炔醚, 都可以适用该加成反应.对该加成反应的应用也进行了研究, 例如, 使用特戊酸和炔醚得到的氢-酰氧化产物148, 可以在Ni(PCy3)2Cl2 (4 mol%)的催化下与芳基硼酸149发生Suzuki-Miyaura偶联反应, 制备三取代的烯醚类化合物150 (Scheme 23, B).该工作为炔醚和羧酸之间的加成反应提供了非常温和条件, 为之后该反应的应用提供了基础.
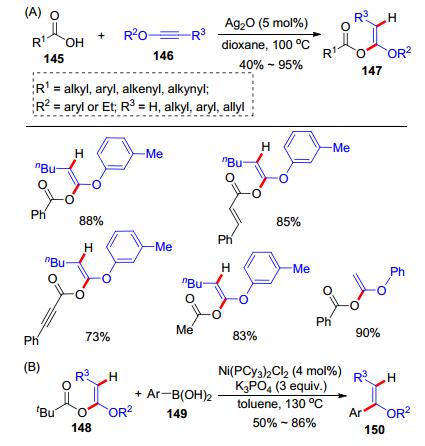
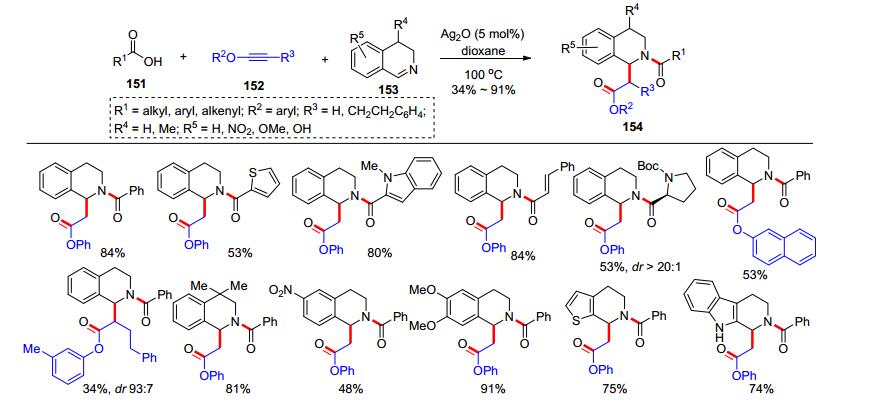
2018年, 结合前期在炔酰胺参与的多组分反应的工作基础, 我们课题组[34]发展了炔醚、羧酸和二氢异喹啉(DHIQs)的三组分反应, 高效合成了四氢异喹啉(THIQs)类化合物(Scheme 24).在氧化银(5 mol%)催化下, 羧酸151和炔醚152于二氧六环中加热原位生成α-烷氧基烯醇酯, 该中间体可以与二氢异喹啉153在100 ℃下反应, 高效制备了四氢异喹啉类产物154.羧酸、炔醚以及二氢异喹啉的底物普适性都良好.不足的是, 当使用内炔醚时, 反应的产率相比端炔醚大大降低.随后, 我们应用该方法做了一些合成应用(Scheme 25, A):例如, 四氢异喹啉产物155经过脱去苄氧羰基(Cbz)保护后, 可以直接发生分子内环化形成七元杂环化合物156; 产物157经过还原硝基成氨基后可以环化成八元杂环化合物158; 而咔唑类产物159在1, 8-二氮杂二环十一碳-7-烯(DBU)的作用下发生分子内缩合反应串联β-消除反应, 得到吲哚并五元环类产物161.为了探究机理, 我们开展了一系列的控制实验, 发现α-烷氧基烯醇酯中间体和二氢异喹啉不需要任何催化剂作用就可以反应.于是, 我们提出了可能的反应历程(Scheme 25, B):在氧化银催化下, 羧酸151和炔醚152原位生成α-烷氧基烯醚162; 然后, 二氢异喹啉153对162的羰基发生加成反应形成中间体163; 最后, 163发生分子内重排反应即可得到产物154.该反应是基于炔醚的第一个多组分反应, 也为优势骨架四氢异喹啉类化合物的合成提供了实用的方法.
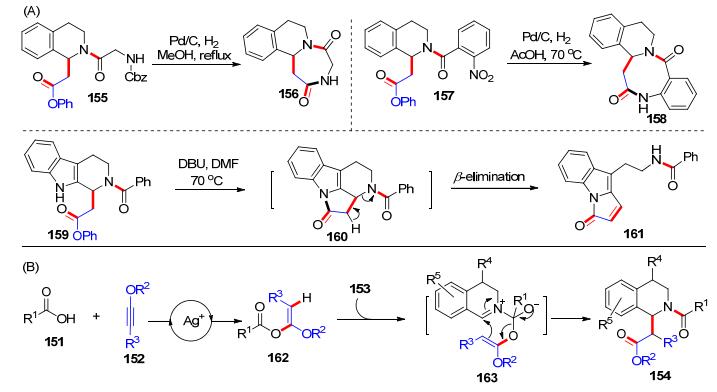
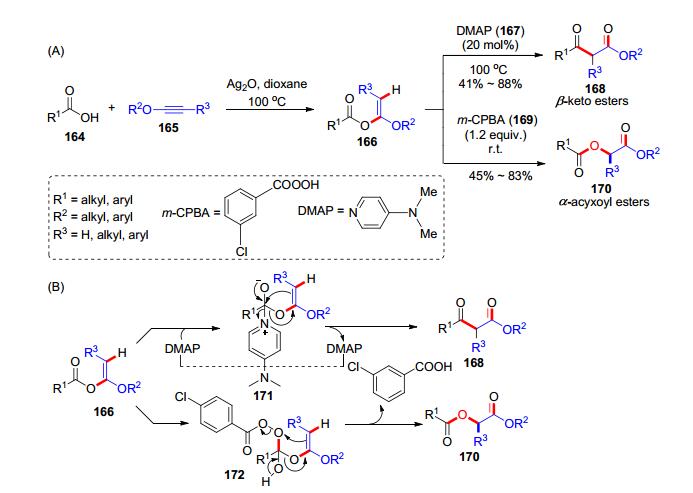
之后, 我们课题组[35]又继续探究了炔醚和羧酸加成反应在有机合成中的应用, 发现在氧化银催化下, 羧酸164和炔醚165形成α-烷氧基烯醚166后, 在催化量的4-二甲氨基吡啶(DMAP, 167, 20 mol%)作用下, 可以发生重排得到β-羰基酯类化合物168; 166也可以与间氯过氧苯甲酸(m-CPBA, 169)发生氧化重排反应, 得到α-酰氧基酯类产物170 (Scheme 26, A).以上两类反应的底物普适性良好, 适用于各类羧酸和炔醚.可能的反应历程(Scheme 26, B)为:在氧化银作用下, 羧酸164和炔醚165原位生成α-烷氧基烯醚166, 加入DMAP后, 可以对羰基进行加成得到中间体171, 随后171发生分子内的重排反应, 释放出产物168, DMAP则完成了循环; 而当α-烷氧基烯醚166碰到m-CPBA, 可能经过缩合过程生成中间体172, 随后发生分子内重排生成α-酰氧基酯类产物170和过氧苯甲酸, 这一过程与经典的Baeyer- Villiger反应有点类似.上述反应, 是我们课题组从炔醚和羧酸的氢-酰氧化反应出发, 通过合理设计优化成功实现的三组分或者一锅法重排反应的方法学, 丰富了炔醚化学和杂环化合物的合成研究.
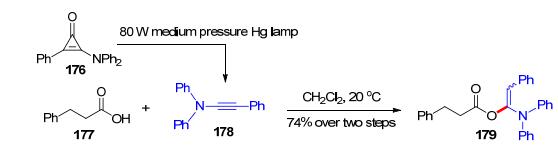
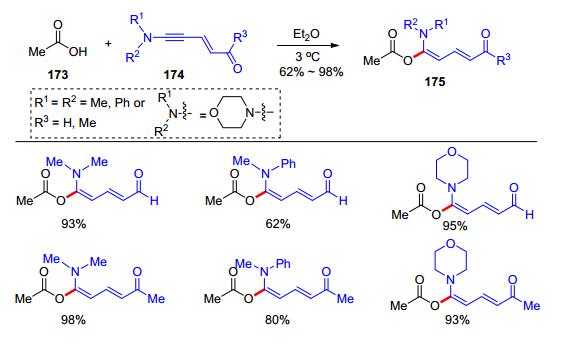
除了炔酰胺和炔醚, 其他的富电子炔烃也可以与羧酸发生加成反应.炔胺是较早报道的富电子炔烃之一, 但由于性质极其不稳定, 大大限制了它们在有机化学中的应用, 因而关于炔胺的反应研究仅有少数的例子. 1996年, Neuenschwander等[36]制备了几个不同结构的炔胺174, 并分别尝试了其与醋酸173的反应, 发现两者在乙醚中于3 ℃下反应, 就可以快速生成α-酰氧基烯胺产物175 (Scheme 27). 2018年, Kunishima课题组[37]在研究环丙烯酮和羧酸的反应机理时, 做了以下实验:将环丙烯酮176在汞灯作用下原位生成炔胺178后, 与苯丙酸177反应, 以74%的收率得到了α-酰氧基烯胺179 (Scheme 28).以上反应无需任何催化剂, 反应在较低温度下进行, 说明炔胺的活性比炔酰胺和炔醚都要高, 这也反映了炔胺类化合物的不稳定性.
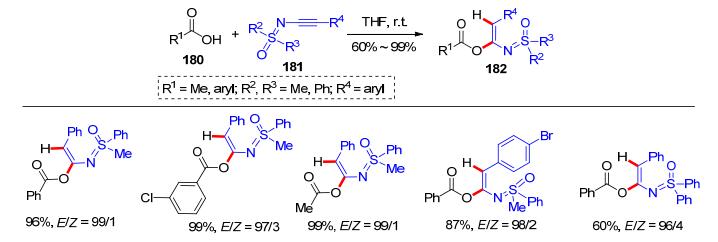
N-炔基砜亚胺(N-alkynyl sulfoximines)是由Bolm研究组[38]设计合成的一类富电子炔烃.他们研究组[39]发现在四氢呋喃(THF)中, 羧酸180在室温下可以与该类炔烃181反应, 得到以E式为主的氢-酰氧化产物182 (Scheme 29).该反应不需要任何的催化剂并具有较高的反应收率.各类脂肪羧酸和芳香羧酸都适用于该加成反应, 并具有良好的区域选择性和立体选择性.
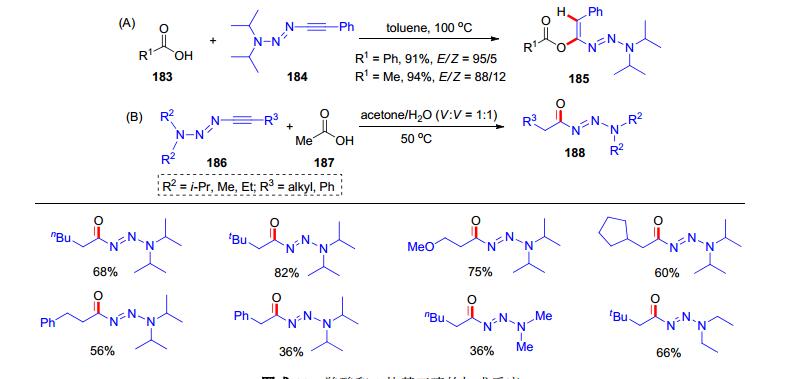
2015年, Severin课题组[1k]报道了一类1-炔基三氮烯(1-alkynyltriazenes)的制备, 在氨基锂试剂与笑气(N2O)发生加成之后, 继续与炔基格式试剂反应可以制备该类炔烃. 1-炔基三氮烯也呈现了富电子炔烃的特点[40]. 2015年, Severin课题组[41]发现1-炔基三氮烯和炔酰胺有着类似的反应活性:羧酸183和1-炔基三氮烯184在高温下可以发生加成反应, 生成氢-酰氧化产物185, 反应收率高, 区域选择性和立体选择性也较好(Scheme 30, A).此外, 丙酮和水的混合溶剂中, 1-炔基三氮烯186和醋酸187可以发生加成反应, 并水解为酰基三氮烯188 (Scheme 30, B), 这类酰基三嗪化合物类被认为是一种酰基化试剂, 可以用于各类反应[42].
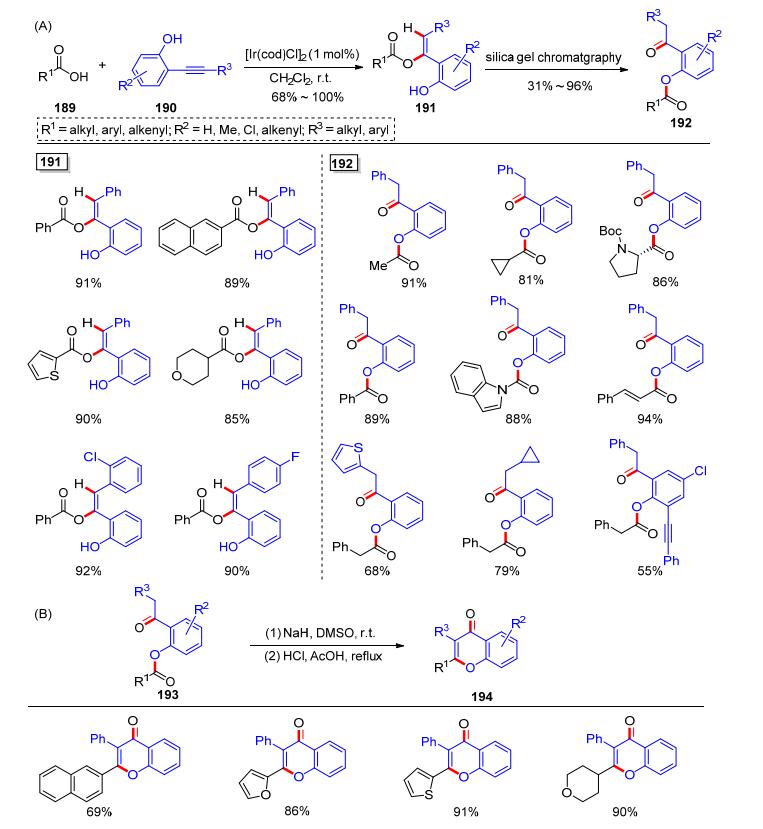
2-炔基苯酚类化合物也可以被认为是一类富电子炔烃, 因为羟基的供电子效应可以通过苯环共轭传递到炔烃上.我们课题组[43]在研究2-炔基苯酚的过程中, 发现在[Ir(cod)Cl]2 (1 mol%)的催化下, 羧酸189和2-炔基苯酚190反应, 可以高效实现氢-酰氧化制备单一E型的羧酸烯醇酯191, 并具有良好的区域选择性、立体选择性和产率, 产物只需重结晶或者简单的浓缩即可得到纯品191.有趣的是, 如果对该反应进行硅胶柱层析, 只能得到2-酰氧基苯乙酮类化合物192, 192是通过191的分子内酰基迁移反应产生的(Scheme 31, A).因此, 该加成反应后可以通过改变纯化方式, 选择性地制备两种不同的产物.另外, 得到的2-酰氧基苯乙酮类化合物也可以继续作为Baker-Venkataraman重排反应的原料, 进行各类反应转化.比如, 将化合物193在氢化钠(NaH)作用下发生重排, 直接在盐酸和醋酸中回流, 可以制备黄酮类母核化合物194 (Scheme 31, B).
综上所述, 富电子炔烃和羧酸的氢-酰氧化反应取得了较大的研究进展; 尤其炔酰胺、炔醚和羧酸的加成反应, 已经在多肽合成、多组分反应、一锅法反应、重排反应以及自由基反应等领域被广泛使用.对于富电子炔烃和羧酸的加成反应及其合成应用的发展前景, 我们认为以下几点值得关注: (1)继续探索炔硫醚、2-炔基苯胺等富电子炔烃和羧酸的氢-酰氧化反应; (2)设计新型的富电子炔烃, 探索它们和羧酸的氢-酰氧化反应及应用; (3)将已经开发的富电子炔烃和羧酸的加成反应, 应用于更多的有机合成中.我们相信, 以富电子炔烃和羧酸的加成反应为基础, 开展的新型有机反应研究是一个绿色合成的重要研究方向, 在未来会迎来更大的发展.
(a) Janousek, Z.; Viehe, H. G.; Collard, J. Angew. Chem., Int. Ed. 1972, 11, 917.
(b) Tu, Y.; Zeng, X.; Wang, H.; Zhao, J. Org. Lett. 2018, 20, 280.
(c) Coste, A.; Karthikeyan, G.; Couty, F.; Evano, G. Angew. Chem., Int. Ed. 2009, 48, 4381.
(d) Frederick, M. O.; Mulder, J. A.; Tracey, M. R.; Hsung, R. P.; Huang, J.; Kurtz, K. C. M.; Shen, L. C.; Douglas, C. J. J. Am. Chem. Soc. 2003, 125, 2368.
(e) Mansfield, S. J.; Campbell, C. D.; Jones, M. W.; Anderson, E. A. Chem. Commun. 2015, 51, 3316.
(f) Souto, J. A.; Becker, P.; Iglesias, A.; Muniz, K. J. Am. Chem. Soc. 2012, 134, 15505.
(g) Zhang, X. J.; Zhang, Y. S.; Huang, J.; Hsung, R. P.; Kurtz, K. C. M.; Oppenheimer, J.; Petersen, M. E.; Sagamanova, I. K.; Shen, L. C.; Tracey, M. R. J. Org. Chem. 2006, 71, 4170.
(h) Jouvin, K.; Coste, A.; Bayle, A.; Legrand, F.; Karthikeyan, G.; Tadiparthi, K.; Evano, G. Organometallics 2012, 31, 7933.
(i) Jouvin, K.; Bayle, A.; Legrand, F.; Evano, G. Org. Lett. 2012, 14, 1652.
(j) Kanemoto, K.; Yoshida, S.; Hosoya, T. Org. Lett. 2019, 21, 3172.
(k) Kiefer, G.; Riedel, T.; Dyson, P. J.; Scopelliti, R.; Severin, K. Angew. Chem., Int. Ed. 2015, 54, 302.
(l) Pena, J.; Talavera, G.; Waldecker, B.; Alcarazo, M. Chem.-Eur. J. 2017, 23, 75.
(a) Wang, X. N.; Yeom, H. S.; Fang, L. C.; He, S. H.; Ma, Z. X.; Kedrowski, B. L.; Hsung, R. P. Acc. Chem. Res. 2014, 47, 560.
(b) Pan, F.; Shu, C.; Ye, L. W. Org. Biomol. Chem. 2016, 14, 9456.
(c) Wang, Y.; Song, L. J.; Zhang, X. H.; Sun, J. W. Angew. Chem., Int. Ed. 2016, 55, 9704.
(d) Zhu, C.; Feng, J.; Zhang, J. Chin. J. Org. Chem. 2017, 37, 1165(in Chinese). (朱超泽, 冯见君, 张俊良, 有机化学, 2017, 37, 1165.)
(e) Liu, J.; Chakraborty, P.; Zhang, H.; Zhong, L.; Wang, Z.; Huang, X. ACS Catal. 2019, 9, 2610.
(a) Marion, F.; Courillon, C.; Malacria, M. Org. Lett. 2003, 5, 5095.
(b) Banerjee, B.; Litvinov, D. N.; Kang, J.; Bettale, J. D.; Castle, S. L. Org. Lett. 2010, 12, 2650.
(c) Wang, L.; Lu, C. R.; Yue, Y. N.; Feng, C. Org. Lett. 2019, 21, 3514.
(a) Shen, C.-H.; Li, L.; Zhang, W.; Liu, S.; Shu, C.; Xie, Y.-E.; Yu, Y.-F.; Ye, L.-W. J. Org. Chem. 2014, 79, 9313.
(b) Liu, R. H.; Winston-McPherson, G. N.; Yang, Z. Y.; Zhou, X.; Song, W. Z.; Guzei, I. A.; Xu, X. F.; Tang, W. P. J. Am. Chem. Soc. 2013, 135, 8201.
(c) Davies, P. W.; Cremonesi, A.; Martin, N. Chem. Commun. 2011, 47, 379.
(a) Ding, S. T.; Jia, G. C.; Sun, J. W. Angew. Chem., Int. Ed. 2014, 53, 1877.
(b) Tian, X. H.; Song, L. N.; Rudolph, M.; Rominger, F.; Oeser, T.; Hashmi, A. S. K. Angew. Chem., Int. Ed. 2019, 58, 3589.
(c) DeKorver, K. A.; Li, H. Y.; Lohse, A. G.; Hayashi, R.; Lu, Z. J.; Zhang, Y.; Hsung, R. P. Chem. Rev. 2010, 110, 5064.
(d) Zhou, B.; Li, L.; Zhu, X. Q.; Yan, J. Z.; Guo, Y. L.; Ye, L. W. Angew. Chem., Int. Ed. 2017, 56, 4015.
(e) Zeng, L.; Lai, Z.; Zhang, C.; Xie, H.; Cui, S. Org. Lett. 2020, 22, 2220.
(a) Zhang, W. H.; Ready, J. M. J. Am. Chem. Soc. 2016, 138, 10684.
(b) Zhang, Y.; Hsung, R. P.; Zhang, X.; Huang, J.; Slafer, B. W.; Davis, A. Org. Lett. 2005, 7, 1047.
(c) Couty, S.; Liegault, B.; Meyer, C.; Cossy, J. Tetrahedron 2006, 62, 3882.
(d) Couty, S.; Meyer, C.; Cossy, J. Tetrahedron Lett. 2006, 47, 767.
(e) Alayrac, C.; Schollmeyer, D.; Witulski, B. Chem. Commun. 2009, 1464.
(a) Cao, W.; Chen, P.; Wang, L.; Wen, H.; Liu, Y.; Wang, W. S.; Tang, Y. Org. Lett. 2018, 20, 4507.
(b) Compain, G.; Jouvin, K.; Mingot, A. M.; Evano, G.; Marrot, J.; Thibaudeau, S. Chem. Commun. 2012, 48, 5196.
(c) Métayer, B.; Compain, G.; Jouvin, K.; MartinMingot, A.; Bachmann, C.; Marrot, J.; Evano, G.; Thibaudeau, S. J. J. Org. Chem. 2015, 80, 3397.
(d) Zeng, X. J.; Lu, Z. C.; Liu, S. W.; Hammond, G. B.; Xu, B. J. Org. Chem. 2017, 82, 13179.
(e) Li, X.; Sun, Y.; Zhang, L.; Peng, B. Chin. J. Org. Chem. 2016, 36, 2530(in Chinese). (李晓锦, 孙艳, 张磊, 彭勃, 有机化学, 2016, 36, 2530.)
(a) Kanemura, S.; Kondoh, A.; Yasui, H.; Yorimitsu, H.; Oshima, K. Bull. Chem. Soc. Jpn. 2008, 81, 506.
(b) Yasui, H.; Yorimitsu, H.; Oshima, K. Chem. Lett. 2008, 37, 40.
(c) Kim, S. W.; Um, T. W.; Shin, S. Chem. Commun. 2017, 53, 2733.
(a) Tolchinskii, S. E.; Dogadina, A. V.; Maretina, I. A.; Petrov, A. A. Zh. Org. Khim. 1980, 16, 1141.
(b) Yoo, H. J.; Youn, S. W. Org. Lett. 2019, 21, 3422.
(a) Rentsch, C.; Slongo, M.; Stadelma, W.; Neuensch, M. Chimia 1973, 27, 70.
(b) Zhang, Y.; DeKorver, K. A.; Lohse, A. G.; Zhang, Y. S.; Huang, J.; Hsung, R. P. Org. Lett. 2009, 11, 899.
(c) DeKorver, K. A.; Johnson, W. L.; Zhang, Y.; Hsung, R. P.; Dai, H. F.; Deng, J.; Lohse, A. G.; Zhang, Y. S. J. Org. Chem. 2011, 76, 5092.
(a) Bianchini, C.; Meli, A.; Peruzzini, M.; Zanobini, F.; Bruneau, C.; Dixneuf, P. H. Organometallics 1990, 9, 1155.
(b) Chen, J.-F.; Li, C. Org. Lett. 2018, 20, 6719.
(c) Goossen, L. J.; Paetzold, J.; Koley, D. Chem. Commun. 2003, 706.
(d) Hua, R. M.; Tian, X. J. Org. Chem. 2004, 69, 5782.
(e) Jeschke, J.; Gabler, C.; Lang, H. J. Org. Chem. 2016, 81, 476.
(f) Lumbroso, A.; Vautravers, N. R.; Breit, B. Org. Lett. 2010, 12, 5498.
(g) Rotem, M.; Shvo, Y. Organometallics 1983, 2, 1689.
(h) Wang, Y.; Wang, Z.; Li, Y.; Wu, G.; Cao, Z.; Zhang, L. Nat. Commun. 2014, 5, 3470.
(i) Dupuy, S.; Gasperini, D.; Nolan, S. P. ACS Catal. 2015, 5, 6918.
Smith, D. L.; Goundry, W. R. F.; Lam, H. W. Chem. Commun. 2012, 48, 1505.
Xu, S.; Liu, J.; Hu, D.; Bi, X. Green Chem. 2015, 17, 184.
(a) Hu, L.; Xu, S.; Zhao, Z.; Yang, Y.; Peng, Z.; Yang, M.; Wang, C.; Zhao, J. J. Am. Chem. Soc. 2016, 138, 13135.
(b) Hu, L.; Zhao, J. Synlett 2017, 28, 1663.
Yang, J.; Wang, C.; Xu, S.; Zhao, J. Angew. Chem., Int. Ed. 2019, 58, 1382.
Wang, X.; Yang, Y.; Zhao, Y.; Wang, S.; Hu, W.; Li, J.; Wang, Z.; Yang, F.; Zhao, J. J. Org. Chem. 2020, 85, 6188.
Yang, M.; Wang, X.; Zhao, J. ACS Catal. 2020, 10, 5230.
Huang, B.; Zeng, L.; Shen, Y.; Cui, S. Angew. Chem., Int. Ed. 2017, 56, 4565.
Shen, Y.; Huang, B.; Zeng, L.; Cui, S. Org. Lett. 2017, 19, 4616.
(a) Quinone Methides, Ed.: Rokita, S. E., Wiley, New York, 2009.
(b) Yang, B.; Gao, S. Chem. Soc. Rev. 2018, 47, 7926.
Chen, R.; Liu, Y.; Cui, S. Chem. Commun. 2018, 54, 11753.
Shen, Y.; Li, Q.; Xu, G.; Cui, S. Org. Lett. 2018, 20, 5194.
Shen, Y.; Wang, C.; Chen, W.; Cui, S. Org. Chem. Front. 2018, 5, 3574.
(a) Pusterla, I.; Bode, J. W. Angew. Chem., Int. Ed. 2012, 51, 513.
(b) Doll, M. K. H. J. Org. Chem. 1999, 64, 1372.
(c) Gooβen, L. J.; Rudolphi, F.; Oppel, C.; Rodríguez, N. Angew. Chem., Int. Ed. 2008, 47, 3043.
(d) Shang, R.; Fu, Y.; Li, J.-B.; Zhang, S.-L.; Guo, Q.-X.; Liu, L. J. Am. Chem. Soc. 2009, 131, 5738.
Chen, R.; Zeng, L.; Huang, B.; Shen, Y.; Cui, S. Org. Lett. 2018, 20, 3377.
Habert, L.; Retailleau, P.; Gillaizeau, I. Org. Biomol. Chem. 2018, 16, 7351.
(a) Qin, T.; Cornella, J.; Li, C.; Malins, L. R.; Edwards, J. T.; Kawamura, S.; Maxwell, B. D.; Eastgate, M. D.; Baran, P. S. Science 2016, 352, 801.
(b) Cornella, J.; Edwards, J. T.; Qin, T.; Kawamura, S.; Wang, J.; Pan, C. M.; Gianatassio, R.; Schmidt, M.; Eastgate, M. D.; Baran, P. S. J. Am. Chem. Soc. 2016, 138, 2174.
(c) Toriyama, F.; Cornella, J.; Wimmer, L.; Chen, T. G.; Dixon, D. D.; Creech, G.; Baran, P. S. J. Am. Chem. Soc. 2016, 138, 11132.
(d) Qin, T.; Malins, L. R.; Edwards, J. T.; Merchant, R. R.; Novak, A. J. E.; Zhong, J. Z.; Mills, R. B.; Yan, M.; Yuan, C.; Eastgate, M. D.; Baran, P. S. Angew. Chem., Int. Ed. 2019, 58, 112.
(e) Qin, T.; Malins, L. R.; Edwards, J. T.; Merchant, R. R.; Novak, A. J. E.; Zhong, J. Z.; Mills, R. B.; Yan, M.; Yuan, C. X.; Eastgate, M. D.; Baran, P. S. Angew. Chem., Int. Ed. 2017, 56, 260.
(f) Edwards, J. T.; Merchant, R. R.; McClymont, K. S.; Knouse, K. W.; Qin, T.; Malins, L. R.; Vokits, B.; Shaw, S. A.; Bao, D. H.; Wei, F. L.; Zhou, T.; Eastgate, M. D.; Baran, P. S. Nature 2017, 545, 213.
Huang, B.; Zeng, L.; Shen, Y.; Cui, S. Chem. Commun. 2017, 53, 11996.
(a) Wasserman, H. H.; Wharton, P. S. J. Am. Chem. Soc. 1960, 82, 661.
(b) Wasserman, H. H.; Wharton, P. S. Tetrahedron 1958, 3, 321.
Kita, Y.; Akai, S.; Yoshigi, M.; Nakajima, Y.; Yasuda, H.; Tamura, Y. Tetrahedron Lett. 1984, 25, 6027.
Kita, Y.; Akai, S.; Ajimura, N.; Yoshigi, M.; Tsugoshi, T.; Yasuda, H.; Tamura, Y. J. Org. Chem. 1986, 51, 4150.
Kita, Y.; Akai, S.; Yamamoto, M.; Taniguchi, M.; Tamura, Y. Synthesis 1989, 334.
Yin, J.; Bai, Y. H.; Mao, M. Y.; Zhu, G. G. J. Org. Chem. 2014, 79, 9179.
Zeng, L.; Huang, B.; Shen, Y.; Cui, S. Org. Lett. 2018, 20, 3460.
(a) Zeng, L.; Lai, Z.; Cui, S. J. Org. Chem. 2018, 83, 14834.
(b) Zeng, L.; Sajiki, H.; Cui, S. Org. Lett. 2019, 21, 6423.
Berger, D.; Neuenschwander, M. Helv. Chim. Acta 1996, 79, 192.
Mishiro, K.; Yushima, Y.; Kunishima, M. J. Org. Chem. 2018, 83, 13595.
(a) Wang, H.; Cheng, Y.; Becker, P.; Raabe, G.; Bolm, C. Angew. Chem., Int. Ed. 2016, 55, 12655.
(b) Priebbenow, D. L.; Becker, P.; Bolm, C. Org. Lett. 2013, 15, 6155.
Pirwerdjan, R.; Becker, P.; Bolm, C. Org. Lett. 2015, 17, 5008.
(a) Kossler, D.; Perrin, F. G.; Suleymanov, A. A.; Kiefer, G.; Scopelliti, R.; Severin, K.; Cramer, N. Angew. Chem., Int. Ed. 2017, 56, 11490.
(b) Jeanbourquin, L. N.; Scopelliti, R.; Tirani, F. F.; Severin, K. Org. Lett. 2017, 19, 2070.
(c) Suleymanov, A. A.; Scopelliti, R.; Tirani, F. F.; Severin, K. Adv. Synth. Catal. 2018, 360, 4178.
(d) Tan, J. F.; Bormann, C. T.; Perrin, F. G.; Chadwick, F. M.; Seyerin, K.; Cramer, N. J. Am. Chem. Soc. 2019, 141, 10372.
Perrin, F. G.; Kiefer, G.; Jeanbourquin, L.; Racine, S.; Perrotta, D.; Waser, J.; Scopelliti, R.; Severin, K. Angew. Chem., Int. Ed. 2015, 54, 13393.
Landman, I. R.; Acuna-Bolomey, E.; Scopelliti, R.; Fadaei-Tirani, F.; Seyerin, K. Org. Lett. 2019, 21, 6408.
Zeng, L.; Chen, R.; Zhang, C.; Xie, H.; Cui, S. Chem. Commun. 2020, 56, 3093.

图式 2 钯催化炔酰胺和羧酸的加成反应
Scheme 2 Pd-Catalyzed addition reaction between ynamides and carboxylic acids

图式 3 炔酰胺和羧酸的无催化加成反应
Scheme 3 Catalyst-free addition reaction between ynamides and carboxylic acids

图式 7 炔酰胺实现无消旋构建硫肽键的应用
Scheme 7 Synthetic application of racemic-free synthesis of thiopeptides using ynamides

图式 9 炔酰胺介导大环内酯化的合成应用
Scheme 9 Synthetic application of ynamide-mediated macrolactonization

图式 11 β-氨基酰胺的四组分合成反应机理
Scheme 11 Mechanism of four-component synthesis of β- amino amides

图式 13 炔酰胺、羧酸及邻羟基苄醇参与的三组分反应
Scheme 13 Three-component reaction of ynamides, carboxylic acids and 2-hydroxybenzyl alcohols

图式 14 炔酰胺和炔酸的加成磺酰基迁移反应
Scheme 14 Addition and sulfonyl migration of ynamides and alkynyl acids

图式 15 炔酰胺和炔酸的加成磺酰基迁移反应的应用和机理
Scheme 15 Applications and mechanism about addition and sulfonyl migration of ynamides and alkynyl acids

图式 17 炔酰胺和α-酮酸的加成重排反应及反应机理
Scheme 17 Reaction and mechanism of addition/rearrangement between ynamides and α-keto acids

图式 19 NHPI, 炔酰胺和芳基硼的加成-自由基偶联反应
Scheme 19 Addition and radical coupling of NHPI, ynamides and arylboronic acids

图式 20 NHPI, 炔酰胺和芳基硼的加成-自由基偶联反应可能的机理
Scheme 20 Mechanism about addition and radical coupling of NHPI, ynamides and arylboronic acids

图式 21 Hg催化炔醚和羧酸的加成反应
Scheme 21 Hg-Catalyzed addition reaction between ynol ethers and carboxylic acids

图式 22 炔醚用于酸酐、酰胺和酯的合成
Scheme 22 Synthesis of anhydrides, amides and esters using ynol ethers

图式 23 Ag催化炔醚和羧酸的加成反应
Scheme 23 Ag-Catalyzed addition reaction between ynol ethers and carboxylic acids

图式 25 三组分合成四氢异喹啉的应用及可能机理
Scheme 25 Applications and mechanism about three-component synthesis of tetrahydroisoquinolines

图式 26 一锅法制备β-羰基酯和α-酰氧基酯的反应及机理
Scheme 26 Reactions and mechanism of one-pot synthesis of β-keto-esters and α-acyxoyl esters

图式 28 羧酸和原位生成炔胺的加成反应
Scheme 28 Addition reaction between carboxylic acids and in situ generated ynamines

图式 29 羧酸和N-炔基砜亚胺的加成反应
Scheme 29 Addition reaction between acetic acids and N-alkynyl sulfoximines

图式 30 羧酸和1-炔基三嗪的加成反应
Scheme 30 Addition reaction between carboxylic acid and 1-alkynyltriazenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:











