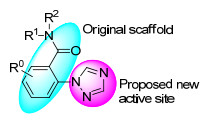
图 1.
芳甲酰胺类活性化合物的结构
Figure 1.
Structures of aryl formamide active compounds

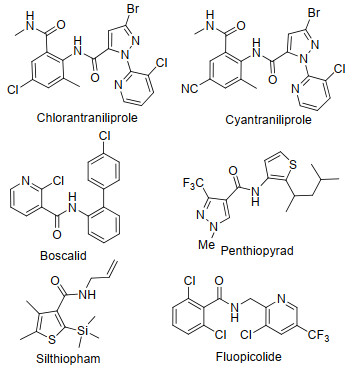
芳甲酰胺类衍生物是具有广泛生物活性的一类化合物, 多年来备受关注.苯甲酰衍生物在医药领域发挥着重要的作用, 取代的苯甲酰胺在精神分裂症抑制[1]、癌细胞的控制[2-3]、对感染HIV-1潜伏期细胞的清除[4]等方面发挥良好活性.一些结构的苯甲酰胺作为鱼尼丁受体(RyR)抑制剂具有优良的杀虫活性[5-8], 已经商品化的有氯虫苯甲酰胺(Chlorantraniliprole)[9]、溴氰虫酰胺(Cyantraniliprole)[10]等(图 1).在除草剂领域, 苯甲酰胺衍生物可以抑制乙酰乳酸合成酶(AHAS), 从而发挥除草活性[11].近年来, 芳甲酰胺类衍生物在杀菌剂领域的研究也比较活跃, 一系列的芳甲酰胺化合物被开发为杀菌剂[12], 如商品化的品种有啶酰菌胺(Boscalid)[13]、氟啶酰菌胺(Fluopicolide)[14]、吡噻菌胺(Penthiopyrad)[15]、硅噻菌胺(Silthiopham)[16]等(图 1).
小麦全蚀病是危害小麦的一种顽固土传病害[17], 大多数杀菌剂对其防治效果不够理想, 在目前的杀菌剂品种中, 硅噻菌胺是少数能够有效防治小麦全蚀病的芳甲酰胺类杀菌剂, 但品种单一.为了解决小麦全蚀病防治品种单一的问题, 并结合芳甲酰胺衍生物在不同生物学领域良好的生物活性表现, 课题组近来也开展了芳甲酰胺类化合物的合成与活性研究[18-19], 发现具有不同结构的噻吩甲酰胺衍生物对小麦全蚀病具有优异的抑制活性, 然而有关取代的苯甲酰胺衍生物对防治小麦全蚀病的活性研究还涉及得较少; 作为该工作的进一步延伸, 本文拟设计合成一系列取代的苯甲酰胺衍生物, 研究它们的结构与活性关系, 以期获得对小麦全蚀病具有抑制活性的新先导化合物.
另一方面, 1, 2, 4-三唑官能团在三唑类杀菌剂中扮演着活性位点的角色[20], 1, 2, 4-三唑引入到芳甲酰胺的结构中可能衍生出一类新的杀菌活性化合物, 为此, 设计了具有较新颖结构的1, 2, 4-三唑苯甲酰胺衍生物, 如图 2所示.
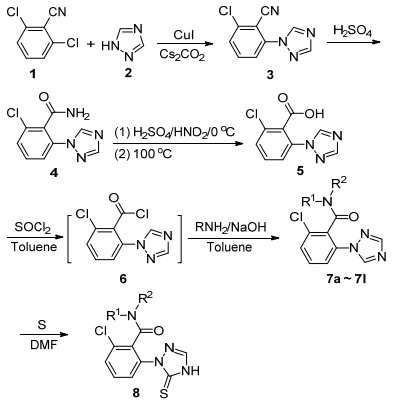
为了获得这些化合物, 本工作组设计了如Scheme 1所示的合成路线, 并通过该路线顺利地合成了14个未见文献报道的1, 2, 4-三唑苯甲酰胺衍生物, 分别考察了它们对小麦全蚀病病原菌(Gaeumannomyces graminis var. tritici)和小麦根腐病病原菌(Fusarium pseudocerea- lum)的抑制活性, 初步分析了所合成化合物的结构活性关系.
4 R1=H, R2=H; 7a R1=H, R2=allyl; 7b R1=H, R2=isopropyl; 7c R1=H, R2=cyclopropylmethyl; 7d R1=H, R2=2-chlorbenzyl; 7e R1=H, R2=4-chlorobenzyl; 7f R1=H, R2=4-methoxybenzyl; 7g R1=Et, R2=Et; 7h R1=H, R2=Et; 7i R1=H, R2=benzene; 7j R1=H, R2=2-chlorobenzene; 7k R1=H, R2=2-fluorobenzene; 7l R1=H, R2=2-cyanobenzene; 8 R1=H, R2=2-chlorobenzyl
在目标化合物的合成过程中, 苯环上三唑官能团的引入具有一定的挑战, 同时涉及到原料的选取.由于苯环1, 2, 4-三唑的引入通常可以通过碳-氮键的催化偶联来实现[21], 因此目标化合物的逆合成分析如Scheme 2所示.
首先尝试以容易获得的2, 6-二氯苯甲酸为原料, 考察通过催化偶联引入1, 2, 4-三唑的可能性, 然而在选定的条件下[21], 几乎没有目标产物生成.考虑到羧基对反应的可能影响, 考察了以2, 6-二氯苯甲酸酯为原料来实现偶联的可能性, 尝试了醋酸钯、三苯基膦钯[22]、碘化亚铜、溴化亚铜[21]等与不同的碱组合的催化体系, 也没有获得理想的催化偶联效果, 却得到了脱羰基化产物.可能是2, 6-二氯苯甲酸酯的碳-氯键与1, 2, 4-三唑的偶联活性低[23], 酯基又不能承受选定的反应条件等.拟通过改变底物提高其反应活性以促进反应的进行, 选择了以2, 6-二氯苯腈为原料的合成路线, 取得了进展.针对该合成方法进行了催化剂选择和反应条件优化, 结果如表 1所示.
 下载:
导出CSV
下载:
导出CSV
 | ||||||
| Entry | Promoter | Catalysis | T/℃ | Time/h | 3:3'b | Yield/%c |
| 1 | K2CO3 | — | 110 | 20 | 1:1 | 40 |
| 2 | Cs2CO3 | — | 110 | 20 | 1:1.6 | 30 |
| 3 | Cs2CO3 | CuI/Md | 110 | 12 | 3.4:1 | 63 |
| 4 | Cs2CO3 | CuI/Md | 85 | 12 | 3.5:1 | 65 |
| 5 | Cs2CO3 | CuI /Md | 65 | 12 | 3.2:1 | 52 |
| 6 | Cs2CO3 | CuBr/Md | 85 | 12 | 3:1 | 60 |
| a 1 equiv. of 2, 6-dichlorobenzonitrile, 1.3 equiv. of 1, 2, 4-triazole and alkali carbonates were used in 3 mL of N, N-dimethylformamide (DMF). bThe molar ratio of 3:3' was measured by HPLC. c Isolated yields of 3 based on 2, 6-dichlorobenzonitrile. d 0.05 equiv. of copper salt and M (8-hydroxyquino- line-N-oxide). | ||||||
2, 6-二氯苯甲腈与1, 2, 4-三唑偶联表现出了相对较高的反应活性, 在以N, N-二甲基甲酰胺为溶剂, 碳酸钾为碱, 在110 ℃的条件下反应20 h, 原料大部分发生转化, 遗憾的是反应的选择性较差, 1, 2, 4-三唑单取代物3和二取代产物3'同时生成, 生成量几乎相等, 目标产物只得到40%的收率(Entry 1).以碳酸铯为碱时, 1, 2, 4-三唑二取代产物3'占优(Entry 2).为了提高反应的选择性, 尝试了在铜盐及促进剂存在下的碳-氮偶联反应[24].结果发现, 以Cs2CO3为碱、CuI/8-羟基喹啉氮氧化物为催化剂的反应体系, 在110 ℃反应12 h, 原料基本完全转化, 反应的选择性具有明显改善, 1, 2, 4-三唑单取代物3和二取代产物3'的比率提高到3.4︰1, 目标产物的收率达到中等水平(63%, Entry 3).为了进一步提高选择性, 把温度降低到85 ℃, 反应亦可顺利进行, 收率稍有改善, 达到65%的水平(Entry 4).进一步将反应温度降至65 ℃, 反应转化不完全.变换催化剂的种类为溴化亚铜, 反应收率及选择性在所选条件下没有进一步改善(Entry 6).较高的1, 2, 4-三唑和碱的用量, 会降低反应的选择性, 而较低的1, 2, 4-三唑和碱的用量导致2, 6-二氯苯腈转化率降低.归纳比较优惠的反应条件为:以N, N-二甲基甲酰胺(DMF)为溶剂, 1.3 equiv. Cs2CO3为碱, 0.05 equiv. CuI/8-羟基喹啉氮氧化物为催化剂, 在85 ℃反应12 h.
首先尝试在碱性条件下直接将氰基水解为酸的方法[25], 然而, 氢氧化钠、氢氧化钾和甲醇钠等在回流条件下都未能较好地实现氰基向羧酸的转化.转而考察在硫酸存在下将氰基水解为酸的方法[26], 遗憾的是反应仅停留在中间体4的步骤.有文献报道, 酰胺可以在NaNO2/H2SO4的存在下平稳地水解为羧酸[27], 于是采取两步法来实现氰基向羧酸的转化, 取得了进展.首先中间体3在浓度为80%的硫酸水溶液中在100 ℃反应10 h, 实现了氰基向酰胺的完全转化得到中间体4.然后, 在低温下(0 ℃左右)向含有中间体4的80%硫酸水溶液中加入5 equiv.亚硝酸钠水溶液后反应1 h, 使酰胺氮原子发生重氮化, 接着再在100 ℃反应2 h, 以84%的收率得到中间体2-氯-6-(1, 2, 4-三唑-1-基)苯甲酸(5).
目标化合物7a~7l的合成参照文献[19]进行.首先, 合成的2-氯-6-(1, 2, 4-三唑-1-基)苯甲酸(5)在氯化亚砜的存在下, 转化为相应的2-氯-6-(1, 2, 4-三唑-1-基)苯甲酰氯(6), 苯甲酰氯6不需分离直接在缚酸剂氢氧化钠的存在下, 分别与各种取代的胺缩合得到2-氯-6-(1, 2, 4-三唑-1-基)苯甲酰胺衍生物7a~7l.由于苯甲酰氯6在各种取代胺的存在下, 对10%氢氧化钠水溶液不敏感, 使用低浓度的氢氧化钠水溶液作为缚酸剂, 反应的后处理更加容易.化合物8则由化合物7d在硫磺的存在下在N, N-二甲基甲酰胺中通过加热硫化而得[28].
目标化合物的表征主要关注1, 2, 4-三唑官能团在结构中是否存在, 由于1, 2, 4-三唑官能团的两个质子和两个碳原子分别在1H NMR和13C NMR中以比较特征的信号在低场出现, 因此, 在目标化合物中1, 2, 4-三唑官能团都清晰可辨, 其它质子和碳原子信号(除7k外)也都与结构完全一致.至于化合物7k的结构表征, 高分辨质谱[M+H]+对应的质荷比为317.0603, 与理论值317.0605符合得很好.其1H NMR化学位移在δ 10.51处的质子信号对应酰胺氮原子的质子, 而δ 8.90和8.21处的质子信号分别对应三唑官能团的两个质子; 化学位移δ在7.73~7.69和7.26~7.19处的两组质子信号对应两个苯环的7个质子, 质子信号与结构也符合得较好.由于氟原子的存在, 化合物7k的13C NMR谱中存在着F-C偶合现象, 氟原子与含氟苯环的6个碳原子分别发生偶合, 致使含氟苯环的每个碳原子都显示两个信号, 并呈现出不同的偶合常数, 如δ 156.0和153.5处的碳信号, 对应与氟直接相连的碳原子, 偶合常数为245.5 Hz; δ 126.9和126.8处的碳信号, 对应氟原子邻位与氮原子相连的碳原子, 偶合常数为7.4 Hz.谱图呈现的这种偶合裂分现象表明氟原子的存在, 也体现了谱图与结构的一致性.
为了评估三唑苯甲酰胺衍生物的结构活性关系, 采用平皿菌丝生长速率法[29]测定了所合成新化合物4和7a~7l对小麦全蚀病病原菌(Gaeumannomyces graminis var. tritici)的抑制活性, 测试结果如表 2所示.起初, 噻吩甲酰胺氮原子上的取代基为烯丙基和烷基时, 表现出较高活性的结果[18], 将烯丙基和具有代表性的含有2~4个碳的简单烷基引入到三唑苯甲酰胺的酰胺氮原子上, 但其活性表现较低(7a~7c, 7g~7h), 在100 mg/L的浓度下, 其抑制率最高仅为30%左右.当酰胺氮原子上的取代基变成取代的苯甲基时(7d~7f), 其活性总体上具有增加的趋势.其中7f在100 mg/L的浓度下, 对小麦全蚀病病原菌的抑制率超过了50%.这进一步支持优化酰胺氮原子上的取代基, 当酰胺氮原子上的取代基变为苯基时, 其活性有明显提高, 化合物7i在100 mg/L的浓度下, 对小麦全蚀病的供试病原菌抑制率达到了80%;在50和25 mg/L的浓度下, 其抑制率分别为63.7%和43.6%, 接近对照硅噻菌胺的水平.于是利用现有的取代苯胺又合成了7j~7l, 活性测试结果表明, 它们也表现出了相对较高的活性, 在100 mg/L的浓度下分别达到61.7%, 69.7%和65.4%的抑制率.作为对比, 酰胺氮原子上无取代基时, 如化合物4, 其活性不及取代基为苯的活性.总体分析, 所合成三唑苯甲酰胺衍生物对小麦全蚀病病原菌均具有一定的抑制活性, 当酰胺氮原子上无取代基或为烷基单取代、双取代时, 均不利于活性的产生, 酰胺氮原子上的取代基为各种取代的苯甲基时, 活性增加, 尤以取代的苯基时活性更好, 值得进一步的优化研究.
下载:
导出CSV
| Compd. | Gaeumannomyces graminis var. tritici | Fusarium pseudocerealum | |||||||
| 100 mg/L | 50 mg/L | 25 mg/L | 100 mg/L | 50 mg/L | 25mg/L | ||||
| 4 | 46.0 | 20.2 | 16.0 | 10.8 | 7.7 | 4.6 | |||
| 7a | 19.6 | 14.2 | 5.8 | 4.8 | 3.9 | 1.7 | |||
| 7b | 12.5 | 9.6 | 7.5 | 12.1 | 6.1 | 5.6 | |||
| 7c | 10.4 | 5.8 | 3.8 | 27.5 | 16.7 | 6.9 | |||
| 7d | 44.7 | 21.1 | 13.2 | 6.2 | 4.4 | 3.4 | |||
| 7e | 36.4 | 34.2 | 12.4 | 10.4 | 7.7 | 5.0 | |||
| 7f | 50.9 | 21.9 | 12.3 | 21.9 | 16.4 | 10.1 | |||
| 7g | 30.3 | 25.4 | 17.5 | 33.8 | 30.9 | 29.4 | |||
| 7h | 32.1 | 17.1 | 7.3 | 20.6 | 19.9 | 17.5 | |||
| 7i | 80.0 | 63.7 | 43.6 | 12.8 | 9.4 | 7.3 | |||
| 7j | 61.7 | 34.7 | 15.3 | 39.6 | 30.2 | 17.6 | |||
| 7k | 69.7 | 53.3 | 32.0 | 31.6 | 24.4 | 16.0 | |||
| 7l | 65.4 | 50.4 | 25.8 | 44.6 | 34.2 | 22.9 | |||
| 8 | — | — | — | 28.1 | 24.6 | 21.9 | |||
| Silthiopham | 92.3 | 66.7 | 40.5 | — | — | — | |||
| Prothioconazole | 100 | 97.3 | 94.1 | 100 | 100 | 100 | |||
| a “—” No detection. | |||||||||
为了进一步了解1, 2, 4-三唑苯甲酰胺衍生物的活性范围, 本工作还测试了它们对小麦根腐病病原菌假禾谷镰孢菌(Fusarium pseudocerealum)的抑制活性, 令人遗憾的是, 所合成的1, 2, 4-三唑苯甲酰胺衍生物, 酰胺氮原子上的取代基无论是烷基还是芳基, 在设计浓度(100, 50, 25 mg/L)下虽然都表现出了一定的活性, 但活性都较低, 只有7l表现出了相对较好的活性, 在100 mg/L的浓度下给出大约45%的抑制率.由于三唑硫酮衍生物[30]对假禾谷镰孢菌(Fusarium pseudocerealum)具有优异的抑制活性, 本工作又合成了三唑硫酮取代的苯甲酰胺衍生物8, 但该化合物没有表现出期待的活性.根据目前三唑苯甲酰胺衍生物结构与活性的表现, 进一步的结构优化是值得尝试的, 相关工作在进行中.
设计了以2, 6-二氯苯腈和1, 2, 4-三唑为原料合成1, 2, 4-三唑苯甲酰胺衍生物的工艺方法, 优化了该方法中2, 6-二氯苯腈和1, 2, 4-三唑催化偶联的反应条件, 提高了反应的选择性.开发了中间体1, 2, 4-三唑取代的苯腈两步法水解为羧酸的方法, 顺利地获得了中间体1, 2, 4-三唑取代的苯甲酸.通过1, 2, 4-三唑取代的苯甲酸的酰化与胺化, 合成了14个三唑苯甲酰胺衍生物.活性测试表明, 除化合物8外, 大部分化合物对供试小麦全蚀病病原菌都表现出一定的抑制活性, 尤以化合物7i表现最好, 在100 mg/L的浓度下, 抑制率达到了80%.在50和25 mg/L的浓度下, 抑制率接近对照硅噻菌胺的水平.初步结构活性关系分析表明, 所合成1, 2, 4-三唑苯甲酰胺衍生物酰胺氮原子芳基取代的活性优于酰胺氮原子烷基取代的活性, 这为活性化合物结构的进一步设计提供了一定的参考.
反应使用HPLC (Agilent-1400)检测和分析; 1H NMR、13C NMR用核磁共振仪(Agilent-NMR-vnmrs 400)测试, 以CDCl3或DMSO-d6为溶剂, TMS为内标; 质谱用Bruker micrOTOF-QII质谱仪测试; 熔点用WRS-1A数字熔点仪测试; 病原菌使用HPG-280H人工气候箱培养.所有国产或进口AR或CP级试剂未经纯化直接使用.
供试的小麦全蚀病病原菌(Gaeumannomyces graminis var. tritici)由南京农业大学植物保护学院实验室提供; 小麦根腐病病原菌假禾谷镰孢菌(Fusarium pseudocerealum)由河南农业大学植物保护学院病理实验室提供.
将0.17 g (1 mmol) 2, 6-二氯苯甲腈, 0.09 g (1.3 mmol)工业级1, 2, 4-三唑, 0.43 g (1.3 mmol)无水碳酸铯和9.5 mg (0.05 mmol) CuI及8.1 mg (0.05 mmol) 8-羟基喹啉-N-氧化物配体加入到20 mL的小反应瓶中, 然后量取3 mL的N, N-二甲基甲酰胺加入到反应瓶中, 在油浴中加热到85 ℃搅拌反应12 h.薄层色谱(TLC)检测反应完成后, 负压滤除碳酸钾以及未反应的1, 2, 4-三唑, 滤液水洗萃取后浓缩, 用薄层色谱(氯仿/乙醇, V:V=100:1)分离得到2-氯-6-(1H-1, 2, 4-三唑-1-基)苯甲腈(3) 133 mg, 收率79%.亚白色固体, m.p. 170~172 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.79 (s, 1H), 8.20 (s, 1H), 7.74~7.69 (m, 2H), 7.64~7.62 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 153.3, 143.3, 140.3, 139.2, 134.5, 129.8, 123.1, 113.3, 107.7. HRMS (ESI) calcd for C9H6ClN4 [M+H]+ 205.0281, found 205.0275.
2, 6-二(1H-1, 2, 4-三唑-1-基)苯甲腈(3'):亚白色固体, m.p. 245~246 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.81 (s, 2H), 8.24 (s, 2H), 7.97~7.93 (m, 1H), 7.87~7.85 (m, 2H); 13C NMR (100 MHz, CDCl3) δ: 153.5, 143.5, 140.5, 135.2, 125.1, 113.1. HRMS (ESI) calcd for C11H8N7 [M+H]+ 238.0841, found 238.0833.
将2-氯-6-(1H-1, 2, 4-三唑-1-基)苯甲腈6.1 g (30 mmol)加入到三口烧瓶中, 然后加入30 g 80%的硫酸(0.24 mol)水溶液, 加热到100 ℃回流反应10 h, 反应呈暗红色均相溶液, TLC检测反应完全, 加水调节pH至中性, 用乙酸乙酯萃取干燥, 负压蒸出溶剂得到粗品为灰色固体, 经硅胶柱色谱(氯仿/乙醇, V:V=100:1)分离得到纯品2-氯-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(4) 5.3 g, 收率79%.灰色固体, m.p. 128~130 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.52 (s, 1H), 8.11 (s, 1H), 7.58~7.56 (m, 1H), 7.53~7.49 (m, 2H), 5.86 (brs, 2H); 13C NMR (100 MHz, CDCl3) δ: 165.7, 152.7, 144.2, 135.2, 132.1, 131.5, 131.1, 130.5, 124.0. HRMS (ESI) calcd for C9H8ClN4O [M+H]+ 223.0387, found 223.0383.
将14.3 g (0.064 mol) 2-氯-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺加入到100 mL的三口反应瓶中, 将三口瓶置于冰浴中, 在0~10 ℃范围内, 慢慢加入79 g 80%的硫酸(0.64 mol)水溶液, 滴完后开始滴加已降温至0~10 ℃的22.1 g (0.32 mol)亚硝酸钠配制成的水溶液, 使反应始终维持在0~10 ℃之间, 滴加完毕后保温反应1 h后.然后再在100 ℃下反应2 h, TLC检测反应完全为止.反应液用乙酸乙酯萃取、干燥、负压蒸出溶剂后得到淡黄色2-氯-6-(1H-1, 2, 4-三唑-1-基)苯甲酸固体12 g, 收率84%.黄色固体, m.p. 122~124 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.03 (s, 1H), 8.26 (s, 1H), 7.74~7.64 (m, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 165.7, 152.9, 144.9, 135.0, 131.7, 131.2, 130.4, 130.3, 123.6. HRMS (ESI) calcd for C9H7ClN3O2 [M+H]+ 224.0227, found 224.0221.
将1.4 g (6.3 mmol) 2-氯-6-(1H-1, 2, 4-三唑-1-基)苯甲酸投入到盛有20 mL甲苯的带有回流和气体吸收装置的三口反应瓶中, 在80 ℃条件下缓慢加入1.5 g (12.6 mmol)的氯化亚砜, 加完后继续反应2 h.负压脱出溶剂和过量的氯化亚砜后, 向反应瓶中补加10 mL新鲜甲苯, 接着在45 ℃向反应瓶中滴加0.5 g (8.7 mmol)烯丙胺, 然后向反应液中加入3 g (7.5 mmol) 10%氢氧化钠水溶液, 再在85 ℃温度下反应2 h, 降温静置分层, 有机相用温水洗涤(5 mL×3), 干燥后过滤, 滤液负压蒸出溶剂, 用硅胶柱色谱(氯仿/乙醇, V:V=100:1)分离得到浅黄色固体.其它酰胺化合物按相似的方法合成与分离.
N-烯丙基-2-氯-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7a):收率60%.黄色固体, m.p. 130~131 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.44 (s, 1H), 8.02 (s, 1H), 7.48~7.42 (m, 3H), 5.79 (brs, 1H), 5.72~5.62 (m, 1H), 5.09~5.05 (m, 2H), 3.89~3.86 (t, J=6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 162.9, 151.6, 143.3, 134.4, 131.8, 131.4, 131.1, 129.9, 129.4, 123.0, 116.4, 41.3. HRMS (ESI) calcd for C12H12ClN4O [M+H]+ 263.0700, found 263.0696.
2-氯-N-异丙基-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7b):收率60%.白色固体, m.p. 162~164 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.53 (s, 1H), 8.09 (s, 1H), 7.55~7.48 (m, 3H), 5.61 (brs, 1H), 4.19~4.13 (m, 1H), 1.11~1.09 (d, J=8.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 163.1, 152.6, 144.3, 135.3, 132.5, 132.4, 130.8, 130.4, 124.0, 42.3, 22.3. HRMS (ESI) calcd for C12H14ClN4O [M+H]+ 265.0856, found 265.0850.
2-氯-N-(环丙基甲基)-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7c):收率55%.浅灰色固体, m.p. 128~129 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.53 (s, 1H), 8.08 (s, 1H), 7.56~7.49 (m, 3H), 5.93 (brs, 1H), 3.19~3.16 (m, 2H), 0.88~0.84 (m, 1H), 0.49~0.46 (m, 2H), 0.16~0.15 (d, J=4.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 163.9, 152.6, 144.3, 135.4, 132.4, 132.2, 130.8, 130.4, 124.0, 45.0, 10.3, 3.5. HRMS (ESI) calcd for C13H14ClN4O [M+H]+ 277.0856, found 277.0852.
2-氯-N-(2-氯苄基)-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7d):收率64%.浅黄色固体, m.p. 155~157 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.33 (s, 1H), 7.83 (s, 1H), 7.49~7.28 (m, 4H), 7.25~7.17 (m, H), 6.97~6.94 (t, J=5.6 Hz, 1H), 4.54~4.53 (d, J=6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 164.1, 152.3, 144.0, 135.2, 134.3, 133.5, 132.5, 131.9, 130.8, 130.3, 129.5, 129.2, 127.1, 123.7, 41.86. HRMS (ESI) calcd for C16H13Cl2N4O [M+H]+ 347.0466, found 347.0462.
2-氯-N-(4-氯苄基)-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7e):收率58%.灰白色固体, m.p. 143~145 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.35 (s, 1H), 7.85 (s, 1H), 7.48~7.39 (m, 3H), 7.32~7.22 (m, 4H), 6.83~.80 (t, J=5.8 Hz, 1H), 4.56~4.54 (d, J=6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 164.0, 152.4, 144.2, 135.5, 135.2, 133.5, 132.4, 132.0, 130.9, 130.4, 129.2, 128.8, 123.8, 43.2. HRMS (ESI) calcd for C16H12Cl2N4O [M+H]+ 347.0466, found 347.0458.
2-氯-N-(4-甲氧基苄基)-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7f):收率68%.浅黄色固体, m.p. 152~154 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.51 (s, 1H), 8.01 (s, 1H), 7.53~7.44 (m, 3H), 7.10~7.08 (d, J=8.4 Hz, 2H), 6.84~6.81 (d, J=8.8 Hz, 2H), 6.15 (brs, 1H), 4.43~4.42 (d, J=5.6 Hz, 2H), 3.79 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.8, 159.2, 152.6, 144.2, 135.4, 132.5, 132.1, 130.9, 130.4, 129.4, 128.8, 123.9, 114.1, 55.3, 43.6. HRMS (ESI) calcd for C17H16ClN4O2 [M+H]+ 343.0962, found 343.0954.
2-氯-N, N-二乙基-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7g):收率56%.黄色固体, m.p. 135~137 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.60 (s, 1H), 8.07 (s, 1H), 7.54~7.47 (m, 3H), 3.50~3.43 (m, 2H), 3.09~2.93 (m, 2H), 1.13~1.10 (t, J=6.8 Hz, 3H), 0.89~0.85 (t, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 164.3, 152.5, 144.3, 134.8, 131.8, 131.5, 130.3, 130.1, 123.8, 42.9, 39.1, 13.3, 12.2. HRMS (ESI) calcd for C13H16ClN4O [M+H]+ 279.1013, found 279.1009.
2-氯-N-乙基-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7h):收率45%.黄色固体, m.p. 128~130 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.52 (s, 1H), 8.08 (s, 1H), 7.54~7.48 (m, 3H), 5.89 (brs, 1H), 3.37~3.34 (m, 2H), 1.10~1.06 (t, J=7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.9, 152.6, 144.3, 135.3, 132.4, 132.3, 130.8, 130.4, 123.9, 35.0, 14.4. HRMS (ESI) calcd for C11H12ClN4O [M+H]+ 251.0700, found 251.0692.
2-氯-N-苯基-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7i):收率63%.黄色固体, m.p. 140~143 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.53 (s, 1H), 8.04 (s, 1H), 7.80 (s, 1H), 7.56~7.45 (m, 5H), 7.36~7.32 (m, 2H), 7.19~7.16 (dd, J=7.2, 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 162.1, 152.7, 144.2, 136.7, 135.4, 132.7, 132.1, 131.2, 130.6, 129.2, 125.5, 123.8, 120.6. HRMS (ESI) calcd for C15H12ClN4O [M+H]+ 299.0700, found 299.0694.
2-氯-N-(2-氯苯基)-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7j):收率73%.淡黄色固体, m.p. 125~127 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 10.38 (s, 1H), 8.91 (s, 1H), 8.25 (s, 1H), 7.76~7.67 (m, 3H), 7.56~7.48 (m, 2H), 7.38~7.34 (m, 1H), 7.27~7.23 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 162.8, 152.8, 145.2, 135.7, 134.4, 132.3, 132.0, 131.7, 130.3, 130.1, 128.0, 127.9, 127.7, 127.5, 123.9. HRMS (ESI) calcd for C15H11Cl2N4O [M+H]+ 333.0310, found 333.0302.
2-氯-N-(2-氟苯基)-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7k):收率66%.淡黄色固体, m.p. 169~171 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 10.51 (s, 1H), 8.90 (s, 1H), 8.21 (s, 1H), 7.73~7.69 (m, 4H), 7.26~7.19 (m, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 162.7, 154.7 (d, J=245.5 Hz), 152.8, 145.1, 135.7, 132.3, 132.0, 131.7, 130.3, 126.9 (d, J=7.4 Hz), 125.5 (d, J=5.9 Hz), 125.4 (d, J=6.2 Hz), 124.8 (d, J=3.6 Hz), 123.9, 116.2 (d, J=19.0 Hz). HRMS (ESI) calcd for C15H11ClFN4O [M+H]+ 317.0605, found 317.0603.
2-氯-N-(2-氰苯基)-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7l):收率60%.亚白色固体, 熔点179~181 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 10.93 (s, 1H), 8.97 (s, 1H), 8.22 (s, 1H), 7.84~7.71 (m, 5H), 7.57~7.55 (d, J=7.6 Hz, 1H), 7.42~7.38 (m, 1H); 13C NMR (100 MHz, DMSO-d6) δ: 163.0, 152.9, 145.1, 139.7, 135.7, 134.3, 134.0, 132.2, 131.9, 131.6, 130.3, 126.8, 126.1, 123.5, 116.9, 107.6. HRMS (ESI) calcd for C16H11ClN5O [M+ H]+ 324.0652, found 324.0644.
取0.24 g (0.7 mmol) 2-氯-N-(2-氯苄基)-6-(1H-1, 2, 4-三唑-1-基)苯甲酰胺(7d)加入到三烧瓶中, 加入10 mL N, N-二甲基甲酰胺(DMF)到反应瓶中, 然后加入0.23 g (7 mmol)硫粉在160 ℃回流反应12 h, 在反应过程中, 随着温度升高, 硫粉逐渐溶解, 溶液颜色加深.通过TLC检测直至反应完全.冷却负压过滤未反应的硫粉, 然后用水洗, 乙酸乙酯萃取, 有机相用无水硫酸钠干燥后浓缩, 残余物用薄层制备色谱[氯仿/甲醇, V:V=90:1]分离得到8 0.11 g, 收率41%.浅黄色固体, m.p. 76~77 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.68 (s, 1H), 7.57~7.56 (m, 1H), 7.49~7.48 (m, 3H), 7.33~7.31 (m, 1H), 7.20~7.18 (m, 2H), 7.05 (brs, 1H), 4.66~4.65 (d, J=6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 167.1, 164.0, 139.2, 135.4, 134.7, 134.4, 133.7, 132.5, 131.5, 130.8, 130.7, 129.4, 129.2, 127.2, 127.1, 41.9. HRMS (ESI) calcd for C16H13Cl2N4OS [M+H]+ 379.0187, found 379.0181.
合成的1, 2, 4-三唑苯甲酰胺衍生物的生物活性采用菌丝生长速率法测试[29].首先, 称取200 g去皮后的新鲜土豆, 用1 L蒸馏水加热煮沸20 min, 用纱布滤出未煮烂的土豆, 向滤液中加入葡萄糖20 g和琼脂15 g, 充分搅拌待其完全溶解后, 用蒸馏水定容到1 L.接着在120 ℃下湿热灭菌20 min, 冷却制得马铃薯葡萄糖琼脂(PDA)培养基.
然后, 以二甲亚砜(DMSO)为溶剂配制适当浓度的供试样品, 并加入适当数量的表面活性剂吐温备用.移取适当浓度的1 mL供试样品溶液, 和熔化并冷却至50 ℃的9 mL PDA培养基充分混合, 然后倒入无菌培养皿中冷却至室温.把在PDA平板上培养7 d的小麦全蚀病病原菌用直径5 mm的打孔器制作成菌饼, 然后将菌饼以倒扣的方式接种在含药的PDA平板中央, 以仅含DMSO的平板培养基为对照.每个药液浓度设4个重复, 在20~25 ℃培养箱中避光培养5 d, 调查菌落直径, 计算相对抑制率[相对抑制率(%)=(空白对照菌落直径-药剂处理菌落直径)/空白对照菌落直径×100%].假禾谷镰孢菌活性测试采用相似的方法.
辅助材料(Supporting Information) 所涉及目标化合物及中间体的1H NMR和13C NMR数据.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Xu, M. S.; Guo, S.; Yang, F. P.; Wang, Y.; Wu, C. H.; Jiang, X. R.; Zhao, Q. J.; Chen, W. M.; Tian, G. H.; Zhu, F. Q.; Xie, Y. C.; Hu, T. W.; Wang, Z.; He, Y.; Shen, J. S. Arch. Pharm. Chem. Life Sci. 2019, 352, e1800306. doi: 10.1002/ardp.201800306
Lu, A.; Luo, H.; Shi, M.; Wu, G.; Yuan, Y.; Liu, J.; Tang, F. Bioorg. Med. Chem. Lett. 2011, 21, 4924. doi: 10.1016/j.bmcl.2011.06.001
Zang, J.; Liang, X. W.; Huang, Y. X.; Jia, Y. P.; Li, X. Y.; Xu, W. F.; Chou, C. J.; Zhang, Y. J. J. Med. Chem. 2018, 61, 5304. doi: 10.1021/acs.jmedchem.8b00384
Huang, L.; Lai, W. H.; Zhu, L.; Li, W.; Wei, L.; Lee, K. H.; Xie, L.; Chen, C. H. ACS Med. Chem. Lett. 2018, 9, 268. doi: 10.1021/acsmedchemlett.8b00012
史建俊, 任贵华, 吴宁捷, 刘幸海, 许天明, 谭成侠, 有机化学, 2017, 37, 2131. doi: 10.6023/cjoc201701019Shi, J.-J.; Ren, G.-H.; Wu, N.-J.; Liu, X.-H.; Xu, T.-M.; Tan, C.-X. Chin. J. Org. Chem. 2017, 37, 2131(in Chinese). doi: 10.6023/cjoc201701019
Zhang, J. F.; Xu, J. Y.; Wang, B. L.; Li, Y. X.; Xiong, L. X.; Li, Y. Q.; Ma, Y.; Li, Z. M. J. Agric. Food Chem. 2012, 60, 7565. doi: 10.1021/jf302446c
Chen, K.; Liu, Q.; Ni, P. J.; Zhu, H. J.; Lia, Y. F.; Wang, Q. Pest Manage. Sci. 2015, 71, 1503. doi: 10.1002/ps.3954
王梦梦, 张青青, 岳凯, 李庆山, 徐凤波, 有机化学, 2017, 37, 1774. doi: 10.6023/cjoc201612030Wang, M.-M.; Zhang, Q.-Q.; Yue, K.; Li, Q.-S.; Xu, F.-B. Chin. J. Org. Chem. 2017, 37, 1774(in Chinese). doi: 10.6023/cjoc201612030
Lahm, G. P.; Stevenson, T. M.; Selby, T. P.; Freudenberger, J. H.; Cordova, D.; Flexner, L.; Bellin, C. A.; Dubas, C. M.; Smith, B. K.; Hughes, K. A.; Hollingshaus, J. G.; Clark, C. E.; Benner, E. A. Bioorg. Med. Chem. Lett. 2007, 17, 6274. doi: 10.1016/j.bmcl.2007.09.012
Hughes, K. A.; Lahm, G. P.; Selby, T. P.; Stevenson, T. M. WO 2004067528, 2004[Chem. Abstr. 2004, 141, 190786].
李文明, 王建国, 李永红, 王素华, 李正名, 高等学校化学学报, 2010, 31, 1574.Li, W.-M; Wang, J.-G.; Li, Y.-H.; Wang, S.-H.; Li, Z.-M. Chem. J. Chin. Univ. 2010, 31, 1574(in Chinese).
杨吉春, 张金波, 柴宝山, 刘长令, 农药, 2008, 47, 6. doi: 10.3969/j.issn.1006-0413.2008.01.002Yang, J.-C.; Zhang, J.-B.; Chai, B.-S.; Liu, C.-L. Agrochemicals 2008, 47, 6(in Chinese). doi: 10.3969/j.issn.1006-0413.2008.01.002
Eicken, K.; Goetz, N.; Harreus, A.; Ammermann, E.; Lorenz, G.; Rang, H. EP 545099, 1993[Chem. Abstr. 1993, 119, 160132].
Moradi, W. A.; Schlegel, G.; Schnatterer, A.; Volz, F. WO 2016173998, 2016[Chem. Abstr. 2016, 165, 571164].
Yoshikawa, Y.; Tomitani, K.; Katsuta, H.; Kawashima, H.; Takahashi, T.; Inami, S.; Yanase, Y.; Takashi, A.; Shimotori, H.; Tomura, N. JP 09301974, 1997[Chem. Abstr. 1997, 128, 22908].
Phillion, D.; Wong, S. C.; Shortt, B. US 5486621, 1996[Chem. Abstr. 1996, 124, 253325].
Freeman, J.; Ward, E. Mol. Plant Pathol. 2004, 5, 235. doi: 10.1111/j.1364-3703.2004.00226.x
谢桂英, 靳文波, 赵艳芹, 程绎南, 孙炳剑, 孙淑君, 汪梅子, 位丹丹, 李洪连, 有机化学, 2014, 34, 1124. doi: 10.6023/cjoc201401036Xie, G.-Y.; Jin, W.-B.; Zhao, Y.-Q.; Cheng, Y.-N.; Sun, B.-J.; Sun, S.-J.; Wang, M.-Z.; Wei, D.-D.; Li, H.-L. Chin. J. Org. Chem. 2014, 34, 1124(in Chinese). doi: 10.6023/cjoc201401036
靳文波, 谢桂英, 孙淑君, 赵艳芹, 程绎南, 孙炳剑, 李洪连, 有机化学, 2014, 34, 2376. doi: 10.6023/cjoc201406006Jin, W.-B.; Xie, G.-Y.; Sun, S.-J.; Zhao, Y.-Q.; Cheng, Y.-N.; Sun, B.-J.; Li, H.-L. Chin. J. Org. Chem. 2014, 34, 2376(in Chinese). doi: 10.6023/cjoc201406006
Banerjee, S.; Ganguly, S.; Sen, K. K. J. Adv. Pharm. Educ. Res. 2013, 3, 102.
Antilla, J. C.; Baskin, J. M.; Barder, T. E.; Buchwald, S. L. J. Org. Chem. 2004, 69, 5578. doi: 10.1021/jo049658b
Hosseini-Sarvari, M.; Razmi, Z. RSC Adv. 2014, 4, 44105. doi: 10.1039/C4RA06486K
胡帅帅, 郭海昌, 蒋华江, 郑人华, 应用化学, 2011, 28, 1179. doi: 10.3724/SP.J.1095.2011.00647Hu, S.-S.; Guo, H.-C.; Jiang, H.-J.; Zheng, R.-H. Chin. J. Appl. Chem. 2011, 28, 1179(in Chinese). doi: 10.3724/SP.J.1095.2011.00647
Yang, K.; Qiu, Y. T.; Li, Z.; Wang, Z. Y.; Jiang, S. J. Org. Chem. 2011, 76, 3151. doi: 10.1021/jo1026035
Lee, S. H.; Kim, M. J.; Lee, S. H.; Kim, J.; Park, H. J.; Lee, J. Eur. J. Med. Chem. 2011, 46, 2662. doi: 10.1016/j.ejmech.2011.03.052
Miyamoto, H.; Ueda, H.; Otsuka, T.; Aki, S.; Tamaoka, H.; Tominaga, M.; Nakagawa, K. Chem. Pharm. Bull. 1990, 38, 2472. doi: 10.1248/cpb.38.2472
Tsukamoto, I.; Koshio, H.; Akamatsu, S.; Kuramochi, T.; Saitoh, C.; Yatsu, T.; Yanai-Inamura, H.; Kitada, C.; Yamamoto, E.; Sakamoto, S.; Tsukamoto, S. Bioorg. Med. Chem. 2008, 16, 9524. doi: 10.1016/j.bmc.2008.09.039
Jautelat, M.; Erdman, D. DE 19744706, 1999[Chem. Abstr. 1999, 130, 267441].
Wang, L.; Zhang, Y.; Wang, D.; Wang, M.; Wang, Y.; Feng, J. J. Agric. Food Chem. 2018, 66, 81. doi: 10.1021/acs.jafc.7b03913
Jautelat, M.; Tiemann, R.; Dutzmann, S.; Haensler, G.; Stenzel, K. DE 19528046, 1996[Chem. Abstr. 1996, 125, 114638].

图式 1 目标化合物的合成路线
Scheme 1 Synthetic routes of target compounds
4 R1=H, R2=H; 7a R1=H, R2=allyl; 7b R1=H, R2=isopropyl; 7c R1=H, R2=cyclopropylmethyl; 7d R1=H, R2=2-chlorbenzyl; 7e R1=H, R2=4-chlorobenzyl; 7f R1=H, R2=4-methoxybenzyl; 7g R1=Et, R2=Et; 7h R1=H, R2=Et; 7i R1=H, R2=benzene; 7j R1=H, R2=2-chlorobenzene; 7k R1=H, R2=2-fluorobenzene; 7l R1=H, R2=2-cyanobenzene; 8 R1=H, R2=2-chlorobenzyl
表 1 2-氯-6-(1, 2, 4-三唑-1-基)苯甲腈的合成与条件优化a
Table 1. Synthesis of 2-chloro-6-(1H-1, 2, 4-triazol-1-yl)ben- zonitrile and conditions optimizing
| ||||||
| Entry | Promoter | Catalysis | T/℃ | Time/h | 3:3'b | Yield/%c |
| 1 | K2CO3 | — | 110 | 20 | 1:1 | 40 |
| 2 | Cs2CO3 | — | 110 | 20 | 1:1.6 | 30 |
| 3 | Cs2CO3 | CuI/Md | 110 | 12 | 3.4:1 | 63 |
| 4 | Cs2CO3 | CuI/Md | 85 | 12 | 3.5:1 | 65 |
| 5 | Cs2CO3 | CuI /Md | 65 | 12 | 3.2:1 | 52 |
| 6 | Cs2CO3 | CuBr/Md | 85 | 12 | 3:1 | 60 |
| a 1 equiv. of 2, 6-dichlorobenzonitrile, 1.3 equiv. of 1, 2, 4-triazole and alkali carbonates were used in 3 mL of N, N-dimethylformamide (DMF). bThe molar ratio of 3:3' was measured by HPLC. c Isolated yields of 3 based on 2, 6-dichlorobenzonitrile. d 0.05 equiv. of copper salt and M (8-hydroxyquino- line-N-oxide). | ||||||
 下载: 导出CSV
下载: 导出CSV
表 2 1, 2, 4-三唑苯甲酰胺衍生物的抑菌活性测试(抑制率/%)a
Table 2. Inhibitory activity (Inhibition rate/%) of 1, 2, 4-triazole benzamide derivatives
| Compd. | Gaeumannomyces graminis var. tritici | Fusarium pseudocerealum | |||||||
| 100 mg/L | 50 mg/L | 25 mg/L | 100 mg/L | 50 mg/L | 25mg/L | ||||
| 4 | 46.0 | 20.2 | 16.0 | 10.8 | 7.7 | 4.6 | |||
| 7a | 19.6 | 14.2 | 5.8 | 4.8 | 3.9 | 1.7 | |||
| 7b | 12.5 | 9.6 | 7.5 | 12.1 | 6.1 | 5.6 | |||
| 7c | 10.4 | 5.8 | 3.8 | 27.5 | 16.7 | 6.9 | |||
| 7d | 44.7 | 21.1 | 13.2 | 6.2 | 4.4 | 3.4 | |||
| 7e | 36.4 | 34.2 | 12.4 | 10.4 | 7.7 | 5.0 | |||
| 7f | 50.9 | 21.9 | 12.3 | 21.9 | 16.4 | 10.1 | |||
| 7g | 30.3 | 25.4 | 17.5 | 33.8 | 30.9 | 29.4 | |||
| 7h | 32.1 | 17.1 | 7.3 | 20.6 | 19.9 | 17.5 | |||
| 7i | 80.0 | 63.7 | 43.6 | 12.8 | 9.4 | 7.3 | |||
| 7j | 61.7 | 34.7 | 15.3 | 39.6 | 30.2 | 17.6 | |||
| 7k | 69.7 | 53.3 | 32.0 | 31.6 | 24.4 | 16.0 | |||
| 7l | 65.4 | 50.4 | 25.8 | 44.6 | 34.2 | 22.9 | |||
| 8 | — | — | — | 28.1 | 24.6 | 21.9 | |||
| Silthiopham | 92.3 | 66.7 | 40.5 | — | — | — | |||
| Prothioconazole | 100 | 97.3 | 94.1 | 100 | 100 | 100 | |||
| a “—” No detection. | |||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们